

Abstract
Telomerase – an enzyme that endows cells with unlimited proliferative potential – is differentially expressed in cancer cells and in normal cells. Although most primary human cells lack telomerase, the enzyme is upregulated in more than 90% of invasive breast cancers. As a result, much of breast cancer development occurs before telomerase is reactivated during a critical transition from a telomerase-negative to a telomerase-positive state. During this transition, the telomere shortening that accompanies cell division may either prevent or facilitate tumorigenesis by activating checkpoints and impairing chromosomal stability. In mature cancers, telomerase probably serves a crucial role in tumor progression and maintenance by stabilizing telomeres and supporting the immortal growth of breast cancer cells.
Keywords: breast cancer, genomic instability, mouse model, telomerase, telomere
Introduction
Telomeres serve as nucleoprotein caps that protect the ends of linear eukaryotic chromosomes. They are comprised of nucleotide repeats (TTAGGG in mammals) and are bound by a complex array of proteins that stabilize formation of a looped and protected chromosome end [1]. This highly packaged telomere structure (t-loop) serves two critical functions: it sequesters the chromosome end from cellular machinery that might mistake it for a broken chromosome and cause checkpoint activation; and it prevents access of recombination enzymes that can physically join chromosomes together. In this way, intact telomere structure is required for continued cell proliferation and viability, and is critical for maintaining chromosomal stability. As cells divide in the absence of telomerase – the enzyme that synthesizes telomere repeats – telomeres shorten progressively because DNA polymerase cannot fully replicate the extreme ends of chromosomes.
In primary human fibroblasts, progressive telomere shortening associated with proliferation in culture leads to two fundamentally different responses. First, after 60–80 cell divisions fibroblasts encounter replicative senescence, a state of permanent growth arrest and altered morphology that requires intact p53 and Rb (retinoblastoma susceptibility gene product) tumor suppressor pathways. Inactivation of both p53 and Rb with viral oncoproteins or antisense strategies allows cells to bypass senescence and, after extended proliferation, enter crisis. Crisis is characterized by widespread apoptosis and chromosomal instability as the full impact of critical telomere shortening and chromosomal end-to-end fusions are seen [2]. Ectopic expression of telomerase reverse transcriptase (TERT), the protein catalytic subunit of telomerase, can reconstitute telomerase activity, lengthen telomeres, and bypass both senescence and crisis, endowing human fibroblasts with immortal proliferative properties [3].
Human mammary epithelial cells enter crisis after p16INK4A loss
Primary human mammary epithelial cells (HMECs) and some other epithelial cells do not abide by this simple two-step paradigm. HMECs experience premature growth arrest after a much smaller number of cell divisions than is necessary for replicative senescence. Unlike replicative senescence, this growth arrest is not bypassed by ectopic TERT expression, indicating that telomere shortening is not the trigger for this arrest. Selection for HMEC clones that escape from this arrest is associated with epigenetic changes, including reduced p16INK4A expression, and therefore compromise of the Rb pathway [4]. After epigenetic silencing of p16INK4A, HMECs proliferate for an additional 30–50 population doublings before encountering a telomere-based barrier to continued cell division. This latter growth arrest has features of both replicative senescence and crisis; HMECs exhibit a senescent morphology, but show elevated rates of apoptosis and evidence for chromosomal instability [5]. That this barrier is telomere-based is evidenced by the formation of chromosome end-to-end fusions and the observation that ectopic TERT expression immortalizes HMECs in this phase and prevents onset of growth arrest [6].
It remains unclear whether the behavior of HMECs in arresting prematurely and entering crisis after epigenetic loss of p16INK4A is indicative of how these cells behave in vivo or instead reflects growth conditions in culture [5,7]. Careful analyses of growth properties of human fibroblasts and HMECs have shown that continued proliferation after checkpoint inactivation (via T antigen expression and p16INK4A loss, respectively) results in eventual loss of telomere capping function and impaired chromosomal stability. Telomeres that become so short that they can no longer protect the chromosome end are said to be critically short or dysfunctional. Experiments in telomerase-deficient mice have shown that such unchecked telomere dysfunction can drive breast carcinogenesis.
Telomere-based crisis promotes mammary carcinogenesis in telomerase-deficient mice
Telomerase is comprised of a heterodimer of TERT and telomerase RNA component (TERC), a required RNA encoding the telomere sequence that is reverse transcribed during telomere synthesis. Inactivation of mouse TERC (mTERC) via homologous recombination in embryonic stem cells resulted in viable mTERC-/- mice that lacked telomerase activity, but were phenotypically normal because they retained long telomeres [8]. Telomeres in mice are much longer than telomeres in humans (40–60 kb versus 5–15 kb). By interbreeding mTERC-/- mice for multiple generations, telomeres shortened progressively and became critically short in generations four to six. In these late generations, mTERC-/- mice became infertile and showed proliferative defects and increased apoptosis in organ systems with high turnover [9]. These defects in cell proliferation and survival occur when telomere uncapping triggers activation of the tumor suppressor p53, which responds to a variety of cellular stresses, including DNA damage, oncogene activation, and hypoxia. Loss of p53 in late-generation mTERC-/- mice blunted the apoptotic response to telomere dysfunction and improved cell survival [10]. Thus, the integrity of the p53 checkpoint is a crucial determinant of the cellular response to telomere dysfunction. Late-generation telomerase-deficient mice can serve as a useful in vivo model to explore the impact of telomere attrition on tumorigenesis.
Late-generation mTERC-/- mice with intact p53 pathways are highly tumor resistant, which is consistent with predictions based on observations of senescence/crisis responses in primary human cells that telomere shortening would suppress tumorigenesis. Telomere uncapping in late-generation mTERC-/- p53+/+ mice resulted in impaired tumor formation when these mice were treated with chemical carcinogens [11], or studied on an INK4A-/-[12] or APC+/-[13] tumor-prone background. To determine the impact of dysfunctional telomeres unchecked by p53 activation on tumorigenesis in vivo, compound mTERC-/- p53-deficient mice were generated and monitored for spontaneous tumor development. In contrast to the models with intact p53-dependent DNA damage response pathways, telomere uncapping in late-generation mTERC-/- p53-/- or p53+/- mice significantly accelerated the rate of tumor formation [14]. In addition to accelerating tumorigenesis, telomere dysfunction dramatically altered the spectrum of tumor types in p53+/- mice. Whereas p53+/- mice with intact telomeres developed predominantly lymphomas and sarcomas, late-generation mTERC-/- p53+/- mice succumbed primarily to epithelial cancers, including carcinomas of the breast, skin, and gastrointestinal tract. Therefore, telomere-based crisis (i.e. telomere shortening that proceeds unchecked by p53 activation) can promote spontaneous transformation in mammary tissue and other epithelial compartments.
Telomere dysfunction, translocations and copy number changes
To determine the mechanism by which unchecked telomere uncapping caused murine breast cancers, chromosome structure was analyzed in breast carcinomas from late-generation mTERC-/- p53+/- mice. Spectral karyotype analysis revealed that telomere shortening promoted formation of translocations between nonhomologous chromosomes. These translocations were of the nonreciprocal type and were not seen in cancers from p53-deficient mice with long telomeres [14]. Unlike classical balanced translocations, nonreciprocal translocations (NRTs) lead to copy number changes in the involved chromosomal regions. Comparative genome hybridization (CGH) analysis of breast cancers and other carcinomas from late-generation mTERC-/- p53+/- mice revealed widespread regions of amplification or deletion, significantly altering gene copy number [15]. These regions presumably harbor important oncogenes and tumor suppressor genes. Dysfunctional telomeres promote these genomic alterations through cycles of chromosomal fusion–bridge–breakage. Telomere uncapping results in exposure of the chromosome end to recombination machinery, generating an end-to-end fusion. The presence of two centromeres renders these dicentric chromosomes unstable, because each centromere can attach to opposite poles of the mitotic spindle during mitosis. The dicentric chromosome is stretched and broken during anaphase, and these broken chromosomes can generate NRTs or a new dicentric chromosome, which can fuel another round of fusion–bridge–breakage. Therefore unrestricted telomere dysfunction in p53-deficient cells promotes cancer in part by generating NRTs and widespread gene copy number changes.
Genomic alterations and telomere dynamics in human breast cancer
These genomic alterations seen in breast cancers driven by telomere dysfunction closely resemble those seen in human breast cancers. Human breast cancers, as well as other carcinomas, harbor NRTs and gene copy number changes. Both known oncogenes and novel oncogenes have been identified in regions of amplification identified by CGH. Chromosomes become destabilized very early in human breast cancer development. Copy number changes are evident in usual ductal hyperplasia and atypical ductal hyperplasia by CGH [16,17]. These aberrations become more abundant by the ductal carcinoma in situ (DCIS) stage, but then appear similar in number in invasive breast cancers. In fact, these clonal gene copy number changes are shared to a large extent among DCIS, invasive cancer, and metastatic lesions in the same patient [18,19]. These data suggest that chromosomal instability is impaired early in tumorigenesis, but only temporarily because the genome appears much more stable during progression. How the breast cancer genome becomes destabilized early in tumorigenesis and then restabilized after the DCIS stage remains unknown. One possible explanation is that incipient cancers, dividing before telomerase reactivation, sustain significant telomere shortening and circumvent the p53 checkpoint, leading to cycles of chromosomal fusion–bridge–breakage that fuel chromosomal instability and carcinogenesis (Fig. 1). In support of this hypothesis, telomeres shorten during carcinogenesis and then become stabilized at a shorter length in invasive breast cancers [20].
Figure 1.
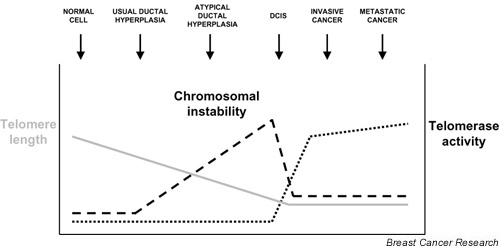
Telomere dynamics and chromosomal instability during human breast cancer development. Telomeres shorten during human tumorigenesis (gray line) and then become stabilized at the ductal carcinoma in situ (DCIS) or invasive stage. Telomerase is expressed at low or undetectable levels in normal cells, but becomes activated in 80–90% of DCIS lesions and remains highly active in invasive and metastatic breast cancers (dotted black line). Comparative genome hybridization analyses have shown that chromosomal imbalances or gene copy number changes begin early in breast cancer, in usual ductal hyperplasia and atypical ductal hyperplasia. Copy number aberrations increase in DCIS and remain similar in number in invasive cancer and metastatic cancer. Chromosomal instability (dashed black line) therefore begins in usual ductal hyperplasia and increases through DCIS development. The rate of accumulation of chromosomal aberrations slows significantly at the DCIS stage and this stabilization of the breast cancer genome coincides temporally with reactivation of telomerase.
Although most human breast cancers may harbor chromosomal rearrangements, not all of these cancers contain p53 mutations. In fact, only approximately 30% human breast cancers have shown p53 mutations, suggesting that gene copy number changes can accumulate in p53 wild-type cancer cells. These data suggest that mutations in other components of the p53 pathway, or in separate pathways that control DNA damage checkpoints, may create a permissive environment for chromosomal rearrangements. Further work will be necessary to determine what role telomere dysfunction plays in formation of chromosomal rearrangements in human breast cancer and what other mechanisms might contribute to destabilizing the breast cancer genome.
Two distinct roles for telomerase reactivation in cancer
Whereas normal mammary tissue lacks significant telomerase activity, telomerase is expressed in 80–90% of DCIS lesions and in 90% of invasive breast cancers [21]. Telomerase reactivation therefore coincides temporally with the relative stabilization of the cancer genome. If telomere dysfunction aids in formation of genomic rearrangements that facilitate cancer formation, then telomerase reactivation later in the course of carcinogenesis probably favors tumor progression. Re-establishing telomere maintenance is probably necessary to stabilize telomeres, to 'lock in' genomic alterations that are necessary for tumorigenesis, and to allow significant clonal expansion. Can telomerase reactivation serve other functions beyond re-establishing telomere maintenance? Experiments designed to address this question have yielded mixed results. In primary human fibroblasts and retinal pigment epithelial cells, telomerase reconstitution led to immortal proliferation but did not alter normal regulatory and checkpoint circuits [22,23]. In contrast, ectopic expression of telomerase in HMECs that have epigenetically silenced p16INK4A resulted in decreased sensitivity to the inhibitory effects of transforming growth factor-β [24]. Transgenic models of TERT overexpression in wild-type mice with long telomeres suggest that telomerase can encourage neoplastic growth even in the absence of critically short telomeres. Expression of TERT in a broad variety of mouse tissues resulted in spontaneous breast cancers at long latency [25]. Directing TERT expression to mouse keratinocytes rendered them more sensitive to the transforming effects of chemical carcinogens [26]. These data suggest that telomerase may have two functions: one in stabilizing short telomeres and averting the deleterious consequences of telomere dysfunction for proliferation and survival; and a second activity that promotes proliferation independent of telomere length.
Conclusion
Primary human fibroblasts and HMECs in culture readily enter telomere-based crisis when p53 and/or Rb checkpoints are impaired. The chromosomal instability associated with unchecked telomere dysfunction can drive breast carcinogenesis in vivo in telomerase-deficient mice, suggesting that telomere-based crisis in humans may similarly enhance rates of epithelial tumorigenesis. This mechanism may underlie the formation of common chromosomal aberrations and copy number changes seen in human breast cancers. Telomerase reactivation serves to restore telomere function, reduce chromosomal instability, and promote tumor maturation. Emerging evidence indicates that telomerase probably serves additional functions in promoting tumorigenesis. Telomerase inhibition remains a promising target for pharmacologic inhibition in the treatment of human malignancies.
Abbreviations
CGH = comparative genome hybridization; DCIS = ductal carcinoma in situ; HMEC = human mammary epithelial cell; NRT = nonreciprocal translocation; (m)TERC = (mouse) telomerase RNA component; Rb = retinoblastoma susceptibility gene product; TERT = telomerase reverse transcriptase.
Acknowledgments
Acknowledgements
SEA is supported by NIH KO8CA82176 and is a V Foundation Scholar and a Rita Allen Scholar.
References
- Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Weinberg RA. Senescence: does it all happen at the ends? Oncogene. 2002;21:627–630. doi: 10.1038/sj.onc.1205062. [DOI] [PubMed] [Google Scholar]
- Foster SA, Wong DJ, Barrett MT, Galloway DA. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol. 1998;18:1793–1801. doi: 10.1128/mcb.18.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature. 2001;409:633–637. doi: 10.1038/35054579. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway D, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, Wright WE. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001;15:398–403. doi: 10.1101/gad.859201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/S0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Samper E, Flores JM, Blasco MA. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet. 2000;26:114–117. doi: 10.1038/79089. [DOI] [PubMed] [Google Scholar]
- Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- O'Hagan RC, Chang S, Maser RS, Mohan R, Artandi SE, Chin L, DePinho RA. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell. 2002;2:149–155. doi: 10.1016/S1535-6108(02)00094-6. [DOI] [PubMed] [Google Scholar]
- Aubele M, Cummings M, Walsch A, Zitzelsberger H, Nahrig J, Hofler H, Werner M. Heterogeneous chromosomal aberrations in intraductal breast lesions adjacent to invasive carcinoma. Anal Cell Pathology. 2000;20:17–24. doi: 10.1155/2000/930246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong G, DeVries S, Chew KL, Cha I, Ljung BM, Waldman FM. Genetic changes in paired atypical and usual ductal hyperplasia of the breast by comparative genomic hybridization. Clin Cancer Res. 2001;7:2410–2414. [PubMed] [Google Scholar]
- Buerger H, Otterbach F, Simon R, Poremba C, Diallo R, Decker T, Riethdorf L, Brinkschmidt C, Dockhorn-Dworniczak B, Boecker W. Comparative genomic hybridization of ductal carcinoma in situ of the breast-evidence of multiple genetic pathways. J Pathol. 1999;187:396–402. doi: 10.1002/(SICI)1096-9896(199903)187:4<396::AID-PATH286>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, DeVries S, Chew K, Goodson WH, III, Ljung BM, Thor A, Waldman FM. Genetic alterations in primary breast cancers and their metastases: direct comparison using modified comparative genomic hybridization. Genes Chromosomes Cancer. 1997;19:267–272. doi: 10.1002/(SICI)1098-2264(199708)19:4<267::AID-GCC9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Odagiri E, Kanada N, Jibiki K, Demura R, Aikawa E, Demura H. Reduction of telomeric length and c-erbB-2 gene amplification in human breast cancer, fibroadenoma, and gynecomastia. Relationship to histologic grade and clinical parameters. Cancer. 1994;73:2978–2984. doi: 10.1002/1097-0142(19940615)73:12<2978::aid-cncr2820731215>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AB, Wahl GM, Tlsty TD, Chiu CP. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Garbe J, Levine G, Lichtsteiner S, Vasserot AP, Yaswen P. Expression of the telomerase catalytic subunit, hTERT, induces resistance to transforming growth factor beta growth inhibition in p16INK4A(-) human mammary epithelial cells. Proc Natl Acad Sci USA. 2001;98:4498–4503. doi: 10.1073/pnas.071483998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, Alson S, Tietze MK, Sharpless N, Ye S, Greenberg R, Castrillion D, Horner J, Weiler S, Carrasco R, DePinho RA. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci USA. 2002;99:8191–8196. doi: 10.1073/pnas.112515399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Suarez E, Samper E, Ramirez A, Flores JM, Martin-Caballero J, Jorcano JL, Blasco MA. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001;20:2619–2630. doi: 10.1093/emboj/20.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]