

Abstract
Neutrophil migration protects the body against foreign invasion. Sequestration and activation of neutrophils, however, require stringent regulation because they may also cause tissue damage by the release of lysosomal enzymes and reactive oxygen species. The activity of various chemoattractants [e.g., leukotriene B4 (LTB4), interleukin-8, and complements] has been documented by in vitro assays, whereas in vivo data have been limited mostly to histology. To examine in an in vivo model the chemotactic activity and subsequent tissue infiltration and the role of a specific chemoattractant, LTB4, we used a rat renal ischemia-reperfusion injury model. Fluorescence-labeled Chinese hamster ovary (CHO) cells stably expressing the LTB4 receptor (CHO-BLT) were able to accumulate along with neutrophils in the postischemic kidney, in contrast to vector control CHO cells. Furthermore, LTB4 antagonists that protect against the decrease in renal function and diminish the tissue myeloperoxidase activity also led to the marked decrease in the number of CHO-BLT cells and neutrophils. Thus, LTB4 alone appears sufficient to cause cells to migrate into postischemic tissues, and its dominant role in reperfusion injury has been demonstrated. The utilization of transfectants to pinpoint the role of LTB4 in these in vivo experiments suggests their potential use with other ligands and/or in other pathological conditions.
During the blood reperfusion phase after tissue ischemia, tremendous additional tissue injury ensues as a consequence of neutrophil infiltration. Various chemokines and a lipid mediator, leukotriene B4 (LTB4), activate neutrophils, changing their shape and promoting their binding to endothelium by inducing cell-adhesion molecules. Once neutrophils migrate into the ischemic area, they release reactive oxygen species, proteases, elastase, myeloperoxidase (MPO), cytokines, and various other mediators (1, 2), all of which are involved in tissue injury. Activated neutrophils induce endothelial cell damage, resulting in impaired vascular dilatory responses, which further exacerbate ischemic tissue injury. Recently, LTB4 receptor (BLT)-transgenic mice were developed and subjected to lung ischemia-reperfusion injury. Compared with nontransgenic mice, the BLT-transgenic mice showed more than a 2.5-fold increase in polymorphonuclear neutrophil (PMN) infiltration into lung after ischemia reperfusion (3). Although there is evidence that inhibition of the extent of neutrophil infiltration is protective against tissue injury (4–6), the chemoattractant(s) responsible in ischemic acute renal failure (iARF) has yet to be identified.
The renal ischemic-reperfusion animal model is well studied (7) and has been valuable in examining the likely pathophysiological mechanisms underlying clinical settings, such as posttransplantation renal injury, acute renal failure after cardiopulmonary bypass, and renal involvement in multiple organ failure after hypovolemia. To determine whether LTB4 alone is capable of causing cells to infiltrate into the postischemic kidney, we infused BLT-transfected cells in this in vivo model and found that LTB4 appears to be the dominant early mediator in it. The use of an in vivo system as a chemotaxis chamber may provide a more physiological method for examining chemotactic mediators in tissue inflammation.
Materials and Methods
BLT Antagonists.
Two kinds of specific BLT antagonists were used in this study. ONO-4057 was generously donated by Ono Pharmaceutical, Osaka. U-75302 was purchased from Biomol (Plymouth Meeting, PA). For feeding purposes, ONO-4057 was mixed with rat chow at the concentration of 7 g/kg, which was continued for more than 4 wk before the start of the experiments to evaluate prefeeding efficacy. The concentration and duration of ONO-4057 feeding were deemed sufficient on the basis of a previously reported study from our laboratory (8).
Surgical Procedures.
All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals issued by the National Institutes of Health. Unilateral renal pedicle crossclamping for 45 min and contralateral nephrectomy were performed by using male Sprague–Dawley rats (150–180 g; Japan Clea Laboratory, Tokyo), as previously described (9). For the chronological evaluation of blood urea nitrogen (BUN) and serum creatinine (Cr), blood sampling was repeatedly performed via rat tail vein at the midpoint of the collection interval.
Chinese Hamster Ovarian (CHO) Cell in Vivo Infusion Protocol.
CHO-BLT cells were established as previously described (10). The cell migratory effect of CHO-BLT on other kinds of chemotactic factors (e.g., C5a, interleukin-8, and formyl-methionyl-leucyl-phenylalanine) was examined by migration assay (10), and no crossreactivity was found (data not shown). Twenty-four hours after surgery, an arterial line was placed in the left jugular artery, and both abdominal sutures and surgical staples were removed. The tip of the arterial line went as far as the supradiaphragm. CHO-BLT cells (1 × 107/ml) were incubated with 5 μM chloromethyl fluorescein diacetate (CMFDA; Molecular Probes) for 1 h at 37°C, lifted with 0.25% trypsin-EDTA and thoroughly washed with PBS. Then 2 × 106 cells in 1 ml of PBS were injected into the renal circulation over 2 min via the arterial line immediately after clamping the abdominal aorta below the left renal artery. After this injection, the abdominal arterial clamp was immediately released. The arterial line was flushed with 1 ml of heparinized saline. Sham-operated rats, in which one kidney was removed 24 h before the CHO-cell injection study, served as controls. After 2 h, the abdominal arterial clamp was again placed below the left renal artery, and a small incision was made in the inferior vena cava. The renal circuit was then thoroughly washed with 40 ml of PBS, subsequently infused with 20 ml of 4% paraformaldehyde in PBS, and the kidney was embedded in paraffin. A portion of harvested kidney was saved in 10% buffered formalin for paraffin section, and one 2-μm section was processed. CHO cells expressing FLAG-tagged BLT at the amino terminus, CHO-FLAG-BLT, were also generated and examined by using the same protocol as described above.
MPO Assay.
The MPO assay was performed as previously reported (11, 12) with minor modifications. Briefly, harvested kidneys were minced and homogenized in 3 ml of 0.5% hexadecyltrimethylammonium bromide in 50 mM potassium phosphate buffer, pH 6.0, by using 10 strokes of Dounce homogenizer on ice. After three freeze/thaw cycles, homogenates were centrifuged at 40,000 × g for 15 min, and the MPO activity of the supernatant was measured by incubating 20 μl of diluted sample with 180 μl of 50 mM potassium phosphate buffer, pH 6.0, containing 0.157 mg/ml o-dianisidine dihydrochloride (Sigma) and 0.0005% of hydrogen peroxide. Absorbance was measured at 460 nm (Spectra; Tecan). The MPO activity was quantified by comparison to dilutions of MPO (Calbiochem) and expressed as units per milligram with the adjustment by protein concentration measured by Bradford assay (Bio-Rad Protein Assay, Bio-Rad).
Estimation of Injected CHO Cell Population.
The number of infused CHO cells was set at 2 × 106 cells on the basis of the following assumptions. In a normal rat, neutrophil numbers are about 8,000/μl or 50% of white blood cells. The estimated circulating blood volume of a rat this size would be 5 ml, and the renal blood flow should be 20% in 1 min. In ischemia reperfusion, the neutrophil percent would be expected to increase about 50% or 4,000/μl. Thus, the estimated neutrophil increment in the renal circulation should be 4 × 106, and 50% of this increment was added by CHO cells in the present study.
Immunohistochemistry.
After routine deparaffinization and rehydration, kidney sections were preheated in 60°C citrate-buffered medium for 5 min by using a microwave and left at room temperature for another 20 min. The nonspecific biotin-binding sites of deparaffinized sections were blocked with a biotin-blocking system (Dako). Sections were incubated for 60 min with a primary monoclonal antibody, anti-FLAG M5 (Eastman Kodak), followed by the biotin-conjugated anti-mouse IgG and by alkaline phosphatase-labeled streptavidin (Dako), as previously described (13).
Neutrophil infiltration into the corticomedullary lesion was determined by polymer-conjugated antineutrophil monoclonal antibody EPOS NP57 (Dako), following a previously reported method (14) with minor modifications. Frozen sections were fixed by 4% paraformaldehyde for 30 min. After blocking the nonspecific tissue peroxidase by using 0.3% hydrogen peroxide for 15 min, sections were incubated with 1/10 diluted Block Ace (Yukijirushi, Sapporo, Japan) for 30 min. Sections were incubated overnight with EPOS NP57 at 4°C, followed by 3, 3′-diaminobenzidine tetrahydrochloride Liquid System (Dako) for 5 min. After washing in three changes of distilled water, sections were immersed at room temperature for 2 min in copper nitrate (Nakarai Chemical, Kyoto) solution (2.5 mg/ml). Sections were counterstained with methylene green solution (Wako Pure Chemical, Osaka). NP57-stained neutrophils were examined under ×40 objective and quantified as neutrophils per unit area (0.0625 mm2), following a previously reported method (15).
Morphologic Evaluation of Kidneys.
Formalin-fixed sections were stained with hematoxylin-eosin and scored as previously reported (13), by using well-established criteria (16).
Statistical Analysis.
The results are expressed as the mean ± SEM. The data in Table 1, neutrophil count, and morphologic evaluation were analyzed by one-way ANOVA with the Bonferroni procedure as a post hoc test for correction. The chronological analyses of both BUN and serum Cr were performed by using the repeated measures ANOVA with the Bonferroni test. A P-value less than 0.01 was considered significant.
Table 1.
BUN and serum Cr levels in rats subjected to 45 min of iARF: Efficacy of pretreatment with BLT antagonist ONO-4057
| BUN, mg/dl | Serum Cr, mg/dl | |
|---|---|---|
| Control (n = 7) | 12.7 ± 1.2 | 0.33 ± 0.04 |
| Sham operation (n = 7) | 13.2 ± 0.3 | 0.33 ± 0.02 |
| Ischemia reperfusion (n = 8) | 110.5 ± 10.0* | 2.55 ± 0.32* |
| ONO-4057 pretreatment + ischemia reperfusion† (n = 8) | 31.3 ± 1.4‡ | 0.73 ± 0.051‡ |
| Ischemia reperfusion + ONO-4057§ (n = 6) | 65.0 ± 6.8¶ | 1.15 ± 0.12¶ |
BUN and serum Cr were obtained on postoperative day 1. *P < 0.001 (sham operation vs. ischemia reperfusion).
ONO-4057 (7 g/kg rat chow) was fed for 4 wk.
P < 0.001 (ischemia reperfusion vs. ONO-4057 pretreatment + ischemia reperfusion).
ONO-4057 300 mg/kg was subcutaneously injected 15 min after the initiation of renal reperfusion.
P < 0.001 (ischemia reperfusion vs. ischemia reperfusion + ONO 4057) and P < 0.005 (ONO-4057 pretreatment + ischemia reperfusion vs. ischemia reperfusion + ONO-4057).
Results
Involvement of Neutrophils in Renal Ischemia-Reperfusion Injury.
In the present study, iARF was initiated by 45-min renal artery clamping, as previously described (9, 17). To determine renal function, blood samples were obtained immediately before the administration of CHO-BLT. Compared with sham-operated animals, postischemic rats had impaired renal function. BUN and serum Cr levels of iARF animals obtained 24 h after the clamp release were 110.5 ± 10.0 and 2.55 ± 0.32 mg/dl, respectively, whereas those of the control animals were 13.2 ± 0.3 and 0.33 ± 0.02 mg/dl, respectively (Table 1; P < 0.001; n = 8, vs. sham). As an indicator of neutrophil infiltration, the renal MPO activity was measured at 24 h postreperfusion in coded kidney tissue. The MPO activity of the whole kidney homogenate was markedly greater in the iARF group than that of the sham-operated group (Fig. 1a; P < 0.001). The number of NP57-positive neutrophils was significantly increased in the iARF group (Fig. 1b). Neutrophil infiltrates were also observed histologically at the peritubular interstitium of the outer medulla at higher magnification in the iARF animals (see below).
Figure 1.

(a) MPO activity of rat kidney homogenates obtained 1 day after 45 min of iARF. The number of animals in each group was six; asterisk denotes P < 0.001. (b) Number of neutrophils in corticomedullary lesion. Asterisk denotes P < 0.01 vs. sham group. The BLT antagonist, ONO-4057 (7 g/kg rat chow), was prefed for 4 wk (a and b).
In Vivo Cell Migration Assay.
An approach was used to show cell infiltration in vivo. CHO-BLT cells prelabeled with CMFDA were directly infused into the renal circulation of iARF rats 24 h after clamp release. Then, 2 h after CHO-BLT infusion, renal blood was thoroughly washed out, and the kidneys were removed. Prelabeled CHO-BLT cells were prominently present at the outer medulla in iARF kidneys, but not in sham-operated kidneys (Fig. 2a). CHO-BLT cells were also injected 2, 6, and 10 h after the clamp release. Cell infiltration was not observed at these earlier time points (data not shown). Mock-transfected CHO cells did not infiltrate to the interstitium of kidneys obtained from sham-operated or iARF animals, suggesting that the cell accumulation is a receptor-dependent process. This observation was further confirmed by the use of CHO-FLAG-BLT. In the ischemic kidney, CHO-FLAG-BLT cells immunostained with the M5 anti-FLAG antibody could also be detected at the peritubular space of the corticomedullary lesion. They were located between tubules (Fig. 3a). Such staining was not observed in sham-operated kidneys (Fig. 3b).
Figure 2.

Localization of CMFDA prelabeled CHO cells 2 h after renal injection on postoperative day 1 (a) and efficacy of BLT antagonist preincubation. (b) CHO-BLT cells were preincubated with either 100 μM ONO-4057 or 10 μM U-75302 for 15 min immediately before renal injection. The concentration of BLT antagonists was determined from the optimum concentrations used to inhibit cell migration in vitro, as previously reported (9). (c) Animals were prefed with rat chow containing ONO-4057 at the concentration of 7 g/kg for more than 4 wk.
Figure 3.
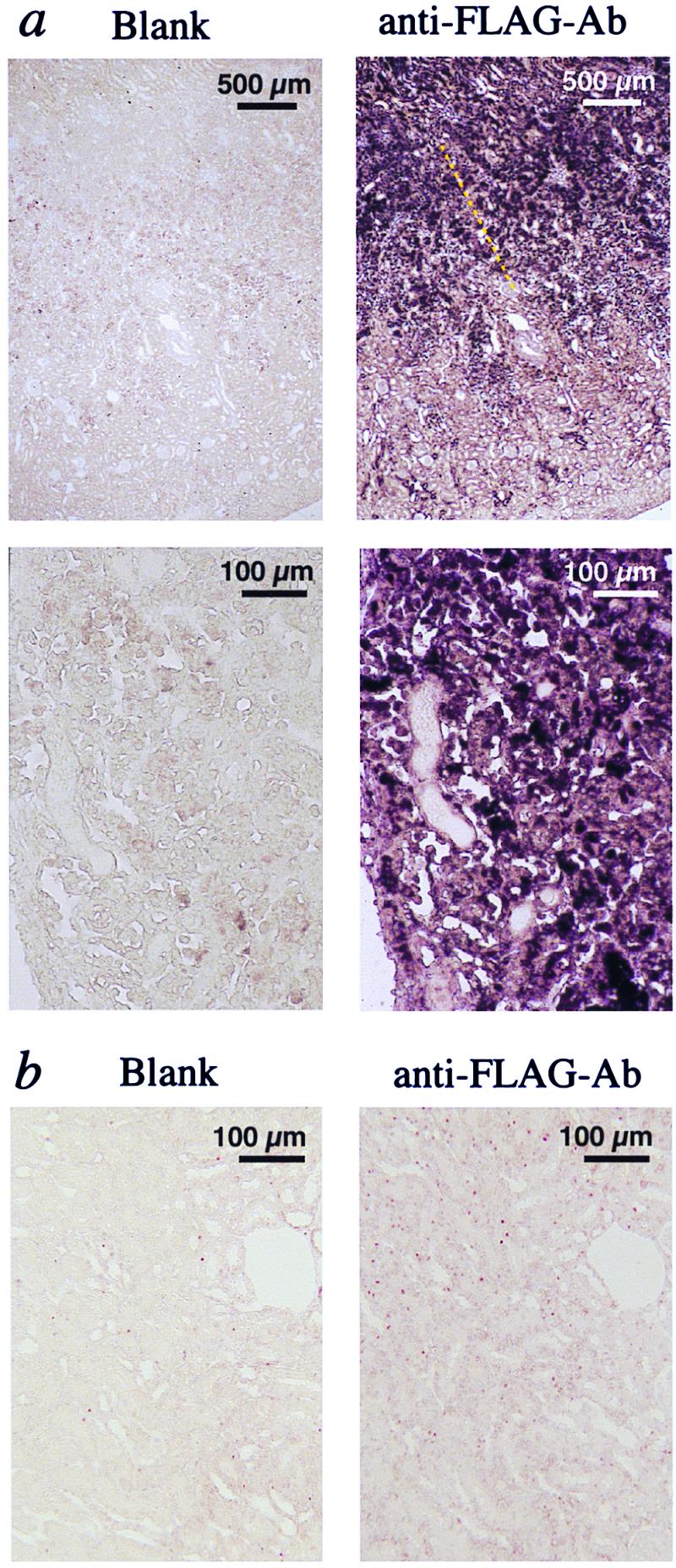
Immunohistochemical analysis. (a) Ischemic kidney injected with CHO-FLAG-BLT cells. (b) Sham-operated kidney obtained 2 h after CHO-FLAG-BLT renal injection.
Effect of BLT-Antagonist Pretreatment on CHO-BLT Infiltration.
Two series of experiments were performed to investigate further the LTB4-induced CHO-BLT cell infiltration. In the first experiment, CHO-BLT cells were preincubated with the BLT antagonists, either 100 μM ONO-4057 or 10 μM U-75302, for 15 min immediately before renal injection. As shown in Fig. 2b, the accumulation of CHO-BLT in iARF animals was markedly reduced by blocking the BLT receptors on the CHO cells, even in the presence of reduced renal function (Cr 2.0 mg/dl). Further, when animals were prefed with rat chow containing ONO-4057 for more than 4 wk, CHO-BLT infiltration was virtually abolished (Fig. 2c). BUN and serum Cr levels 24 h after reperfusion in ONO-4057 prefed animals were also examined (Table 1). BUN and serum Cr levels of iARF animals prefed with ONO-4057 (ONO-4057 pretreatment + ischemia reperfusion) were 31.3 ± 1.4 and 0.73 ± 0.05 mg/dl, respectively (P < 0.001; n = 8, vs. iARF). The renal MPO activity of these animals was remarkably diminished (iARF + ONO-4057; Fig. 1a). The NP57-stained neutrophil count was also significantly reduced in kidney obtained from ONO-4057 prefed rats compared with control ischemia (Fig. 1b).
Effect of BLT-Antagonist Injection after Renal Ischemia Reperfusion Injury.
In light of the above observations, we questioned whether BLT antagonism would be effective even if given after the renal ischemia-reperfusion injury. To address this concern, ONO-4057 at the concentration of 300 mg/kg was subcutaneously injected 15 min after the initiation of renal reperfusion injury with the release of clamping, and blood was drawn on postoperative day 1. BUN and serum Cr levels of this model (ischemia reperfusion + ONO-4057 in Table 1) were 65.0 ± 6.8 and 1.15 ± 0.12 mg/dl, respectively (P < 0.001; n = 6, vs. iARF; and P < 0.005; n = 6, vs. ONO-4057 pretreatment + ischemia reperfusion). Although BLT antagonism was less effective than prophylaxis, it was partially and still significantly effective in the preservation of renal function, even when used after the initiation of renal ischemia reperfusion. The chronological effectiveness of the BLT antagonist was evaluated further. ONO-4057 at the concentration of 300 mg/kg was subcutaneously injected 15 min after the initiation of renal reperfusion injury with the release of clamping, then followed by an additional injection of the same dosage at 24 h after the clamp release. Vehicle was injected to control ischemia and sham animals. In addition, ONO-4057 at the concentration of 7 g/kg was fed with rat chow in the ONO-4057 group after the clamp release. Fig. 4 summarizes the dynamics of Cr and BUN in the postischemic period. The group treated with ONO-4057 showed a significantly lesser retention of Cr and BUN (P < 0.01) compared with iARF group.
Figure 4.

Dynamics of serum Cr (S-Cr) and BUN in rats subjected to 45 min of renal ischemia. Closed circle, closed square, and open triangle denote sham group, iARF group, and iARF with ONO-4057 posttreated group, respectively. Number of animals in each group was six. B indicates “before the initiation of ischemia reperfusion injury.” Asterisk denotes P < 0.01 vs. iARF group.
Morphological Evaluation.
Morphological analysis of ischemia-reperfusion kidney obtained from ONO-4057 prefed rats (ONO-4057 + iARF; Fig. 5) demonstrated much milder tubular damage and fewer neutrophil infiltrates than did kidney from iARF animals. Histological scoring of injury to the nephron, by using criteria reported previously (16), was performed to elucidate the mode of protection afforded by BLT antagonism (Fig. 6). The major registered effects of ONO-4057 were in prevention of tubular necrosis, reduction of vascular infiltration and interstitial inflammation, and diminution of loss of the brush border and cast formation. It can be inferred that the BLT-antagonist protection of renal function against iARF results from the prevention of neutrophil infiltration to both vascular and interstitial spaces, and that the markers of tubular epithelial injury (e.g., loss of brush border, tubular necrosis, and cast formation) were decreased in the BLT-antagonist-treated kidneys.
Figure 5.
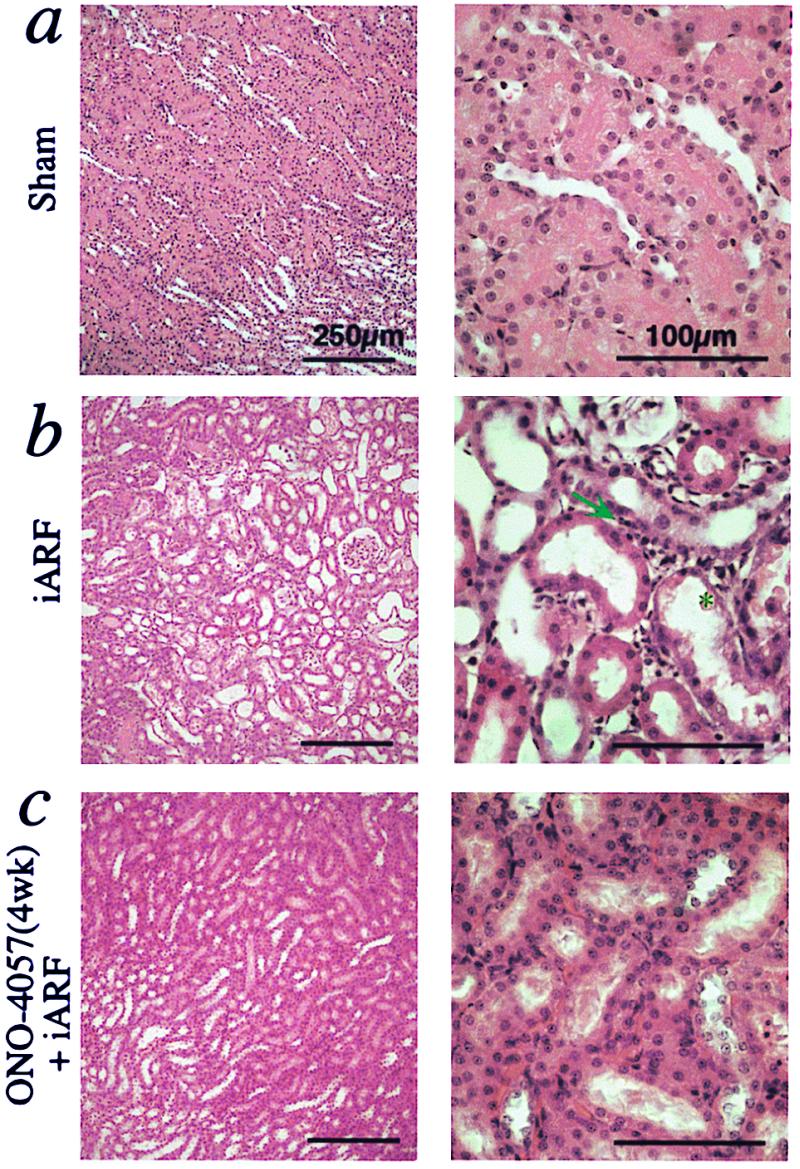
BLT antagonists morphologically ameliorate ischemic renal injury (hematoxylin and eosin staining). (a) Kidney obtained from sham-operated animal. (b) Ischemic kidney (iARF) obtained on postoperative day 1. Neutrophil infiltration was seen in the interstitial spaces (indicated by arrow). Brush border loss, tubular dilatation, exfoliation of tubular epithelial cells (asterisk), and denuded tubular basement membrane were evident. (c) Kidney sections from a BLT antagonist-pretreated animal.
Figure 6.

Histological scoring of intensity of ischemia reperfusion injury in kidneys treated with BLT antagonist. Scoring was performed on seven kidneys for sham, eight kidneys for control ischemia (iARF) and ONO-4057 pretreatment + iARF (Pre-Tx), and six kidneys for iARF + ONO-4057 posttreatment (Post-Tx). Asterisk denotes P <0.01 vs. control ischemia. Tub. nec., tubular necrosis; Vasc. infilt., vascular infiltration; Int. inf., interstitial inflammation. L-BB indicates loss of brush borders, and “Cast” indicates cast formation. Mean score was calculated for 13 categories of scoring, as previously described (16).
Discussion
Recent studies have addressed the role of PMNs as mediators of radical-based injury during ischemia reperfusion or preservation reperfusion of various tissues (18–20). Sequestered and activated neutrophils release lysosomal hydrolytic enzymes, lipid mediators, and reactive oxygen species that protect against invading organisms and help remove dead cells but also damage surrounding viable tissues. The involvement of neutrophils in renal ischemia-reperfusion injury had been suggested by histological analysis, and PMN infiltration was significant between 24 and 48 h after the initiation of the reperfusion period (15, 21). Although neutrophil recruitment and activation involve a series of redundant and interrelated mechanisms by multiple molecules, the present study focuses on the role of LTB4. The recent cloning of its receptor, BLT, in our laboratory (10, 22, 23) and those of others (24) and the discovery of CHO-BLT cells showing chemotactic activity in vitro (10) provide the opportunity to examine in vivo the role of this critical chemoattractant.
In our acute renal ischemia-reperfusion model, neutrophil migration was identified by histology (Fig. 4b), NP57-positive neutrophil count (Fig. 1b), and measurement of MPO activity in the kidney (Fig. 1a). The essential role of LTB4 in PMN sequestration is shown by CHO-BLT cells comigrating with neutrophils when injected 24 h after injury. These studies are an extension and in vivo application of our previous finding that CHO cells have intrinsic machinery to cause cell migration in vitro when chemoattractant receptors are expressed on their surface (10). Fig. 2a shows direct evidence of CMFDA-prelabeled CHO-BLT infiltrating into the interstitial spaces of ischemia-reperfusion kidneys obtained 24 h after clamp release. When mock-transfected CHO cells were injected, the cellular infiltration was negligible in ischemic kidneys (Fig. 2a). CHO-BLT cells were not detected in the kidneys of sham-operated animals (Fig. 2a). Further, to confirm that it was the BLT-expressing cells that were infiltrating, we generated CHO cells carrying FLAG-tagged BLT at the amino terminus (CHO-FLAG-BLT). The infiltrated cells were immunostained with the M5 anti-FLAG antibody. Serial sections of the kidney specimens showed that cells expressing FLAG-tagged BLT were infiltrating most prominently at the corticomedullary junctions (Fig. 3a), where peritubular blood flow is structurally most easily mitigated by exfoliated tubular epithelial obstruction. The specificity and dominant importance of LTB4 are shown by the use of two different receptor antagonists. The accumulation of CHO cells was virtually abolished when cells were pretreated with BLT antagonists (Fig. 2b), or animals were prefed with one of them (ONO-4057; Fig. 2c). Although Klausner et al. have demonstrated a 1.5-fold increase in LTB4 concentration in renal venous blood at 5 min postischemia (25), we, by using genetically engineered cells, have provided the first, to our knowledge, demonstration of in vivo LTB4-dependent cell migration toward ischemic renal parenchyma. With MPO activity decreased significantly by BLT antagonists (Fig. 1), we surmise that LTB4 plays a dominant role in the PMN sequestration in the renal ischemic-reperfusion model.
LTB4 is produced from membrane phospholipids by a sequential activation of phospholipases, 5-lipoxygenase, and LTA4 hydrolase. In the kidney, the transcellular biosynthesis of LTB4 has been demonstrated (26, 27), and there is accumulating evidence that some of these enzymes are present in it (26–28). Although the sites of the initial biosynthesis of LTB4 in the renal ischemia-reperfusion model remain unclear, the LTB4-biosynthetic capacity of renal parenchymal cells (glomerulus, tubulus, and interstitial cells) has been demonstrated (27, 28). Another potential source for LTB4 biosynthesis is the resident macrophages in the kidney, which Hume and Gordon have demonstrated (29). The molecular mechanism by which LTB4 production is initiated by the sensing of ischemia and/or reperfusion has not yet been determined. LTB4 activates neutrophils and can rapidly up-regulate leukocyte adhesion molecules, especially components of the CDw18 leukocyte surface glycoprotein complex (Mo1/LFA-1/GP 150, 95 or MAC-1, LFA-1 family) (LFA, lymphocyte function-associated antigen; GP, glycoprotein; MAC, membrane attack complex) within 5 to 30 min postischemia/reperfusion (30), which will promote initial PMN infiltration. Perhaps small numbers of PMNs, minutes after reperfusion, are recruited and load LTA4 to tissue transcellularly, which will be immediately converted to LTB4 in the kidney. But once these initial PMNs have infiltrated into the interstitial spaces, the mechanism mentioned above will be multiplied rapidly to promote further PMN migration and create the vicious cycle of tissue injury.
To answer the question of whether renal function is protected or improved by inhibiting PMN sequestration, we analyzed the serum parameters and histology of the study animals. As shown in Table 1, both serum Cr and BUN increases were mitigated when the BLT antagonist (ONO-4057) was administered before the ischemic insult. Even when the BLT antagonist was administered after the initiation of injury, a smaller but still significant protective effect was seen on postoperative day 1. The significant effectiveness of posttreatment was demonstrated until 72 h after the initiation of ischemia-reperfusion injury (Fig. 4). Histologically, the tissue damage was partly rescued by pretreatment of the animals with the BLT antagonist (Fig. 5 b and c). The accumulation of neutrophils in ischemic kidney was further reduced in BLT-antagonist-treated animals (Figs. 1b and 5). The mode of efficacy of the BLT antagonism, which was revealed by histological scoring (Fig. 6), was related to the infiltration of neutrophils and their lethal and sublethal effects on the tubular epithelium. Serum Cr represented only a relatively insensitive marker of renal injury in the early stages. Considering the extent of injury induced during reperfusion, the observed protective effects of BLT antagonism are prominent. Neutrophils migrate rapidly in early inflammation, because they can move at a speed of 100–200 μm/h and are capable of penetrating the peritubular arterial wall. Early neutrophils may migrate into ischemic areas in the first 15 min and, although the number of them is small, they amplify the response by producing leukotrienes or other mediators.
Blocking this anticipated migration of neutrophils by the prophylactic clinical use of BLT antagonists for procedures such as kidney transplantation and cardiopulmonary bypass would be clearly more advantageous than postischemia BLT administration. The use, however, of a BLT antagonist after short episodes of shock to moderate the extent of acute tubular necrosis may have significant effects on mortality and morbidity in severely ill patients. In addition to LTB4, several other molecules [e.g., interleukin-8 (31–33), complement (34–36), and cell-adhesion molecules (37–40)] might be involved in leukocyte recruitment and renal dysfunction. The roles of individual molecules as well as their combined effects in relation to LTB4 functions can now be addressed in future in vivo studies.
In conclusion, the present study clearly demonstrates that LTB4 is one of the principal mediators leading to neutrophil accumulation and renal dysfunction in the rat iARF model. In addition, the study provides a methodology with the potential of using genetically engineered CHO cells to carry receptors of interest to identify and examine the chemotactic ligands in vivo under various pathophysiological conditions.
Acknowledgments
We are grateful to Dr. Dennis Wong (Department of Biochemistry and Molecular Biology, University of Tokyo) for suggestions and to Dr. Frederick Miller (Department of Pathology, State University of New York, Stony Brook, NY) for critical discussions concerning histological interpretation. The work was supported in part by grants from the Ministry of Education, Science, Sports and Culture of Japan (T.S. and A.N.), the Human Science Foundation (T.S.), Japan Intractable Diseases Research Foundation (E.N.), and Mitsui Life Social Welfare Foundation (E.N.).
Abbreviations
- BLT
LTB4 receptor
- CHO
Chinese hamster ovary
- iARF
ischemic acute renal failure
- LTB4
leukotriene B4
- MPO
myeloperoxidase
- PMN
polymorphonuclear neutrophil
- BUN
blood urea nitrogen
- Cr
serum creatinine
- CMFDA
chloromethyl fluorescein diacetate
References
- 1.Kelly K J, Bonventre J V. In: Contemporary Issues in Nephrology. Goligorsky M S, Stein J H, editors. Vol. 30. New York: Churchill Livingstone; 1995. pp. 401–423. [Google Scholar]
- 2.Rabb H, O'Meara Y M, Maderna P, Coleman P, Brady H R. Kidney Int. 1997;51:1463–1468. doi: 10.1038/ki.1997.200. [DOI] [PubMed] [Google Scholar]
- 3.Chiang N, Gronert K, Clish C B, O'Brien J A, Freeman M W, Serhan C N. J Clin Invest. 1999;104:309–316. doi: 10.1172/JCI7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romson J L, Hook B G, Kunkel S L, Abrams G D, Schork A, Luccesi B R. Circulation. 1983;67:1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 5.Korthius R J, Grisham M B, Granger D N. Am J Physiol. 1988;254:H823–H827. doi: 10.1152/ajpheart.1988.254.5.H823. [DOI] [PubMed] [Google Scholar]
- 6.Weiss S J. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 7.Oliver J, MacDowell M, Tracy A. J Clin Invest. 1951;30:1307–1351. doi: 10.1172/JCI102550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakao A, Nosaka K, Ohishi N, Noiri E, Suzuki T, Taniguchi S, Seki G, Watanabe T, Kurokawa K, Shimizu T, et al. Kidney Int Suppl. 1997;63:S236–S238. [PubMed] [Google Scholar]
- 9.Noiri E, Gailit J, Sheth D, Magazine H, Gurrath M, Muller G, Kessler H, Goligorsky M S. Kidney Int. 1994;46:1050–1058. doi: 10.1038/ki.1994.366. [DOI] [PubMed] [Google Scholar]
- 10.Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. Nature (London) 1997;387:620–624. doi: 10.1038/42506. [DOI] [PubMed] [Google Scholar]
- 11.Bradley P P, Priebat D A, Christensen R D, Rothstein G. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- 12.Goulet J L, Snouwaert J N, Latour A M, Coffman T M, Koller B H. Proc Natl Acad Sci USA. 1994;91:12852–12856. doi: 10.1073/pnas.91.26.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noiri E, Peresleni T, Miller F, Goligorsky M S. J Clin Invest. 1996;97:2377–2383. doi: 10.1172/JCI118681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konrurek S J, Brzozowski T, Stachura J, Dembinski A, Majka J. Gut. 1994;35:1189–1196. doi: 10.1136/gut.35.9.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly K J, Williams W W, Jr, Colvin R B, Meehan S M, Springer T A, GutiJrrez-Ramos J-C, Bonventre J V. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelleher S P, Robinette J B, Miller F, Conger J D. Kidney Int. 1987;31:725–730. doi: 10.1038/ki.1987.58. [DOI] [PubMed] [Google Scholar]
- 17.Noiri E, Goligorsky M S, Wang G-J, Wang J, Cabahug C J, Sharma S, Rhodes B A, Som P. J Am Soc Nephrol. 1996;7:2682–2688. doi: 10.1681/ASN.V7122682. [DOI] [PubMed] [Google Scholar]
- 18.Kukielka G L, Hawkins H K, Michael L, Manning A M, Youker K, Lane C, Entman M L, Smith C W, Anderson D C. J Clin Invest. 1993;92:1504–1516. doi: 10.1172/JCI116729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosa Y, Lee W P, Kollias N, Randolph M A, May J W., Jr Plast Reconstr Surg. 1998;101:1586–1596. doi: 10.1097/00006534-199805000-00023. [DOI] [PubMed] [Google Scholar]
- 20.Marubayashi S, Oshiro Y, Maeda T, Fukuma K, Okada K, Hinoi T, Ikeda M, Yamada K, Itoh H, Dohi K. Surgery. 1997;122:45–52. doi: 10.1016/s0039-6060(97)90263-4. [DOI] [PubMed] [Google Scholar]
- 21.Rabb H, Mendiola C C, Saba S R, Dietz J, Smith C W, Bonventre J V, Ramirez G. Biochem Biophys Res Com. 1995;211:67–73. doi: 10.1006/bbrc.1995.1779. [DOI] [PubMed] [Google Scholar]
- 22.Masuda H, Yokomizo T, Izumi T, Shimizu T. Biochem J. 1999;342:79–85. [PMC free article] [PubMed] [Google Scholar]
- 23.Toda A, Yokomizo T, Masuda K, Nakao A, Izumi T, Shimizu T. Biochem Biophys Res Commun. 1999;262:806–812. doi: 10.1006/bbrc.1999.1284. [DOI] [PubMed] [Google Scholar]
- 24.Huang W-W, Garcia-Zepeda E A, Sauty A, Oettgen H C, Rothenberg M E, Luster A D. J Exp Med. 1998;188:1063–1074. doi: 10.1084/jem.188.6.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klausner J M, Paterson I S, Goldman G, Kobzik L, Rodzen C, Lawrence R, Valeri R, Shepro D, Hechtman H B. Am J Physiol. 1989;256:F794–F802. doi: 10.1152/ajprenal.1989.256.5.F794. [DOI] [PubMed] [Google Scholar]
- 26.Badr K F. J Am Soc Nephrol. 1992;3:907–915. doi: 10.1681/ASN.V34907. [DOI] [PubMed] [Google Scholar]
- 27.Brady H R, Lamas S, Papayianni A, Takata S, Matsubara M, Marsden P A. Am J Physiol. 1995;268:F1–F12. doi: 10.1152/ajprenal.1995.268.1.F1. [DOI] [PubMed] [Google Scholar]
- 28.Nakao A, Watanabe T, Ohishi N, Asano K, Taniguchi S, Nosaka K, Noiri E, Suzuki T, Kurokawa K, Shimizu T, et al. Kidney Int. 1999;55:100–108. doi: 10.1046/j.1523-1755.1999.00257.x. [DOI] [PubMed] [Google Scholar]
- 29.Hume D A, Gordon S. J Exp Med. 1983;157:1704–1709. doi: 10.1084/jem.157.5.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimmerman G A, McIntyre T M. J Clin Invest. 1988;81:531–537. doi: 10.1172/JCI113351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsumoto T, Ikeda K, Mukaida N, Harada A, Matsumoto Y, Yamashita J, Matsushima K. Lab Invest. 1997;77:119–125. [PubMed] [Google Scholar]
- 32.Sekido N, Mukaida N, Harada A, Nakanishi I, Watanabe Y, Matsushima K. Nature (London) 1993;365:654–657. doi: 10.1038/365654a0. [DOI] [PubMed] [Google Scholar]
- 33.Kukielka G L, Smith C W, LaRosa G J, Manning A M, Mendoza L H, Daly T J, Hughes B J, Youker K A, Hawkins H K, Michael L H, et al. J Clin Invest. 1995;95:89–103. doi: 10.1172/JCI117680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eppinger M J, Deeb G M, Bolling S F, Ward P A. Am J Pathol. 1997;150:1773–1784. [PMC free article] [PubMed] [Google Scholar]
- 35.Schulman G, Fogo A, Gung A, Badr K, Hakim R. Kidney Int. 1991;40:1069–1074. doi: 10.1038/ki.1991.316. [DOI] [PubMed] [Google Scholar]
- 36.Vakeva A P, Agah A, Rollins S A, Matis L A, Li L, Stahl G L. Circulation. 1998;97:2259–2267. doi: 10.1161/01.cir.97.22.2259. [DOI] [PubMed] [Google Scholar]
- 37.Ma X L, Lefer D J, Lefer A M, Rothlein R. Circulation. 1992;86:937–946. doi: 10.1161/01.cir.86.3.937. [DOI] [PubMed] [Google Scholar]
- 38.Vollmar B, Glasz J, Menger M D, Messmer K. Surgery. 1995;117:195–200. doi: 10.1016/s0039-6060(05)80085-6. [DOI] [PubMed] [Google Scholar]
- 39.Kelly K J, Williams W W, Jr, Colvin R B, Bonventre J V. Proc Natl Acad Sci USA. 1994;91:812–816. doi: 10.1073/pnas.91.2.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linas S L, Whittenburg D, Parsons P E, Repine J E. Kidney Int. 1995;48:1584–1591. doi: 10.1038/ki.1995.451. [DOI] [PubMed] [Google Scholar]