

Abstract
Mutations and deletions that result in the stabilization of β-catenin are frequently found in a number of tumors, including those of the colon, the liver and the ovary, but are less frequently found in breast cancer. To investigate and understand the molecular nature of cell-specific β-catenin signaling, experimental mouse genetics has been employed extensively. Gain-of-function and loss-of-function mutations have provided evidence that β-catenin plays essential roles in development and tumorigenesis. Specifically, the Wnt/β-catenin signaling pathway controls cell fate decisions throughout development, and a unique role in differentiated epithelia has emerged. Not only β-catenin, but also the activation of other components of this pathway in differentiated mammary epithelium and prostate epithelium of transgenic mice can induce neoplasias and transdifferentiation to squamous metaplasias. This suggests that the Wnt/β-catenin pathway is dominant over existing differentiation programs and can impose an epidermal fate or neoplasias onto a variety of cell types. Although there is evidence for a contextual specificity of the Wnt signaling, the experimental systems and designs used in different studies probably influence the cellular responses.
Keywords: β-catenin, cell specification, mammary gland, transdifferentiation
Setting the stage
β-Catenin was originally identified as a component of cell–cell adhesion complexes that are composed of cadherins, α-catenin and β-catenin, and actin. It became clear during the past decade that β-catenin is also a downstream signaling molecule in the Wnt signaling pathway and it controls transcription in concert with TCF/LEF proteins (reviewed in [1]). Derailing this signaling pathway can result in developmental defects or in the neoplastic transformation of cells (Tables 1 and 2). For example, overexpression of β-catenin induces an additional embryonic axis in Xenopus laevis [2]. Also, ablation of the β-catenin gene in the mouse resulted in a lack of mesoderm, a lack of embryonic lethality [3] and an absence and/or mislocation of anterior markers at embryonic day 5.5 [4]. In brain and craniofacial development, β-catenin-null neural crest cells fail to form the connecting parts between the cranial ganglia and the hindbrain, and also fail to form cranial skeletal structures [5].
Table 1.
Gain-of-function of β-catenin signaling
| Organ | Phenotype | References |
| Intestine | Polyps | Harada et al. [17] |
| Skin | Development of extra hair follicles, hair tumor | Gat et al. [33] |
| Mammary gland | Adenocarcinomas | Imbert et al. [10] |
| Michaelson and Leder [9] | ||
| Transdifferentiation, squamous metaplasias | Miyoshi et al. [16,18] | |
| Liver | No neoplasias | Harada et al. [40] |
| Prostate | Epithelial neoplasias | Gounari et al. [39] |
| Squamous transdifferentiation | Unpublished data | |
| Salivery gland | Squamous metaplasias | Gounari et al. [39] |
| Harderian gland | Squamous metaplasias | |
| Tooth | Odontoma |
Table 2.
Loss-of-function of β-catenin signaling
| Organ | Phenotype | Reference |
| Mesoderm patterning | No mesoderm, no head structure | Haegel et al. [3] |
| Huelsken et al. [4] | ||
| Body axis | Absence and/or mislocation of anterior markers at embryonic day 5.5 | Huelsken et al. [4] |
| Skin | Stem cells fail to differentiate into follicular keratinocytes, but adopt an epidermal fate | Heulsken et al. [35] |
| Brain and craniofacial development | Brain malformation and failure of craniofacial development | Brault et al. [5] |
These experiments demonstrated that β-catenin constitutes an essential switch in the establishment of the vertebrate body plan. On the other hand, gain-of-function mutations within β-catenin that generate stabilized β-catenin are well known in cancers of the colon, the liver, the ovary, and the endometrium (reviewed in [6]). Although stabilizing mutations in β-catenin are rarely observed in breast cancer [7], 40% of breast cancers overexpress cyclin D1 and almost of all of them (92%) also had high levels of active β-catenin [8]. This suggests that elevated β-catenin levels correlate with mammary tumorigenesis.
Two distinct approaches of experimental mouse genetics have been used to explore the specific contributions of β-catenin in the transformation of mammary epithelium. These experiments unveiled two faces of β-catenin: its ability to elicit adenocarcinomas, and its ability to induce transdifferentiation into squamous metaplasias (Table 1). The ability of β-catenin to induce different reactions of cells might be the result of its concentration, the time of its activation and the cellular context.
Models on the stage
The role of β-catenin has been investigated using two distinct approaches of experimental mouse genetics. In the first set of experiments, transgenes encoding stabilized β-catenins were expressed under control of the mouse mammary tumor virus-long terminal repeat (MMTV-LTR), a control element that is active in a variety of epithelial and hematopoietic cells. This approach provided information on the cell-specific function of β-catenin when expressed ectopically. The second approach relied on the Cre-loxP recombination system to establish mice in which the endogenous β-catenin gene was altered to produce a stabilized/activated protein. This experimental design ensures that the expression of the stabilized β-catenin remains under the control of its own genetic control elements.
Activated β-catenin can induce adenocarcinomas in mouse mammary tissue
Two studies have demonstrated that the expression of a gene, encoding an N-terminal truncated β-catenin, under the control of an MMTV-LTR induces adenocarcinomas in the mammary gland [9,10]. These investigators expressed β-catenin molecules from which either 89 (ΔN89β-catenin) or 90 (ΔN90β-catenin) N-terminal amino acids had been deleted, which resulted in their stabilization and activation.
In a high expressing ΔN89β-catenin line, all females developed adenocarcinomas of the breast within approximately 4 months of age [10]. In MMTV-ΔN90β-catenin mice [9], the emerging hyperplasias and adenocarcinomas appeared to be similar to those observed in mice ectopically expressing Wnt-1 and Wnt-10b under a similar MMTV-LTR [11,12]. Adenocarcinomas of the breast were detected after 4 months of age but the progression appeared to be slower. This could be the result of strain differences or expression levels at early stages. Both groups concluded that Wnt-induced mammary tumorigenesis is mediated by β-catenin, which directly targets and activates the expression of the cyclinD1 and c-myc genes. Furthermore, this suggests that the β-catenin-cyclin D1/c-myc signaling pathway is common to colon tissue [13-15] and to mammary tissue in its ability to induce neoplasias. Interestingly, ΔN89β-catenin also induced precocious lobuloalveolar development and differentiation in males and in virgin females, which resembled a developmental state normally seen during pregnancy [10]. This obviously raises another curtain and portrays β-catenin as a molecule that can alter the differentiation state of a cell.
Activated β-catenin can alter the fate of mammary epithelium
In the second genetic approach to investigate the role of activated β-catenin in mammary epithelium, the endogenous gene was altered using the Cre-loxP recombination system to produce a stabilized protein [16]. Exon 3 of the endogenous β-catenin gene, which encodes amino acids 5–80, was flanked by loxP sites (ΔExon3 β-catenin mice) [17]. Two distinct transgenic mice expressing Cre recombinase were used for the deletion of exon 3, which results in the translation of a stabilized β-catenin. While Cre expression in whey acidic protein-Cre (WAP-Cre) transgenic mice occurs in differentiating mammary epithelium at around mid pregnancy, expression in MMTV-Cre transgenic mice was already observed in ductal epithelium prior to puberty.
Although it was predictable that these mice should have developed the same lesions as observed in the transgenic mice described earlier, the outcome was surprisingly different. No adenocarcinomas were observed within 6 months after stabilizing β-catenin in ΔExon3 β-catenin mice [16]. Instead, extensive hyperplasias and metaplasias were detected at very early stages. Although the WAP gene promoter is activated during the estrus cycle, this did not result in the establishment of prominent lesions. Around days 10–12 of pregnancy, the WAP-Cre transgene is efficiently activated, and hyperplasias and squamous metaplasias were observed at day 15 of pregnancy. The histological features included ghost cells and fully keratinized structures, which are reminiscent of epidermal differentiation.
The molecular character of these cells was established with immunohistochemical markers, and it was determined that mammary secretory cells expressing stabilized β-catenin lose their differentiation and acquire the fate of epidermis. Markers predictive of mammary epithelium, such as NKCC1 and Npt2b, were lost and indicators of early epidermal differentiation, such as cytokeratin1, were acquired. This suggested that mammary epithelium has a latent program in place to alter its fate to epidermal cells, which can be triggered by β-catenin.
Why does activated β-catenin induce two different phenotypes?
On the face of it, the expression of a stabilized β-catenin under the control of a transgenic promoter or its own promoter should induce similar physiological consequences; after all, it is the same molecule (Fig. 1). It can especially be predicted that ΔExon3 β-catenin and ΔN89β-catenin/ΔN90β-catenin are expressed in the same cell type; namely, the secretory epithelium and not the myoepithelium.
Figure 1.
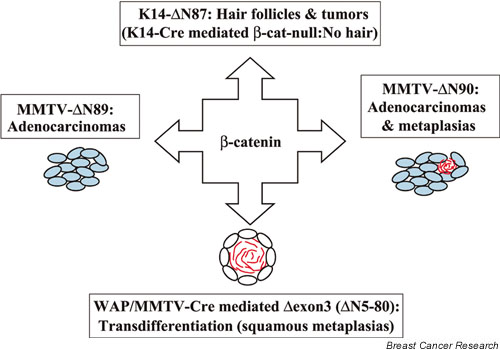
Activated β-catenin altered the fate of epithelial cells. In skin, activated β-catenin induced hair follicles and tumors. In mammary epithelia, activated β-catenin could induce adenocarcinomas (MMTV-ΔN89), squamous metaplasias (WAP/MMTV-ΔExon3) or both (MMTV-ΔN90). MMTV, mouse mammary tumor virus; WAP, whey acidic protein.
However, there are some fundamental differences in the experimental design. First, the amount of β-catenin will be very different because the respective genes were controlled by different promoters. The ΔN89β-catenin and ΔN90β-catenin transgenes were under control of a MMTV-LTR and their expression most probably exceeded that of the endogenous gene. Even the low expressing line with one copy of the transgene developed adenocarcinomas, although with a longer delay. On the other hand, the ΔExon3 β-catenin gene is under control of its endogenous promoter. The accumulated amounts of ΔExon3 β-catenin may be sufficient to induce metaplasias but be insufficient to cause adenocarcinomas.
However, expression levels may not be the only reason for the observed differences. While the deletion of exon 3 results in the loss of amino acids 5–80, the ΔN89β-catenin/ΔN90β-catenin mutants lose an additional 15 amino acids. It is possible that these additional amino acids could contribute to the observed changes (i.e. their loss could contribute to the formation of adenocarcinomas). Although the nature of this difference is not known, we can hypothesize that the two β-catenins exhibit different interactions with other signaling molecules.
Other transgenic mouse models with an activated canonical Wnt signaling pathway
It is established that β-catenin is a central molecule of the Wnt signaling pathway, and a number of transgenic mice have been produced in which different arms of this pathway have been activated/repressed. A comprehensive study using these mice established that both adenocarcinomas and epidermal transdifferentiation can be induced [18,19]. Clearly, the disruption of the Wnt signaling pathway at any level (Wnt-10b, dominant-negative glycogen synthase kinase 3β [GSK3β], ΔExon3 β-catenin, adenomatous polyposis coli [APC] and cyclin D1) [18,19] can induce hyperplasias, adenocarcinomas and squamous metaplasias in mammary tissue (Fig. 2).
Figure 2.
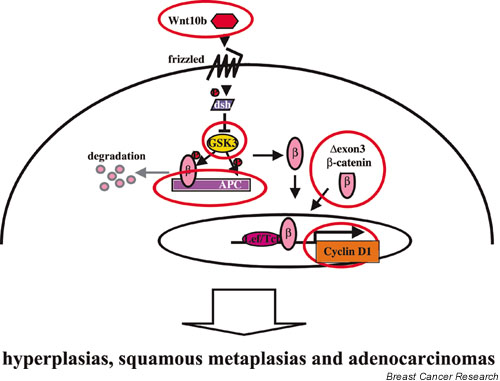
Canonic Wnt signaling pathway molecules can induce a similar phenotype. When each molecule in the canonic Wnt signaling pathway was activated, they could induce hyperplasias, adenocarcinomas and squamous metaplasias. GSK3, dominant negative glycogen synthase kinase 3β; APC, adenomatous polyposis coli; P, phosphorylated form; dsh, dishevelled.
APC is a key molecule in the degradation pathway of β-catenin, and loss of APC function leads to the stabilization of β-catenin [20,21]. Gallagher and colleagues recently established mice in which the APC gene had been inactivated specifically in mammary epithelium using the Cre-loxP recombination system [22]. Exon 14 of the Apc gene was deleted by a Cre transgene under the control of the ovine β-lactoglobulin gene promoter. The observations from this study are directly relevant to the ΔExon3 and ΔN89/ΔN90 β-catenin mouse models [9,10,16,22]. Inactivation of Apc led to the development of metaplasias with concurrent β-catenin activation. No adenocarcinomas were observed. Mice that lack both Apc and Tcf-1 develop acanthoma between 3.5 and 8 weeks of age [22], which display high levels of cytoplasmic and nuclear β-catenin. These results demonstrate synergism between Apc and Tcf-1, and the combined loss leads to distinct neoplastic lesions. Furthermore, they support a previous notion on the threshold levels of β-catenin and a contextual dependency of β-catenin function. The frequency and degree of metaplasias were very different in the various transgenic models, but a common denominator (with the exception of cyclin D1 transgenic mice) was the accumulation of β-catenin.
Other actors supporting β-catenin activation
Besides the canonical Wnt signaling pathway, many players cooperate in the activation of β-catenin. For example, the epidermal growth factor receptor, erbB-2, c-met and Ron [23-26] can trigger tyrosine phosphorylation of β-catenin and can cause its nuclear translocation. Precenilin1 is known as a cause of early onset familial Alzheimer's disease, and it was suggested to be involved in the degradation machinery of β-catenin, similar to axin [27]. Notably, a protein kinase A–precenilin 1–GSK3 complex may be functionally equivalent to the casein kinase 1–axin–GSK3 complex [27,28]. Precenilin 1-null mice exhibit embryonic lethality, but a rescue with a specific transgene revealed skin lesions. In precenilin 1-null skin, β-catenin accumulated and was correlated with hyperplasia without evidence of malignancy [27,29]. Although inhibitor of nuclear factor kappa B kinases (IKKα and IKKβ) interacted with and phosphorylated β-catenin, these two kinases exhibit different effects for β-catenin signaling [30]. While IKKα increased β-catenin-dependent gene expression, IKKβ decreased it. Expression of casein kinase 2α (CK2α) under the control of a MMTV-LTR also induced adenocarcinomas and squamous metaplasias [18,19,31]. Although the role of CK2 in the Wnt signaling pathway has not been fully defined, it is known that CK2α, the catalytic subunit of CK2, and β-catenin are increased in mammary epithelia after the forced expression of Wnt-1 [32]. Precenilin 1-null cells still employ a casein kinase 1–axin–GSK3 pathway to degrade β-catenin, and CK2α transgenic mice displayed adenocarcinomas and squamous metaplasias. These results suggest that these molecules could be able to control the amount of β-catenin and therefore may explain the different phenotypes that were observed. It is further possible that these or other molecules differentially communicate with the various stabilized β-catenin molecules, and thus elicit distinct cellular responses.
β-Catenin and epidermis differentiation
What is the mechanistic basis underlying the transdifferentiation process of mammary epithelia into the epidermal lineage? Over the past few years β-catenin has emerged as a prominent executor that can define lineages in the skin. It had already been shown in 1998 that stabilized β-catenin can induce new hair follicles and trichofolliculoma-like tumors in skin [33]. Follicular (hair) and epidermal stem cells are located in the bulge region [34]. Huelsken and colleagues demonstrated that, in the absence of β-catenin, stem cells can differentiate into the epidermal lineage but not into the hair follicular lineage [35].
It is possible that β-catenin in stem cells cooperates with LEF-1 and/or TCF-3 to control cell fate decision [36]. Surprisingly, expression of ΔN Lef-1, which cannot bind to β-catenin, in skin caused the development of squamous epidermal cysts and skin tumors [37]. This observation was supported by experiments in which the stabilization of β-catenin lacking the C-terminal transactivation domain caused the promotion of hair fates in epidermis and an epidermal fate in hair cells [38]. Although β-catenin plays an important role in hair fate, the molecular signals causing the transdifferentiation process into squamous metaplasias remain elusive. Taken together, these experiments suggest a complex role for β-catenin in that it can alter cell fate during the establishment of a specific lineage, and that this feature is dependent on the cooperation of cell-specific factors.
Conclusion: the cell specificity of β-catenin signaling
Mutations in the β-catenin signaling pathway have been associated with a number of different cancers, notably that of the colon. No clear link between an activated β-catenin signaling pathway and breast cancer has been established. However, an activation of the Wnt/β-catenin pathway in mammary tissue of transgenic mice can result in neoplasias and adenocarcinomas, demonstrating that these cells can read and interpret the respective signals. In contrast, the development of squamous cysts in human disease could be the result of β-catenin signaling as has been observed in transgenic mice.
Expression of stabilized β-catenin has been targeted to many organs in the mouse, and a cell-specific response is now appreciated. Clearly, cells from different germ layers respond to the signaling cues of activated β-catenin. Gounari and colleagues used the ΔExon3 β-catenin mouse model and stabilized the protein in mammary tissue, preputial gland, prostate, salivary gland, harderian gland, and tooth [39] (Table 1). Their experiments suggest that cells derived from both the endoderm and the ectoderm can differentiate into epidermis (except tooth). Stabilization of β-catenin in the intestine caused the development of polyps [17], and neoplasias were observed in the prostate [39]. Similarly, the activation in liver cells did not result in neoplastic foci and only caused hepatomegaly [40]. Taken together, the control of cell specification by β-catenin is not confined to the establishment of the embryonic body plan, but also to cells in the adult.
β-catenin performs different roles on many stages. A burning question focuses on the identity of the supporting actors that confer the specificity. Equally important, the nature of the downstream mediators and executors remains to be discovered. Willert and colleagues recently published an intriguing study from human teratocarcinoma cells that were cultured with Wnt-3A [41]. The 5-fold to 10-fold elevation of β-catenin levels resulted in the induction of approximately 50 genes, some of them having the ability to block or revert the differentiation [41]. As our understanding of the relevant signaling pathways deepens, we will understand how β-catenin can slip into so many costumes and adopt so many different roles.
Competing interests
None declared.
Abbreviations
APC = adenomatous polyposis coli; CK2 = casein kinase 2; GSK3 = glycogen synthase kinase 3; LTR = long terminal repeat; MMTV = mouse mammary tumor virus; WAP = whey acidic protein.
References
- Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997;11:3286–3305. doi: 10.1101/gad.11.24.3286. [DOI] [PubMed] [Google Scholar]
- Heasman J, Crawford A, Goldstone K, Garner-Hamrick P, Gumbiner B, McCrea P, Kintner C, Noro CY, Wylie C. Overexpression of cadherins and underexpression of beta-catenin inhibit dorsal mesoderm induction in early Xenopus embryos. Cell. 1994;79:791–803. doi: 10.1016/0092-8674(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R. Lack of beta-catenin affects mouse development at gastrulation. Development. 1995;121:3529–3537. doi: 10.1242/dev.121.11.3529. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior–posterior axis formation in mice. J Cell Biol. 2000;148:567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Candidus S, Bischoff P, Becker KF, Hofler H. No evidence for mutations in the alpha- and beta-catenin genes in human gastric and breast carcinomas. Cancer Res. 1996;56:49–52. [PubMed] [Google Scholar]
- Lin SY, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, Pestell RG, Hung MC. Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci USA. 2000;97:4262–4266. doi: 10.1073/pnas.060025397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson JS, Leder P. Beta-catenin is a downstream effector of Wnt-mediated tumorigenesis in the mammary gland. Oncogene. 2001;20:5093–5099. doi: 10.1038/sj.onc.1204586. [DOI] [PubMed] [Google Scholar]
- Imbert A, Eelkema R, Jordan S, Feiner H, Cowin P. Delta N89 beta-catenin induces precocious development, differentiation, and neoplasia in mammary gland. J Cell Biol. 2001;153:555–568. doi: 10.1083/jcb.153.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TF, Leder P. Wnt-10b directs hypermorphic development and transformation in mammary glands of male and female mice. Oncogene. 1997;15:2133–2144. doi: 10.1038/sj.onc.1201593. [DOI] [PubMed] [Google Scholar]
- Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55:619–625. doi: 10.1016/0092-8674(88)90220-6. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Shillingford JM, Le Provost F, Gounari F, Bronson R, von Boehmer H, Taketo MM, Cardiff RD, Hennighausen L, Khazaie K. Activation of beta-catenin signaling in differentiated mammary secretory cells induces transdifferentiation into epidermis and squamous metaplasias. Proc Natl Acad Sci USA. 2002;99:219–224. doi: 10.1073/pnas.012414099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999;18:5931–5942. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Rosner A, Nozawa M, Byrd C, Morgan F, Landesman-Bollag E, Xu X, Seldin DC, Schmidt EV, Taketo MM, Robinson GW, Cardiff RD, Hennighausen L. Activation of different Wnt/beta-catenin signaling components in mammary epithelium induces transdifferentiation and the formation of pilar tumors. Oncogene. 2002;21:5548–5556. doi: 10.1038/sj.onc.1205686. [DOI] [PubMed] [Google Scholar]
- Rosner A, Miyoshi K, Landesman-Bollag E, Xu X, Seldin DC, Moser AR, MacLeod CL, Shyamala G, Gillgrass AE, Cardiff RD. Pathway pathology: histological differences between ErbB/Ras and Wnt pathway transgenic mammary tumors. Am J Pathol. 2002;161:1087–1097. doi: 10.1016/S0002-9440(10)64269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin–Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Gallagher RC, Hay T, Meniel V, Naughton C, Anderson TJ, Shibata H, Ito M, Clevers H, Noda T, Sansom OJ, Mason JO, Clarke AR. Inactivation of Apc perturbs mammary development, but only directly results in acanthoma in the context of Tcf-1 deficiency. Oncogene. 2002;21:6446–6457. doi: 10.1038/sj.onc.1205892. [DOI] [PubMed] [Google Scholar]
- Wong NA, Pignatelli M. Beta-catenin – a linchpin in colorectal carcinogenesis? Am J Pathol. 2002;160:389–401. doi: 10.1016/s0002-9440(10)64856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Suzuki K, Tsukatani Y. Induction of tyrosine phosphorylation and association of beta-catenin with EGF receptor upon tryptic digestion of quiescent cells at confluence. Oncogene. 1997;15:71–78. doi: 10.1038/sj.onc.1201160. [DOI] [PubMed] [Google Scholar]
- Shibata T, Ochiai A, Kanai Y, Akimoto S, Gotoh M, Yasui N, Machinami R, Hirohashi S. Dominant negative inhibition of the association between beta-catenin and c-erbB-2 by N-terminally deleted beta-catenin suppresses the invasion and metastasis of cancer cells. Oncogene. 1996;13:883–889. [PubMed] [Google Scholar]
- Danilkovitch-Miagkova A, Miagkov A, Skeel A, Nakaigawa N, Zbar B, Leonard EJ. Oncogenic mutants of RON and MET receptor tyrosine kinases cause activation of the beta-catenin pathway. Mol Cell Biol. 2001;21:5857–5868. doi: 10.1128/MCB.21.17.5857-5868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Soriano S, Xia X, Eberhart C, De Strooper B, Zheng H, Koo E. Presenilin couples the paired phosphorylation of beta-catenin independent of axin. Implications for beta-catenin activation in tumorigenesis. Cell. 2002;110:751–762. doi: 10.1016/s0092-8674(02)00970-4. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10863–10868. doi: 10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti C, Lin KM, Yamamoto Y, Verma U, Verma IM, Byers S, Gaynor RB. Regulation of beta-catenin function by the IkappaB kinases. J Biol Chem. 2001;276:42276–42286. doi: 10.1074/jbc.M104227200. [DOI] [PubMed] [Google Scholar]
- Landesman-Bollag E, Romieu-Mourez R, Song DH, Sonenshein GE, Cardiff RD, Seldin DC. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene. 2001;20:3247–3257. doi: 10.1038/sj.onc.1204411. [DOI] [PubMed] [Google Scholar]
- Song DH, Sussman DJ, Seldin DC. Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells. J Biol Chem. 2000;275:23790–23797. doi: 10.1074/jbc.M909107199. [DOI] [PubMed] [Google Scholar]
- Gat U, DasGupta R, Degenstein L, Fuchs E. De novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell. 1998;95:605–614. doi: 10.1016/s0092-8674(00)81631-1. [DOI] [PubMed] [Google Scholar]
- Taylor G, Lehrer MS, Jensen PJ, Sun TT, Lavker RM. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell. 2000;102:451–461. doi: 10.1016/s0092-8674(00)00050-7. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. Beta-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/S0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- Merrill BJ, Gat U, DasGupta R, Fuchs E. Tcf3 and Lef1 regulate lineage differentiation of multipotent stem cells in skin. Genes Dev. 2001;15:1688–1705. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann C, Owens DM, Hulsken J, Birchmeier W, Watt FM. Expression of DeltaNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development. 2002;129:95–109. doi: 10.1242/dev.129.1.95. [DOI] [PubMed] [Google Scholar]
- DasGupta R, Rhee H, Fuchs E. A developmental conundrum: a stabilized form of beta-catenin lacking the transcriptional activation domain triggers features of hair cell fate in epidermal cells and epidermal cell fate in hair follicle cells. J Cell Biol. 2002;158:331–344. doi: 10.1083/jcb.200204134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounari F, Signoretti S, Bronson R, Klein L, Sellers WR, Kum J, Siermann A, Taketo MM, von Boehmer H, Khazaie K. Stabilization of beta-catenin induces lesions reminiscent of prostatic intraepithelial neoplasia, but terminal squamous transdifferentiation of other secretory epithelia. Oncogene. 2002;21:4099–4107. doi: 10.1038/sj.onc.1205562. [DOI] [PubMed] [Google Scholar]
- Harada N, Miyoshi H, Murai N, Oshima H, Tamai Y, Oshima M, Taketo MM. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer Res. 2002;62:1971–1977. [PubMed] [Google Scholar]
- Willert J, Epping M, Pollack J, Brown P, Nusse R. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev Biol. 2002;2:8. doi: 10.1186/1471-213X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]