

Abstract
By inactivating the gene for l-gulono-γ-lactone oxidase, a key enzyme in ascorbic acid synthesis, we have generated mice that, like humans, depend on dietary vitamin C. Regular chow, containing about 110 mg/kg of vitamin C, is unable to support the growth of the mutant mice, which require l-ascorbic acid supplemented in their drinking water (330 mg/liter). Upon withdrawal of supplementation, plasma and tissue ascorbic acid levels decreased to 10–15% of normal within 2 weeks, and after 5 weeks the mutants became anemic, began to lose weight, and die. Plasma total antioxidative capacities were approximately 37% normal in homozygotes after feeding the unsupplemented diet for 3–5 weeks. As plasma ascorbic acid decreased, small, but significant, increases in total cholesterol and decreases in high density lipoprotein cholesterol were observed. The most striking effects of the marginal dietary vitamin C were alterations in the wall of aorta, evidenced by the disruption of elastic laminae, smooth muscle cell proliferation, and focal endothelial desquamation of the luminal surface. Thus, marginal vitamin C deficiency affects the vascular integrity of mice unable to synthesize ascorbic acid, with potentially profound effects on the pathogenesis of vascular diseases. Breeding the vitamin C-dependent mice with mice carrying defined genetic mutations will provide numerous opportunities for systematic studies of the role of antioxidants in health and disease.
Oxygen-derived free radicals are inevitable byproducts of normal biological functions such as respiration, energy generation, biosynthesis of macromolecules, and protection against infections. Normally, free radicals are swiftly destroyed by various antioxidant enzymes and small antioxidant molecules, which the body produces to prevent free radicals from reacting with lipids, proteins, and nucleic acids. Failure to maintain a coordinated balance between the production of free radicals and defenses against them can lead to cell death, tissue injury, and disease. Thus, much attention has been given in recent years to oxidative stress as a potentially important factor in the pathogenesis of many diseases, including atherosclerosis, cancer, and neurodegenerative diseases (1–4).
Studies of the pathogenesis of human diseases have benefited from the availability of small laboratory animal models for many years. Mutant mice are particularly useful for investigating the interplay of genetic and environmental factors in the pathogenesis of common, but multifactorial, diseases (5). However, there is a major handicap for studies involving the endogenous redox systems of humans and mice: humans (and other primates) lack the ability to synthesize ascorbic acid because of a loss of function in the gene (Gulo) coding for a key synthetic enzyme, l-gulono-γ-lactone oxidase (6), whereas mice have the functional gene. As a consequence, mouse tissues generally have high levels of ascorbic acid, which are only slightly influenced by exogenous vitamin C. In contrast, humans depend entirely on vitamin C derived from the diet. Guinea pigs also have lost this enzyme function and depend on dietary vitamin C (7). Hence severe deficiency of vitamin C causes disease (scurvy) in both primates and guinea pigs. A strain of rats (ODS, Osteogenic Disorder Sionogi, ref. 8) also has a defect in the same gene (9). No mouse strain is known with this deficiency, yet such mice, in which vitamin C intake can be controlled by diet, would be particularly valuable for investigating the interplay between endogenous and exogenous redox systems, genetic factors, and various diseases. To this end, we have generated Gulo −/− mutant mice, and we here describe the effects of vitamin C deficiency on three factors influential in vascular disease: plasma antioxidative capacity, plasma lipoprotein levels, and vascular integrity.
Materials and Methods
Gene Targeting.
Using published sequences of the rat Gulo gene (7), we isolated a phage clone spanning exons 3–7 of the mouse Gulo gene (Fig. 1A). The targeting construct (Fig. 1B) was designed to modify the locus so that a neomycin resistance gene replaces exons 3 and 4. A 6-kb EcoRI fragment containing exons 5–7 provided a 3′ arm of homology, and a 1.4-kb EcoRI/BamHI fragment in intron 2 formed a 5′ arm of homology. The neomycin resistance gene (pMC1neo) and the Herpes simplex thymidine kinase gene (pGKTK) both are oriented oppositely from the transcriptional orientation of the Gulo gene in the construct.
Figure 1.

Targeted modification of the mouse Gulo gene. (A) The endogenous mouse Gulo locus. The thick horizontal bar indicates the phage clone containing exons 3–7, marked by black boxes. (B) The targeting vector. Arrows P1 and P2 are relative positions of primers used to screen embryonic stem cells for the targeting event. (C) The inactivated Gulo locus. [a] and [b] are probes used for Southern blottings. Sizes and restriction enzymes of fragments diagnostic for the modification are indicated. B, BamHI; Bg, BglII; E, EcoRI; H, HindIII; N, NotI; S, SacI; X, XbaI. (D) Northern blot analysis of the liver RNA (20 mg) isolated from the livers of a male and a female of the three Gulo genotypes. The filter was hybridized with a Gulo probe (Upper), washed, and rehybridized with a glyceraldehyde-3-phosphate dehydrogenase gene (Gapdh) probe (Lower). The positions of 28S and 18S RNA are marked.
Mouse embryonic stem cells were cultured as described (10). The PCR primers used for screening homologous recombinants were P1 (5′-AATTGACCCGAGTCCTGGCT-3′) and P2 (5′-CGCGCCTTAATTAAGGATCC-3′), located 5′ to the EcoRI site in the Gulo intron 2 and within the linker of the vector. Clones that generated a 1.5-kb PCR fragment were expanded and confirmed by Southern blot analyses with two EcoRI/SacI fragments of 0.3 kb and 0.8 kb from introns 2 and 7, respectively, as probes.
Mice.
Chimeras were generated as described (10) and mated with C57BL/6 mice. For genotyping by PCR, three primers, P2 described above, P3 (5′-GTCGTGACAGAATGTCTTGC-3′), and P4 (5′-GCATCCCAGTGACTAAGGAT-3′) were used with tail DNA in a single reaction. A 230-bp fragment derived with P2 and P3 from the targeted locus and/or a 330-bp fragment derived with P3 and P4 from the endogenous locus distinguish homozygous, heterozygous, and wild-type animals.
Animals were handled as approved by the Institutional Animal Care and Use Committee. Mice were weaned at 3 weeks of age and fed ad libitum with irradiated mouse chow (PicoLab 5058, PMI Feeds, St. Louis). They had free access to water with or without vitamin C. The average ascorbic acid content of the diet was 110 mg/kg. To supplement vitamin C, drinking water was made to contain 330 mg l-ascorbic acid/liter and 0.01 mM EDTA and changed at least once a week. Heterozygous or homozygous mutant breeding pairs were kept on vitamin C supplementation for the production of homozygous pups, which were weaned onto vitamin C-supplemented water unless specifically stated. Heterozygotes were maintained on regular chow without ascorbic acid supplementation, unless specifically stated.
All mice used in the current experiments were younger than 6 months old and had a mixed genetic background derived from the strains 129 and C57BL/6. Blood was obtained either from the retroorbital sinus or by heart puncture and was anticoagulated with heparin or EDTA. Mice were euthanized with an overdose of Avertin (2,2,2-tribromoethanol) followed by fixation of the vasculature with 4% paraformaldehyde through the left ventricle at a physiological pressure.
Biochemical Measurements.
Ascorbic acid levels were measured by the α,α′-dipyridyl method as described (11). Antioxidative capacities of fresh plasma were determined by an enhanced chemiluminescence assay, based on the potential to quench light emission from a glowing horseradish peroxidase-catalyzed enhanced chemiluminescence reaction (12). The antioxidant activity was expressed by comparison with the quenching activity of a tocopherol-analogue, trolox. Lipid peroxidation was assayed colorimetrically as free malonaldehyde plus 4-hydroxyalkenals by using a kit (Oxis International, Portland, OR) according to the supplier's protocol. Plasma levels of cholesterol and triglycerides were determined enzymatically with kits obtained from Wako BioProducts (Richmond, VA) and Sigma. High density lipoprotein (HDL) cholesterol levels were determined after removing apolipoprotein B-containing particles by magnesium/dextran precipitation method (13). Fractionation of the plasma lipoproteins by ultracentrifugation and SDS gel electrophoresis of apolipoproteins have been described (14). Hematological determinations were made with an Animal Blood Counter (ABC vet, ABX Hematology, Garden Grove CA).
Liver RNA was isolated by using the TRI reagent (Molecular Research Center, Cincinnati). Northern blot analyses were done by conventional methods, using a 450-bp Gulo-cDNA probe generated by reverse transcription–PCR of the mRNA isolated from wild-type liver and primer sequences corresponding to exons 3 and 7. The probe for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, pTRI-GAPDH-mouse, was obtained from Ambion (Austin TX).
The significance of differences between means was calculated by ANOVA (jmp software, SAS, Cary, NC).
Microscopic Examination.
For transmission electron microscopy (TEM), two 0.3-cm sections of the thoracic aorta just distal to the left common carotid artery were processed by using standard protocols and embedded in Epon resin. One-micrometer thick sections were cut, stained with toluidine blue, and examined by light microscopy. Sections with detectable abnormalities were thin-sectioned, stained with uranyl acetate followed by lead citrate, and viewed by using a Phillips 301 TEM at 60 kV.
For scanning electron microscopy, the entire aortic arch from the heart to just proximal to the left common carotid was dehydrated through a graded series of ethanol solutions, treated with hexamethyldisilazane, and allowed to air-dry overnight. The samples were bisected, placed on nickel stubs, coated with a thin layer of carbon and a mixture of gold/palladium, and viewed by using a Leo (Helsingborg, Sweden) model 435VP scanning electron microscope at 20 kV.
To assay vascular permeability, Evans blue (30 mg/kg) was injected into animals through the tail vein 15 min before the sacrifice (15). The ascending aorta was removed and frozen in OCT compound, and cross sections (6 μm) were prepared. Extravasation of macromolecules was evaluated under fluorescent microscopy (Nikon B2A) with an excitation filter at 400–440 nm, a dichromic mirror at 510 nm, and a barrier filter at 520 nm.
Results
Inactivation of the Murine Gulo Gene.
The strategy for inactivating the Gulo gene is shown in Fig. 1. A missense mutation in the Gulo gene of Osteogenic Disorder Sionogi rats that replaces cysteine at position 61 in exon 3 with tyrosine is sufficient to dramatically reduce mRNA stability and enzyme activity (9), and the Gulo gene in guinea pigs has a stop codon at codon 61 in exon 3 in addition to many other defects in exon 6 (7). There therefore can be little doubt that deletion of the sequences containing exons 3 and 4 eliminates all gene function. Even if splicing were to occur between exons 2 and 5, the reading frame is not preserved. On Southern blot analyses, DNA from the modified cells gave a 14-kb SacI fragment and a 9.5-kb HindIII fragment hybridizing to probe a in addition to 5.5-kb SacI and 8-kb HindIII endogenous fragments; probe b hybridized to a 14-kb SacI fragment in addition to the endogenous 9-kb fragment (not shown). Two chimeras generated from targeted cells transmitted the embryonic stem cell genome to their F1 pups.
Gulo-Deficient Mice Depend on Dietary Vitamin C.
When heterozygous F1 animals were mated, homozygous pups (−/−) were born at the expected frequency. The homozygous mice lack Gulo gene expression as shown in the Northern blot hybridization of total RNA isolated from the liver (Fig. 1D). The body weights of homozygous pups born to heterozygous mothers were not different at 21 days of age from their heterozygous and wild-type litter mates. However, after weaning onto regular chow, which contains about 110 mg/kg of ascorbic acid, the homozygotes grew very little, and after 40 days of age they began to lose weight, whereas the body weights of wild types and heterozygotes steadily increased (Fig. 2A). In this experiment, one of the three homozygotes died on the 47th day, but after supplementation of vitamin C in the drinking water at 330 mg/liter the remaining two began to gain weight. This amount of vitamin C supplementation, also used in Osteogenic Disorder Sionogi rats (16), allows homozygous animals to gain weight and reproduce normally, as long as they remain on vitamin C supplementation.
Figure 2.

Body weight changes of Gulo −/− mice. (A) Females from a single litter from a heterozygous pair weaned on regular chow at age 21 days. Mean weights ± SD of one wild type plus three heterozygotes (●) and of three homozygotes (○) are shown. At day 45, vitamin C (330 mg/liter) in the drinking water was given to the animals (stippled circle). Because one homozygote died at day 47, the values at days 51 and 54 are the average weights of two homozygotes. Differences between two groups were statistically significant at all time points. (B) Nine Gulo −/− males were raised on chow with vitamin C supplementation to 8 weeks of age and then supplementation was withdrawn from six of the mice. Mean body weights ± SD for three mice with vitamin C supplementation (stippled square) and for six mice without supplementation (□) are shown, except that the 14th week was for three mice. P < 0.01 at 13-week time point.
In mature mice, withdrawal of vitamin C for longer than 5 weeks causes weight loss (Fig. 2B) and death. By the sixth week the animals had lost an average 30% of their body weight and become anemic as evidenced by a significant reduction in their red blood cell counts (−38%), hemoglobin concentration (−37%), and hematocrit (−35%). At necropsy, hemorrhages often were detected in knee joints and the dorsal margin of the scapula. Some animals had petechia in the intestinal mucosa. External bleeding was not seen in the mutant mice. All major organs were histologically unremarkable, and the immediate cause of death was not apparent.
Thus we found that approximately 0.08 mg/g of body weight/day (with supplementation) of ascorbic acid is sufficient for normal health in the Gulo −/− mice, assuming that an average mouse weighs 25 g and eats 4 g of chow/day and drinks 4.5 ml of water/day (17). However, 0.02 mg/g of body weight/day (on chow diet only) cannot sustain normal body function.
Gulo −/− Homozygotes Have Reduced Tissue and Plasma Ascorbic Acid Levels and a Reduction in Plasma Antioxidant Capacity.
Ascorbic acid levels in the plasma, liver, and brain of the Gulo −/− homozygous mutants maintained on unsupplemented chow for 3–5 weeks are 1.7 ± 0.2 μg/ml, 31 ± 2 μg/g, and 112 ± 14 μg/g, respectively (Fig. 3 A–C). These values are significantly lower (P < 0.001) than those in wild type (11.1 ± 0.5 μg/ml, 186 ± 8 μg/g, and 511 ± 15 μg/g, respectively). The levels in heterozygotes are also slightly lower than normal, although the difference is not statistically significant except in the plasma (9.6 ± 0.3 μg/ml, P < 0.03). Supplementation with 330 mg/liter of vitamin C in water normalizes the plasma levels in heterozygotes and increases the levels in homozygotes to approximately 60% normal. The plasma levels in supplemented homozygous males (5.7 ± 0.3 μg/ml, n = 6) are significantly lower than the levels in females (8.2 ± 0.5 μg/ml, n = 6, P < 0.001), and a similar trend is also present in heterozygotes (10.8 ± 0.7 vs. 12.4 ± 0.5, n = 6, not significant). Plasma ascorbic acid levels do not differ between male and female wild-type mice.
Figure 3.

Ascorbic acid (AA) levels in plasma (A), liver (B), and brain (C). Homozygous (−/−) mice were on chow diet without vitamin C supplement for 3–5 weeks. Data are expressed mean ± SE. P < 0.0001. (D) Plasma total antioxidative capacity expressed as trolox equivalent (μM). P < 0.0001. (E) Correlation between plasma ascorbic acid and total antioxidative capacity of individual animals including all three genotypes.
In parallel to the reduced ascorbic acid levels, the plasma antioxidative capacities in the homozygous mutants (48 ± 7 μM trolox equivalent, P < 0.001) are significantly lower than those in wild-type mice (134 ± 41 μM) and heterozygous mice (112 ± 31 μM). The antioxidative capacities in heterozygotes are also lower than wild-type mice, although this difference is not statistically significant (Fig. 3D). Comparison between plasma vitamin C levels and antioxidative capacity in individual animals demonstrates a significant positive correlation between ascorbic acid levels and plasma antioxidative capacity (r2 = 0.69, P < 0.0001, Fig. 3E). The intercept (37 μM) of the regression line suggests that antioxidants other than ascorbic acid explain 28% of the total antioxidative capacities (134 μM) in the normal mouse plasma.
The levels of lipid peroxidation products measured as free malonaldehyde plus 4-hydroxyalkenals in liver homogenates (80 ± 41 nmol/g of tissue, n = 10) of homozygotes were not statistically different from those in wild-type mice (71 ± 19 nmol/g of tissue, n = 19). The lipid peroxidation in plasma was below detection in both wild type and homozygotes by this method. Thus, despite a reduction in plasma antioxidative capacities, the Gulo −/− mice do not accumulate lipid peroxides in tissues.
Plasma Ascorbic Acid Levels Correlate Negatively with Plasma Total Cholesterol and Positively with HDL-Cholesterol Levels.
Because animal and epidemiological studies have suggested an association between plasma lipid levels and ascorbic acid levels, we tested whether reduced plasma and tissue ascorbic acid levels are correlated with the plasma lipid and lipoprotein distribution in our mice. We found a significantly increased mean plasma cholesterol levels in males (Fig. 4A) and a trend toward it in females (Fig. 4B) with a decrease in the number of the functional Gulo genes. Both Gulo genotype and gender of animals significantly affected the plasma cholesterol levels in mice with the Gulo genotype explaining 7% of the variance (P < 0.01) and gender explaining 8% (P < 0.005). Significant effects on HDL cholesterol levels in 80 animals also were observed as a function of genotype (P < 0.05) and gender (P < 0.02), with each explaining 8% of the variance.
Figure 4.

Plasma cholesterol levels in the Gulo mutant males (A) and females (B). Mean ± SEM. Homozygotes (−/−) were on chow with vitamin C supplement until 3–5 weeks before the determinations. Heterozygotes and wild-type mice were on unsupplemented chow at all times. Correlation between plasma ascorbic acid (AA) and plasma total cholesterol (C) and HDL cholesterol (D) levels in individual animals including all three genotypes. Some homozygotes were with vitamin C supplement and others were unsupplemented. ○, females; ■, males.
Regression analyses of values using individual animals, including all genotypes and with or without supplement, in which both data are available, revealed that plasma levels of ascorbic acid are negatively correlated with total cholesterol levels, but positively with HDL cholesterol levels. The correlations were highly significant with plasma ascorbic acid levels explaining about 10% of the variance in total cholesterol (P < 0.001, Fig. 4C), and 8% of the variance in HDL cholesterol (P < 0.05, Fig. 4D).
Plasma lipoproteins fractionated by ultracentrifugation showed that the cholesterol in low density lipoprotein fraction of homozygotes was higher than in wild type (24% vs. 16% when averaged from two experiments using plasma pooled from at least five each group of mice). HDL fraction in homozygotes was decreased compared with wild type (75% vs. 80%). There were no remarkable differences in apolipoprotein composition in these fractions (not shown). Plasma triglyceride levels are not different among the three genotypes.
Taken together, these data demonstrate that a suboptimal ascorbic acid has a small, but significant, effect on the plasma cholesterol levels and suggests that it increases the levels of atherogenic lipoprotein particles. However, these effects are small, and the Gulo −/− mice are not overtly hypercholesterolemic.
Rupture of Elastic Lamina, Smooth Muscle Cell Proliferation, and Injury of the Luminal Surface in the Thoracic Aorta.
Ascorbic acid is an important cofactor for the hydroxylation of proline and lysine necessary for the crosslinking of collagen and elastin, important structural components of vessel wall (18, 19). Inspection under both light microscopy and TEM of cross sections of the thoracic aorta from animals with low levels of plasma and tissue ascorbic acid revealed marked alterations in the pattern and integrity of the elastic laminae. Prominent breaks and fragmentation of the elastica were located in both the superficial and deep media (Fig. 5B). The endothelial cells overlying the areas of internal elastic lamia disruption were attenuated as a result of the accumulation of basement membrane material, collagen/elastic tissue, and aggregates of smooth muscle cells. Some smooth muscle cells had an altered morphology, were devoid of cytoplasmic filaments, and contained myelin figures indicative of cellular degeneration (not shown). Small clusters of smooth muscle cells, ranging from a few to a diffuse collections of several cells, were present within the subintima below the basement membrane and above the superficial elastic lamina (Fig. 5C). Most of the smooth muscle cells present within the intima were mildly activated as judged by a slight increase in their rough endoplasmic reticulum. Small collections of elastic fibers were located between smooth muscle cells both in the intima and in the deep media, suggesting that reorganization of the elastic lamina is a dynamic process in these mice.
Figure 5.

Abnormalities in the aorta of Gulo −/− mice. (A) A cross section of the descending thoracic aorta from a wild-type mouse showing uninterrupted elastic laminae. Toluidine blue staining, light microscopy, ×400. (B) A cross section of the descending thoracic aorta from a Gulo −/− mouse with disrupted elastic laminae. Light microscopy, ×400. (C) TEM of the area adjacent to that shown in B. A single layer of mildly activated smooth muscle cells (smc) is present in the intima between endothelial cells (ec) and superficial elastic lamina (sel). A break in the elastica is marked by an arrow. Magnification: ×4,000. (D) Scanning electron microscopy of the aortic arch of a wild-type mouse showing a normal luminal surface. lca, left carotid artery; rca, right carotid artery. Magnification: ×20. (E) Scanning electron microscopy of the aortic arch of an Gulo −/− mouse showing multiple longitudinal surface defects (arrows). Magnification: ×20. (F) Higher magnification (×200) of the defect.
On scanning microscopy, the aortic arches from the vitamin C-deficient mice contained focal areas where the normal endothelial cell pattern was absent (Fig. 5E). The foci with loss of normal endothelial architecture were oriented in the direction of blood flow and most often were located in the aortic wall near the takeoff of the carotid arteries. The surface was rough and irregular but devoid of fibrin deposition and attached blood cells, with the exception of occasional red blood cells (Fig. 5F). Cytoplasmic extensions of adjacent endothelial cells were in contact with the edges of the defects, and remnants of endothelial cells were present within the altered areas. Although aortic segments from all 10 Gulo −/− mice evaluated had at least one of such lesions, sections of the aortic arch in none of the wild-type mice showed abnormalities in the arrangements and integrity of the endothelial cells (Fig. 5D).
To determine the effects of these lesions on vascular permeability, wild-type and Gulo −/− mice were injected with Evans blue dye before sacrifice. On examination of frozen sections using fluorescence microscopy, the ascending thoracic as well as abdominal aortas from vitamin C-deficient mice showed patchy areas of dye binding to the elastica (Fig. 6B), indicating that there were changes in endothelial permeability that allowed leakage of macro-molecules (albumin-bound Evans blue) into the vessel wall. The Evans blue staining selectively highlighted the elastic laminae, allowing visualization of the general morphologic pattern and breaks in the elastica and confirming and augmenting the TEM observations. No deposition of Evans blue-albumin was present in the elastica of the wild-type mice (Fig. 6A).
Figure 6.
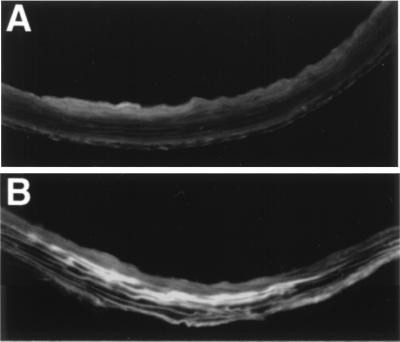
Evans blue extravasation in the ascending aorta of (A) a wild-type mouse and (B) a Gulo −/− mouse. Magnification: ×200. Image processing was used to subtract green autofluorescence, and the brighter area in this black and white figure corresponds to the red fluorescence of Evans blue.
These findings confirm that lower than normal levels of ascorbic acid are detrimental to the maintenance of the normal architectural integrity of the large vessels in the mutant mice.
Discussion
Humans, as a consequence of the loss during evolution of the enzyme, l-gulono-γ-lactone oxidase, are not able to synthesize ascorbic acid, a small water-soluble antioxidant. Mice lacking the same enzyme, like humans, depend on dietary supplementation of vitamin C for survival. Thus, the Gulo −/− mice with 0.02 mg/g of body weight/day of ascorbic acid intake have 10% normal plasma ascorbic acid levels and do not live longer than 6 weeks.
In humans, the Sheffield study carried out on healthy volunteers showed that the minimum protective dose of vitamin C defined by the absence of clinical scurvy is 10 mg/day (20). This result contributed importantly to the currently recommended daily dose of 60 mg/day (0.001 mg/g of body weight/day) for humans. Recent studies by Levine et al. (21) have shown, however, that this daily dose maintains plasma ascorbic acid levels in healthy male volunteers at subsaturation levels of about 25 μM; they recommended an increase in vitamin C intake to 200 mg/day, which saturates plasma level at 70 μM (12 μg/ml). The requirement of more than 0.08 mg/g per day of vitamin C to reach comparable plasma levels in the Gulo −/− mice is probably in part caused by the species differences in metabolic rates and in plasma levels of uric acid; about 5 mg/dl in humans (22) and 0.9 mg/dl in mice (23). Urate serves as a primary radical scavenger and probably helps to maintain plasma level of ascorbic acid by inhibiting iron-catalyzed ascorbate oxidation. These species differences also are reflected in our finding that approximately 72% of the total antioxidative capacity in normal mouse plasma is accounted for by ascorbic acid, whereas ascorbic acid accounts for only 0–24% of plasma antioxidant capacity in healthy human nonsmokers, whereas uric acid contributes 35–65% (22).
Although we do not exclude the possibility that Gulo −/− animals may accumulate oxidative damage in tissues or cells after a prolonged period with insufficient vitamin C, no elevation of lipid peroxidation products measured by plasma or liver malonaldehyde was detectable in homozygotes after 4 weeks of removing vitamin C from the drinking water. This result is not surprising, because these mice still have 35% normal levels of plasma total antioxidative capacity. Whether differences in the levels of other biomarkers can be detected (24) or whether the mutant animals will respond differently from wild-type mice to various stresses, such as severe inflammatory conditions, remains to be determined.
Epidemiological studies have not been conclusive in linking vitamin C to cardiovascular diseases in humans (25). For example, the World Health Organization/MONICA (monitering of trends and determinants in cardiovascular diseases) study showed, on a group level, an association of low plasma vitamin C levels with high cardiovascular mortality (26). On the other hand, in the Health Professionals Follow-up Study, no association could be established (27). In mice with plasma ascorbic acid levels ranging from normal to subnormal levels, we find that plasma ascorbic acid levels in mice are inversely correlated with plasma cholesterol and positively with HDL cholesterol. Similar relationships also have been seen in some, but not all, human studies (26, 28). Although vitamin C deficiency in guinea pigs has been reported to be associated with 20–40% lower levels of HDL cholesterol (29), the contribution of the plasma ascorbic acid levels to the variance in HDL cholesterol levels seen in our experiments was 10%. This contribution, although small, is significant. In addition, the vitamin C-deficient mice have an increase in low density lipoprotein-size particles and a small decrease in HDL, giving them less favorable lipoprotein profiles for cardiovascular protection than wild-type mice. The mechanisms that underlie the observed relationship between the levels of ascorbic acid and lipoproteins in the plasma of mice have not been determined, but a reduction in 7-α-hydroxylase activity has been demonstrated in vitamin C-deficient guinea pigs (30–32). Although further investigation is clearly needed, our results re-emphasize the importance of proper vitamin C intake to maintain a plasma lipoprotein profile that is associated with reduced risk for coronary heart disease.
The most striking pathological finding in our current study is the presence of abnormalities in the aortic walls of the vitamin C-deficient mice. The simplest explanation of this pathology is that the fragmentation of elastic lamina is caused by defects in the crosslinking of collagen and elastin because of the need for vitamin C to generate hydroxylysine and hydroxyproline. Support for this explanation is provided by the observations of similar fragmentation of elastic fibers in mice with the Blotchy allele at the X-linked Mottled locus (33). The Mottled locus codes for a copper-transporting ATPase, which is necessary for lysyl oxidase activity to crosslink collagen and elastin (34). Similar aortic wall changes also have been seen in young rats treated with β-aminopropionitrile, an inhibitor of lysyl oxidase (35, 36). However, although the aortic wall abnormalities in Blotchy mice progress to gross dilatation, aneurysm, and aortic rupture, the abnormalities seen in our vitamin C-deficient mice appear to be more subtle. The lesions in the aortic arch of vitamin C-deficient mice are longitudinal and most frequently are seen near the takeoff of the carotid where the blood flow related shear-stress is high. We did not find platelets, fibrin, or other blood cells in the areas with endothelial alteration. But there is a marked increase in the extravasation of macromolecules into the aortic wall in these areas as evidenced by the distribution of Evans blue in the vitamin C-deficient mice.
In conclusion, we have generated mice lacking l-gulono-γ-lactone oxidase, a key enzyme for ascorbic acid synthesis. The mutant mice, like humans, entirely depend on dietary vitamin C, and they show changes indicating that the integrity of their vasculature is compromised. When combined with the batteries of mutant mice generated in recent years, the Gulo −/− mutant mouse should provide unique opportunities for exploring the interactions between genetic determination and environmental factors, such as oxidative stress, and the effects on these interactions of different levels of vitamin C in the diet.
Acknowledgments
We thank K. Kluckman, A. Staton, E. Boudyguina, and K. Gush for technical help, and Dr. O. Smithies for discussion. This work was supported by grants from the American Heart Association (9650086N) and the National Institutes of Health (HL42630).
Abbreviations
- Gulo,l-gulono-γ-lactone oxidase gene
HDL, high density lipoprotein
- TEM
transmission electron microscopy
References
- 1.Knight J A. Ann Clin Lab Sci. 1995;25:111–121. [PubMed] [Google Scholar]
- 2.Berliner J A, Heinecke J W. Free Radical Biol Med. 1996;20:707–727. doi: 10.1016/0891-5849(95)02173-6. [DOI] [PubMed] [Google Scholar]
- 3.Collins A R. BioEssays. 1999;21:238–246. doi: 10.1002/(SICI)1521-1878(199903)21:3<238::AID-BIES8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 4.Bowling A C, Beal M F. Life Sci. 1995;56:1151–1171. doi: 10.1016/0024-3205(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 5.Smithies O, Maeda N. Proc Natl Acad Sci USA. 1995;92:5266–5272. doi: 10.1073/pnas.92.12.5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. J Biol Chem. 1994;269:13685–13688. [PubMed] [Google Scholar]
- 7.Nishikimi M, Kawai T, Yagi K. J Biol Chem. 1992;267:21967–21972. [PubMed] [Google Scholar]
- 8.Mizushima Y, Harauchi T, Yoshizaki T, Makino S. Experientia. 1984;40:359–361. doi: 10.1007/BF01952551. [DOI] [PubMed] [Google Scholar]
- 9.Kawai T, Nishikimi M, Ozawa T, Yagi K. J Biol Chem. 1992;267:21973–21976. [PubMed] [Google Scholar]
- 10.Bronson S K, Plaehn E G, Kluckman K D, Hagaman J R, Maeda N, Smithies O. Proc Natl Acad Sci USA. 1996;93:9067–9072. doi: 10.1073/pnas.93.17.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omaye S, Turnbull J D, Sauberlich H E. Methods Enzylmol. 1979;62:3–11. doi: 10.1016/0076-6879(79)62181-x. [DOI] [PubMed] [Google Scholar]
- 12.Maxwell S R J, Wiklund O, Bondjers G. Atherosclerosis (Shannon, Irel) 1994;111:79–89. doi: 10.1016/0021-9150(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 13.Warnick G R, Benderson J, Albers J J. Clin Chem. 1982;28:1379–1388. [PubMed] [Google Scholar]
- 14.Zhang S H, Reddick R L, Piedrahita J A, Maeda N. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 15.Lin S-J, Jan K-M, Chien S. Arteriosclerosis. 1990;10:703–709. doi: 10.1161/01.atv.10.5.703. [DOI] [PubMed] [Google Scholar]
- 16.Chan S W, Reade P C. Lab Anim. 1996;30:337–346. doi: 10.1258/002367796780739826. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein S E. In: Biology of the Laboratory Mouse. 2nd Ed. Green E L, editor. New York: Dover; 1966. pp. 337–350. [Google Scholar]
- 18.Pinnell S R. Yale J Biol Med. 1985;58:553–559. [PMC free article] [PubMed] [Google Scholar]
- 19.Peterkofsky B. Am J Clin Nutr. 1991;54:1135S–1140S. doi: 10.1093/ajcn/54.6.1135s. [DOI] [PubMed] [Google Scholar]
- 20.Krebs H A. Proc Nutr Soc. 1953;12:237–246. [Google Scholar]
- 21.Levine M, Conry-Cantilena C, Wang Y, Welch R W, Washko P W, Dhariwal K R, Park J B, Lazarev A, Graumlich J F, King J, Cantilena L R. Proc Natl Acad Sci USA. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nyyssonen K, Porkkala-Sarataho E, Kaikkonen J, Salonen J T. Atherosclerosis (Shannon, Irel) 1997;130:223–233. doi: 10.1016/s0021-9150(96)06064-9. [DOI] [PubMed] [Google Scholar]
- 23.Wu X, Wakamiya M, Vaishnav S, Geske R, Montgomery C, Jr, Jones P, Bradley A, Caskey C T. Proc Natl Acad Sci USA. 1994;91:742–746. doi: 10.1073/pnas.91.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Zwart L L, Meerman J H, Commandeur J N, Vermeulen N P. Free Radical Biol Med. 1999;26:202–226. doi: 10.1016/s0891-5849(98)00196-8. [DOI] [PubMed] [Google Scholar]
- 25.Simon J A. J Am Coll Nutr. 1992;11:107–125. [PubMed] [Google Scholar]
- 26.Gey K F, Brubacher G B, Stahelin H B. Am J Clin Nutr. 1987;45:1368–1377. doi: 10.1093/ajcn/45.5.1368. [DOI] [PubMed] [Google Scholar]
- 27.Rimm E B, Stampfer M J, Ascherio A, Giovannucci E, Colgitz G A, Willett W C. N Engl J Med. 1993;328:1450–1456. doi: 10.1056/NEJM199305203282004. [DOI] [PubMed] [Google Scholar]
- 28.Jacques P F, Sulsky S I, Perrone G A, Schaefer E J. Epidemiology. 1994;5:19–26. doi: 10.1097/00001648-199401000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Satinder, Sarkar A K, Majumdar S, Chakravarti R N. Indian J Med Res. 1987;86:351–360. [PubMed] [Google Scholar]
- 30.Ginter E, Bobek P, Jurcovicova M. In: Ascorbic Acid: Chemistry, Metabolism, and Uses. Seib P A, Tolbert B M, editors. Washington, DC: Am. Chem. Soc.; 1982. pp. 381–393. [Google Scholar]
- 31.Holloway D E, Rivers J M. J Nutr. 1981;111:412–424. doi: 10.1093/jn/111.3.412. [DOI] [PubMed] [Google Scholar]
- 32.Bjorkhem I, Kallner A. J Lipid Res. 1976;17:360–365. [PubMed] [Google Scholar]
- 33.Andrews E J, White W J, Bullock L P. Am J Pathol. 1975;78:199–200. [PMC free article] [PubMed] [Google Scholar]
- 34.Levinson B, Vulpe C, Elder B, Martin C, Verley F, Packman S, Gitschier J. Nat Genet. 1994;4:369–373. doi: 10.1038/ng0494-369. [DOI] [PubMed] [Google Scholar]
- 35.Julian M, Pieraggi M T H, Bouissou H. Pharmacol Res Commun. 1979;11:501–501. doi: 10.1016/s0031-6989(79)80022-3. [DOI] [PubMed] [Google Scholar]
- 36.Sauvage M, Jacob M P, Osborne-Pellegrin M. J Vasc Res. 1997;34:126–136. doi: 10.1159/000159215. [DOI] [PubMed] [Google Scholar]