

Abstract
BETA2/NeuroD is a homologue of the Drosophila atonal gene that is widely expressed during development in the mammalian brain and pancreas. Although studies in Xenopus suggest that BETA2/NeuroD is involved in cellular differentiation, its function in the mammalian nervous system is unclear. Here we show that mutant mice homozygous for a deletion at the BETA2/NeuroD locus fail to develop a granule cell layer within the dentate gyrus, one of the principal structures of the hippocampal formation. To understand the basis of this abnormality, we analyzed dentate gyrus development by using immunocytochemical markers in BETA2/NeuroD-deficient mice. The early cell populations in the dentate gyrus, including Cajal–Retzius cells and radial glia, are present and appear normally organized. The migration of dentate precursor cells and newly born granule cells from the neuroepithelium to the dentate gyrus remains intact. However, there is a dramatic defect in the proliferation of precursor cells once they reach the dentate and a significant delay in the differentiation of granule cells. This leads to malformation of the dentate granule cell layer and excess cell death. BETA2/NeuroD null mice also exhibit spontaneous limbic seizures associated with electrophysiological evidence of seizure activity in the hippocampus and cortex. These findings thus establish a critical role of BETA2/NeuroD in the development of a specific class of neurons. Furthermore, failure to express BETA2/NeuroD leads to a stereotyped pattern of pathological excitability of the adult central nervous system.
The mammalian genome contains many homologues of the Drosophila atonal gene that have been separated into two families based on their expression patterns and function in biological models of neurogenesis (1, 2). One family includes the neuronal determination genes that are expressed in undifferentiated precursors and regulate the decision to proceed down a pathway leading to the birth of particular groups of neurons (1, 2). Null mutations in several of these neuronal determination genes, such as neurogenin1, neurogenin2, and Math1 lead to failed induction of specific subclasses of neurons (3–11). The second group of atonal homologues, termed neuronal differentiation genes, have a putative role in the regulation of specific features of mature neurons (12–16). However, mice having mutations in these genes, including BETA2/NeuroD and NEX-1/Math2, initially were reported not to reveal the dramatic phenotypes expected based on the gain-of-function studies seen in Xenopus (17, 18). The lack of neuronal phenotypes in these mice likely is due to complementation by these highly related gene products and a third close relative, NeuroD2/NDRF.
BETA2/NeuroD was isolated based on its ability to activate the promoter of the insulin gene and was shown independently to induce formation of ectopic neurons in Xenopus embryos (13, 14). A null mutation of this gene produced mice with a diabetic phenotype and perinatal lethality (17). Others have shown that retinal explants from mutants have subtle changes in cell fate within the retina (19). Recently, Lee and colleagues (20) were able to rescue the diabetic phenotype with transgenic BETA2/NeuroD expression under the control of the insulin promoter, allowing some of these mice to survive to adulthood. Analysis of these mice revealed developmental abnormalities in the cerebellum and dentate gyrus (20). Using the originally described BETA2/NeuroD null mouse line, we have found that crossing the mutation into a different genetic background reduces the severity of the diabetes and allows 65% of the homozygous mutant mice to survive. This has enabled us to investigate the molecular pathogenesis of the hippocampal defect from early development through adulthood. In this report, we provide the initial characterization of the steps leading to the developmental failure of dentate granule cell layer formation in this mutant. Furthermore, we identify epilepsy as a major neurological phenotype arising from the loss of BETA2/NeuroD.
Materials and Methods
Generation of BETA2/NeuroD Mutant Mice.
All mice examined in this study were from a hybrid genetic background. The original BETA2/NeuroD null animals were generated on a 129/SvEv genetic background (17). The perinatal lethality because of diabetes was unchanged when these mice were bred into a C57BL/6J background. Surprisingly, when BETA2/NeuroD heterozygotes (C57BL/6J × 129/SvEv hybrid) were crossed into the 129/SvJ background, approximately 60–70% of the BETA2/NeuroD null offspring survived to adulthood. The null mice that did not survive to adulthood died in the first postnatal week because of diabetes. Animals were analyzed in the F1 or F2 generation.
Collection and Processing of Tissue.
Brains dissected at embryonic time points were immersed in 4% paraformaldehyde (PFA) in PBS. Postnatal mice were perfused with PBS followed by 4% PFA. For in situ hybridization and immunohistochemistry (except BrdUrd cell counts), brains were frozen and sectioned coronally at 15–30 μM. For BrdUrd cell counts, brains were processed and embedded in paraffin. Sections were cut at 7 μm for BrdUrd counting. BrdUrd was injected i.p. 3 hr before sacrifice at 150 μg/g body weight.
Probe Preparation and in Situ Hybridization.
The probe for Prox-1 was obtained from Peter Gruss. The NEX-1/Math2 probe was generated by reverse transcription–PCR followed by subcloning into the pCRII vector (Invitrogen); its generation and use are described elsewhere (S.J.P., A.E.C., and D.H.L., unpublished results). Nonradioactive in situ hybridization was performed by using a previously published protocol (8).
Antibodies.
Rabbit anticalretinin and anticalbindin antisera (Chemicon) were used at 1:1,000 dilution. Mouse antiparvalbumin monoclonal ascites (Sigma) was diluted 1:1,000. Rat anti-GFAP monoclonal supernatant was obtained from Virginia M.-Y. Lee. Rat anti-BrdUrd monoclonal ascites (Accurate) was diluted 1:200. Rabbit anti-β-galactosidase (β-gal) antiserum (5 Prime → 3 Prime) was diluted 1:500.
Detection of Apoptotic Cells.
Staining for apoptotic cells was performed by using the Klenow-FragEL DNA fragmentation detection kit (Oncogene Research) on formalin-fixed, paraffin-embedded, coronal 7-μm sections.
Electrographic Recordings.
Mice were anesthetized with Avertin, and 0.005-inch-diameter Teflon-coated silver wire electrodes were positioned through cranial burr holes onto the epidural surface or in a twisted pair configuration placed stereotactically within the dorsal hippocampus. The microconnector was cemented to the skull surface, and the mice were allowed to recover for at least 48 hr. Digital recordings with simultaneous behavioral videotaping were performed on freely moving animals.
Results
BETA2/NeuroD Mutant Mice Lack an Organized Dentate Gyrus.
The gene-targeting strategy and generation of BETA2/NeuroD−/− mice were described previously (17). This strategy resulted in the replacement of the BETA2/NeuroD gene with LacZ containing a nuclear localization signal. When bred with 129/SvJ mice, about 65% of the mutant mice survive to adulthood. They are somewhat hyperglycemic but have no ketonuria, and by 3 weeks of age the insulin level has returned to normal values (H. P. Huang, M.L., and M.-J.T., unpublished results). The adult mutant mice show severe ataxia, hyperactivity, circling, and swaying head movements. In addition, they have episodic, brief freezing spells with tonic posturing that are exacerbated by handling. Histologic analysis of mutant mice reveals striking abnormalities in the hippocampal formation (Fig. 1). Although the pyramidal layers appear normal, the mutant brains lack the dentate granule cell layer and have no organized dentate hilus. Closer examination reveals that a small cap of cells, just separated from the CA3 pyramidal region, contains a rudimentary population of neurons (Fig. 1 a– c). The heterozygotes appear identical to their wild-type littermates. The cerebellar abnormalities responsible for the ataxia in these mice will be described elsewhere (M.L. and M.-J.T., unpublished results). It is unlikely that these histologic defects are due to the loss of insulin because the dentate phenotype of these mice is similar to that reported by Miyata et al. (20). This group's mice have been engineered to have normal insulin levels by bypassing the requirement for NeuroD for the expression of the insulin gene.
Figure 1.
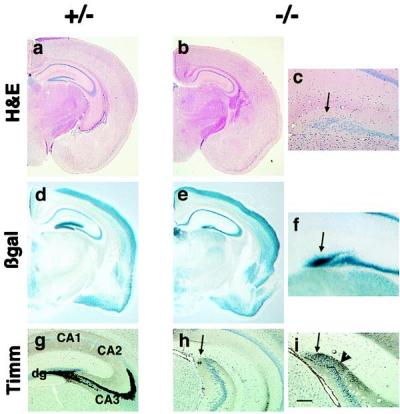
Histologic abnormalities of the dentate gyrus in BETA2/NeuroD mutant mice. Photomicrographs of heterozygote (+/−) and homozygote (−/−) mutant mice are shown. (a– c) Hematoxylin/eosin-stained sections. The arrow in c points to the abnormal cap of cells. (d– f) 5-Bromo-4-chloro-3-indolyl β-d-galactoside-stained sections to reveal expression of β-gal. (g– i) Timm-stained sections. The arrow in h points out a few Timm-labeled fibers in the cell cap. In i, the arrow indicates the Timm-stained cells in the cap and the arrowhead shows a few fibers directed toward CA3. [Bar = 2 mm (a, b, d, and e), 280 μm (c, f, and i), 500 μm (g and h).] dg, dentate gyrus; CA1–3, the various subfields of the pyramidal layer.
The developmental requirement for BETA2/NeuroD in hippocampus and cerebellum is highly selective, because examination of transcriptional activity of the BETA2/NeuroD promoter, as revealed by analysis of β-galactosidase (β-gal) activity, shows widespread expression of BETA2/NeuroD in adult heterozygous mice (Fig. 1d). This demonstrates that most brain regions are likely to be largely unaffected by loss of BETA2/NeuroD. The β-gal staining in the mutant mice shows a similar pattern (including the markedly abnormal dentate cap), indicating that the BETA2/NeuroD promoter activity is unchanged by the induced mutation (Fig. 1 e and f).
The Dentate Cap of Mutant Mice Contains Cells of Various Phenotypes.
To characterize the cells in the dentate cap of adult animals, we used the Timm stain, which labels zinc concentrated in granule cell mossy fiber axons. In wild-type and heterozygote mice, Timm-stained axons project normally from the hilar side of the dentate granule cell layer to the CA3 region of the hippocampus (Fig. 1g). In contrast, only a few Timm-stained axons arising from the abnormal dentate cap in mutant mice were visible (Fig. 1h). These processes seem to be oriented toward CA3 in some cases (Fig. 1i).
To study the cell phenotype in the dentate gyrus further, we examined the dentate cap for other markers expressed in subtypes of neurons found in this region. Calbindin is expressed in dentate granule cells and some hilar interneurons in wild-type mice (Fig. 2a), and the mouse homologue of the Drosophila gene prospero, prox-1 (21), is expressed almost exclusively in dentate granule cells postnatally (Fig. 2c). Interestingly, the disorganized cap of cells in the mutant mice contains both calbindin- and prox-1-expressing cells (Fig. 2 b and d, respectively), suggesting that they have some granule cell characteristics. We next tested whether there are cells in the cap of the mutants that have properties of other hippocampal cell populations. NEX-1/Math2 is expressed in pyramidal neurons throughout the hippocampus but is excluded from the dentate granule cell layer (18). No expression of NEX-1/Math2 was detected in the dentate cap of BETA2/NeuroD mutants, implying that these cells do not adopt a pyramidal cell fate (Fig. 2 e and f). To study the hilar interneuron population, we examined expression of parvalbumin and calretinin. Cells expressing parvalbumin and calretinin were scattered within the dentate cap of the BETA2/NeuroD mutant mice in a disorganized fashion (Fig. 2 g and h; data not shown). Taken together, these observations suggest that the dentate cap in mutant mice contains cells with some phenotypic features of dentate granule neurons and other cells similar to hilar interneurons.
Figure 2.
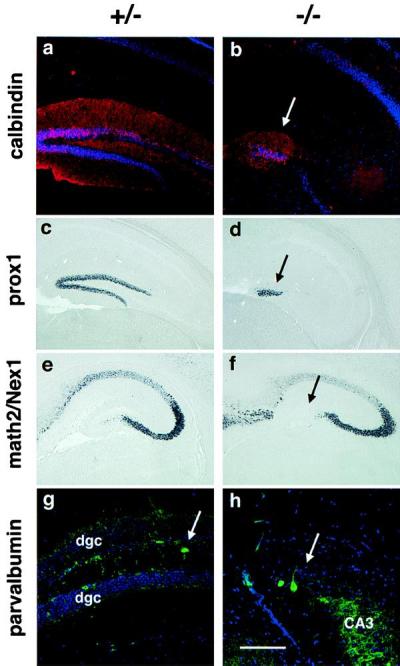
The dentate cap contains cells of various phenotypes. (a and b) Tissue sections from P23 mice labeled with an antiserum specific for calbindin in red and counterstained with a DNA stain in blue. The arrows in b, d, f, and h indicate the position of the cell cap. (c and d) In situ hybridization for prox-1 in sections from P23 mice. (e and f) In situ hybridization for NEX-1/Math2 in sections from P5 mice, showing that the cap cells in the mutant have not been respecified to a pyramidal fate. (g and h) Tissue sections from P23 mice labeled with a mAb specific for parvalbumin in green and counterstained with a DNA stain in blue. The arrow in g points to a parvalbumin-positive interneuron. [Bar = 110 μm (g and h) and 220 μm (a–f).]
BETA2/NeuroD Mutant Mice Have Defects in Granule Cell Differentiation and Precursor Cell Proliferation.
To understand the etiology of the dentate abnormality in the BETA2/NeuroD mutant mice, we sought to determine the developmental stage at which the abnormality becomes apparent. We first examined the distribution of Cajal–Retzius cells and radial glia, because these cell types play a role in the early migration of granule cells and their precursors into the dentate gyrus (22, 23). In BETA2/NeuroD mutant mice, cells resembling Cajal–Retzius cells and the fibers of radial glia are abundant in the embryonic day 18.5 (E18.5) dentate gyrus, and their distribution appears largely normal although we cannot exclude subtle abnormalities in their organization (data not shown).
Next, we asked whether the migration of mitotic precursor cells to the dentate gyrus is abnormal in mutant mice. Mitotic precursor cells migrate from the ventricular zone into the dentate after the anlage is established (24, 25). This is apparent first at E16.5 and continues for several days postnatally (24, 25). Once these precursor cells reach the dentate gyrus they take up residence in a transient proliferative zone in the hilus termed the tertiary matrix (24, 25). Because precursor cells continue to divide while en route to the dentate gyrus, acute (3 hr before sacrifice) BrdUrd labeling observed in the migratory pathway labels migrating dentate precursor cells without labeling already postmitotic migrating neurons. In BETA2/NeuroD mutants at least some mitotic precursor cells are able to migrate to the dentate gyrus when assessed by BrdUrd labeling to mark proliferating, migrating cells (Fig. 3 a and b). However, these precursor cells do not establish themselves in the upper blade of the dentate gyrus as fully as in the heterozygote mice (see arrows in Fig. 3 a and b). In addition, quantitation of BrdUrd-labeled cells in the dentate region of animals shows that the number of BrdUrd-labeled cells is decreased at all ages in the BETA2/NeuroD mutant mice as compared with controls (Fig. 3c), and these differences are even more apparent at later ages. This implies that BETA2/NeuroD function contributes to the proliferation of dentate precursor cells. This is of particular interest because BETA2/NeuroD is thought generally to function once neurons become postmitotic (1, 2).
Figure 3.
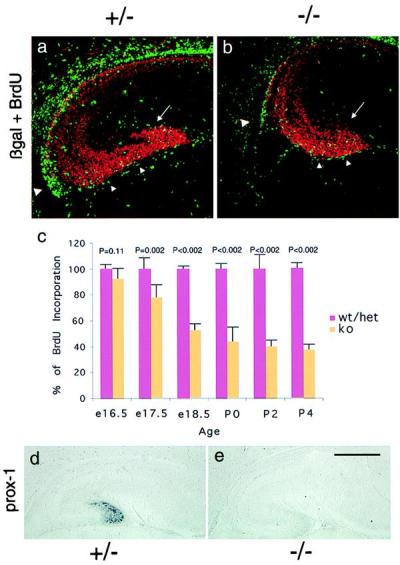
BETA2/NeuroD mutant mice have intact migration of mitotic precursor cells from the neuroepithelium but fail to form the scaffolding of the dentate granule cell layer. (a and b) E18.5 sections stained with anti-BrdUrd antibody (Accurate Chemicals) in green and anti-β-gal antibody (5 Prime → 3 Prime) in red. The large arrowhead labels the site of initiation of migration from the hippocampal neuroepithelium, and the small arrowheads label the subpial migratory route. The arrow shows the forming upper blade of the dentate, which is underpopulated with precursor cells and newly born granule cells in the mutant mouse. (c) BETA2/NeuroD mutant mice have a progressive decline in the number of mitotic precursor cells in the dentate gyrus. At each time point, the number of BrdUrd-labeled nuclei from three sections from each animal were counted. At least three different animals of the indicated genotypes were counted. The average number of labeled nuclei per animal from the heterozygote/wild-type pool was defined as 100% at each time point, and the numbers from the mutant animals were compared with this value. The error bars represent the SEM. The statistical significance of the differences at each time point was calculated by using Student's t test. P values are indicated in the figure. (d and e) Prox-1 in situ hybridization on sagittal sections of E18.5 brain. [Bar = 220 μm (a and b) and 800 μm (d and e).]
The differentiation of dentate granule cells is a complex process that takes place in at least two sites. Some granule cells are born at the ventricular surface and migrate into the dentate anlage along with the precursor cells, whereas the majority of granule cells are born postnatally in the tertiary matrix (24, 25). Granule cells born at the ventricular surface and en route to the dentate are thought to be responsible for forming the primary dentate granule cell layer that serves as the scaffolding for the addition of granule cells born later (24, 25). At E18.5 in heterozygote mice, prox-1 is expressed in the newly differentiated granule cells only after they reach the nascent dentate gyrus (Fig. 3d). However, no prox-1 expression is observed at E18.5 in BETA2/NeuroD-deficient mice (Fig. 3e). This is not likely to be due to a direct upstream effect of BETA2/NeuroD on prox-1 expression, because prox-1 expression in other brain regions is not affected (data not shown). This result indicates that, although the migration of precursor cells away from the neuroepithelium appears qualitatively intact, neurons generated at the ventricular surface fail to express a specific dentate granule cell marker at the appropriate time during migration to the dentate gyrus. This defect in differentiation of the early granule cell population in BETA2/NeuroD mutant mice may account for the abnormal morphology of the initial scaffolding of the dentate granule cell layer.
To pinpoint the location of prospective granule cells in heterozygote and mutant mice, we examined β-gal expression within the hippocampus in E16.5 and E18.5 mutant animals. As early as E16.5, we could see that the upper blade of the nascent dentate gyrus is poorly formed in the mutant mice as compared with the heterozygotes (Fig. 3 a and b). Nevertheless, the migratory pathway from the neuroepithelium in mutant mice has a qualitatively normal pattern of cells expressing β-gal. Thus, BETA2/NeuroD mutant mice have a defect in the formation of the initial scaffolding of the dentate gyrus, reflected by a poorly formed granule cell layer and failure to express prox-1.
Once the scaffolding for the dentate granule cell layer is formed, granule cells are generated in the tertiary matrix from mitotically active precursor cells (24, 25). In heterozygote mice at postnatal day 5 (P5), the granule cell layer is well organized, with calbindin-expressing granule cells in the most superficial layer and more immature granule cells packed tightly below. Abundant BrdUrd-labeled nuclei, reflecting the presence of persistent granule cell precursors, are distributed widely throughout the hilus of heterozygote mice (Fig. 4a). In P5 mutant mice, the dentate gyrus consists only of a small mass of cells expressing calbindin with no orderly granule cell layer (Fig. 4b). Also, there are far fewer mitotic cells (Fig. 4b; see also Fig. 3c), and there is a low level of prox-1 expression confined to the abnormal dentate cap (Fig. 4d). The appearance of a low level of prox-1 expression by this time suggests that the differentiation of granule-like cells in the dentate cap occurs on a delayed schedule.
Figure 4.
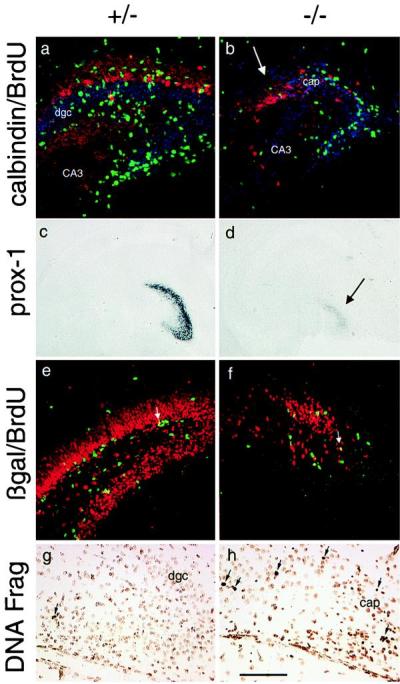
BETA2/NeuroD mutant mice have delayed differentiation of granule cells, a disorganized tertiary matrix, and excess cell death in the dentate gyrus. (a and b) Calbindin (red) and BrdUrd (green) antibody staining with blue DNA counterstain of P5 brain sections. The arrow indicates the cell cap containing cells expressing calbindin. (c and d) Prox-1 in situ hybridization on coronal sections of P5 brain sections. The arrow indicates the cell cap. (e and f) Anti-β-gal (red) and anti-BrdUrd (green) antibody staining of P11 brain sections demonstrates the almost complete involution of the tertiary matrix by this age in the knockout mice compared with the well organized dentate gyrus of heterozygote mice. Arrows point to β-gal/BrdUrd double-labeled cells. (g and h) Labeling of DNA fragmentation in cells. The sections are lightly counterstained with methyl green, and positive signals are the darker, condensed nuclei; see arrows. [Bar = 110 μm (a, b, e, and f), 1,000 μm (c and d), and 75 μm (g and h).] CA3, hilar portion of CA3 pyramidal layer; dgc, dentate granule cell layer; cap, dentate cell cap in mutant mice.
To determine whether the dramatic decrease in the numbers of precursor cells is likely to be a cell autonomous effect of BETA2/NeuroD loss, we used β-gal/BrdUrd double labeling, reasoning that the BETA2/NeuroD promoter would be active in dentate precursor cells if BETA2/NeuroD expression is required as a cell intrinsic determinant. Staining at P11 in the mutant mice reveals that, by the second postnatal week, the tertiary matrix involutes and appears similar to the abnormal structure present in adult mutant mice, and there are far fewer β-gal-labeled cells and BrdUrd-labeled precursors compared with the heterozygotes (Fig. 4 e and f). Interestingly, some cells are double-labeled with BrdUrd and β-gal in both the heterozygote and knockout animals. However, this group represented only a small proportion of the BrdUrd- or β-gal-labeled cells, implying that mitotic cells with activity of the BETA2/NeuroD promoter make up a small fraction of the total precursor cells at this stage (Fig. 4 e and f). This is not a phenomenon simply present in this late stage of dentate gyrus development because the same result was seen in E16.5 heterozygotes and mutants (Fig. 3 a and b). These findings are consistent with a role for BETA2/NeuroD in the differentiation of dentate precursor cells at some, but not all, stages in the granule cell lineage. Because BETA2/NeuroD is expressed in all newly postmitotic granule cells, it seems most likely that BETA2/NeuroD function begins in the latter stages of precursor cell maturation. The decrease in mitotic precursor cells appears to involve the entire pool of BrdUrd-labeled cells, and, therefore, a portion of this effect may not be cell autonomous. One attractive hypothesis for future study is that the intact granule cell layer supports the continued existence of mitotic precursor cells in the tertiary matrix and subgranular zone by supplying trophic or growth factors.
Finally, we wondered whether the marked reduction in the number of dentate granule cells seen postnatally might be due to excess cell death in addition to the defects in granule cell differentiation and proliferation of granule cell precursors. Apoptosis in the dentate gyrus normally occurs at fairly constant levels in the first week of postnatal life (26). We examined several ages of mice between P2 and P14 and found that the number of apoptotic nuclei in the dentate gyrus of the mutant mice is increased during this period (Fig. 4 g and h shows data from P4 mice). However, the specific identity of these dying cells remains uncertain.
Adult Mutant Mice Have Limbic Epilepsy.
Behaviorial observation of adult mutants revealed frequent (one to three episodes per hour), spontaneous behavioral seizures that resemble those seen in rodent models of limbic epilepsy. Typical episodes are brief, lasting 5–15 sec, with sudden focal dystonic posturing, flexor spasm of one or both forelimbs, and versive head movements. Electroencephalogram (EEG) activity during the seizures consists of a stereotyped, rapid-burst discharge, followed by slower-interval synchronized spiking. A similar pattern is seen in the hippocampus (Fig. 5a). Generalized tonic–clonic seizures also occur occasionally, accompanied by either sustained cortical epileptiform discharges or intermittent abnormal synchronous discharges. In the hippocampus, depth EEG activity in BETA2/NeuroD mutant mice shows an intact and reactive theta rhythm activity that is highly correlated with exploratory locomotor activity similar to wild-type mice (Fig. 5b). Video-EEG recordings of typical seizure episodes are available for viewing at http://www.bcm.tmc.edu/neurol/research/neurogenetics/videos/neurod1.rm.
Figure 5.

BETA2/NeuroD mutant mice have seizures and intact theta rhythm. (a) Representative traces showing spontaneous electrographic seizures recorded from three BETA2/NeuroD−/− mice. (1) Typical cortical discharge pattern associated with partial behavioral seizure. (2) Similar event recorded from hippocampus in a second mutant. (3) Continuous cortical spiking during prolonged seizure in a third mutant. (4) Interictal cortical spike discharges without behavioral accompaniment. (b) Hippocampal depth recordings of spontaneous (7.6-Hz) theta rhythm in wild-type (+/+) and BETA2/NeuroD mutant mice. Calibration in a and b is 1 sec.
Discussion
These studies provide direct evidence that the neuronal differentiation gene BETA2/NeuroD is required for formation of the dentate gyrus, the primary input to the hippocampus. Without BETA2/NeuroD, few if any normal granule cells are formed and the dentate gyrus is replaced by a small cap of cells containing neurons of various molecular phenotypes. Some of the cells in the cap are likely to be abnormally differentiated granule cells. This malformation is not due to a hippocampal patterning defect, and the dentate granule lineage appears intact up to the precursor cell stage. The abnormality appears to be due in large part to a defect in the transition from late precursor cells into differentiated granule cells. This affects the earliest-born granule cells, so that after migrating to the dentate gyrus they form a loosely packed group of cells instead of a compact upper blade. This defect then leads to disorganization of the tertiary matrix and a failure to produce the appropriate number of postnatal granule neurons. The dramatic decrease in the number of BrdUrd-labeled precursor cells in the tertiary matrix of the mutant animals is curious considering the small number of mitotic cells expressing BETA2/NeuroD. It seems likely that the primary defect is in late precursor cells that express BETA2/NeuroD and in newly born neurons. Absence of BETA2/NeuroD may have a secondary effect on the environment that allows the maintenance of the tertiary matrix.
It is curious that some cells in the mutant mice are able to partially circumvent the block in dentate granule cell differentiation caused by the loss of BETA2/NeuroD function. One possible explanation for this observation is that normal BETA2/NeuroD function may not be completely cell autonomous. BETA2/NeuroD may be required for the specification of an early subpopulation of granule cells that then organizes the dentate granule cell layer and regulates the birth of additional granule cells independent of continued BETA2/NeuroD function. Thus, incompletely differentiated granule cells are born in small numbers in the mutant and remain in a disorganized structure. It is also possible that other basic helix–loop–helix proteins partially complement BETA2/NeuroD function. NeuroD2/NDRF and NEX-1/Math2 are potential candidates because they are close relatives of BETA2/NeuroD. NeuroD2/NDRF is expressed in the dentate gyrus postnatally but only at low levels prenatally (S.J.P., A.E.C., and D.H.L., unpublished results). Thus, NeuroD2/NDRF is a particularly attractive candidate for the complementing factor, allowing delayed granule cell differentiation because its expression at later times may account for the expression of prox-1 at older ages in the BETA2/NeuroD−/− mice. Indeed, we have observed expression of NeuroD2/NDRF in the dentate cap in BETA2/NeuroD mutant mice only at postnatal times (data not shown).
A recent publication by Miyata and colleagues reports that mice similar to those described here have dentate abnormalities (20). The lethal diabetes in these mice was ameliorated by the expression of BETA2/NeuroD as a transgene under the control of the insulin promoter. The analysis of the hippocampal phenotype in this paper included a description of the absence of an organized dentate gyrus and a defect in dentate precursor cells. However, the authors concluded that there are no dentate granule cells remaining in mutant mice. It is unclear whether residual granule-like cells were not appreciated in their study or whether there are strain-related differences in the two mutant mouse lines. A more in-depth developmental analysis using the various markers employed in our study will be required to determine whether the dentate gyrus abnormalities in the mice described by Miyata and colleagues are equivalent to what we have observed.
Mice with homozygous mutations in the emx2 homeobox gene also have severe hippocampal abnormalities, including a complete lack of the dentate gyrus (27, 28). However, these mice also have marked abnormalities in the pyramidal cell layer. A detailed analysis of the development of the emx2 mutant mice (which die at birth) showed that they have a patterning defect at the rostral telencephalic midline that presumably underlies the hippocampal abnormalities (27, 28). Recently, mice with mutations in the Lim homeobox genes Lhx2 and Lhx5 were shown to lack the entire hippocampus (29, 30). These mutations act early to affect the development of the entire hippocampal neuroepithelium and its derivatives. In contrast, the BETA2/NeuroD mutation affects the generation of a far more specific subset of hippocampal derivatives at the time of terminal differentiation.
The spontaneous seizures present in BETA2/NeuroD mutants with dentate granule cell layer abnormalities is of significant clinical interest. Humans with temporal lobe epilepsy commonly have pathological changes that include disorganization or loss of cells in the dentate gyrus (31, 32), and it remains unknown in such cases whether the dentate abnormalities are the cause or result of seizures. However, recent clinical imaging studies are providing a more complete description of hippocampal structural pathology in certain familial epilepsy syndromes (33, 34). Within these families, hippocampal anomalies can be seen in some individuals who have not yet had any seizures, suggesting that primary hippocampal dysgenesis may be linked to an epileptic phenotype.
Despite the poorly organized dentate gyrus and the presence of a seizure disorder with both limbic and generalized features, the preservation of normal slow (theta) wave activity in the BETA2/NeuroD mutant hippocampus is significant, given the massive loss of granule cell input onto pyramidal cells, the principle cell population of the hippocampus. Theta rhythm is produced by coordinated synchrony between hippocampal pyramidal cells and GABAergic/cholinergic cells of the medial septum and is critically involved in memory functions (35). This result in the BETA2/NeuroD mutant mice thus confirms an earlier finding of intact theta rhythms in adult rats with dentate granule cell agenesis produced by focal neonatal x-irradiation (36). These observations demonstrate that the granule cell network integrates cortical information entering the hippocampus, but is not a generator of this important oscillation.
The BETA2/NeuroD mutant mice represent an example of an experimental model of early dentate gyrus abnormalities and epilepsy. It remains to be determined whether the epilepsy in these mice is related directly to the lack of dentate granule cells, network changes caused by the lack of dentate granule cells, the loss of BETA2/NeuroD expression per se, or other developmental abnormalities associated with this mutation. Nonetheless, this model will allow a genetic dissection of the role of dentate granule cells on functional properties of the hippocampal network and their contribution to human epileptogenesis, learning, and memory.
Acknowledgments
We thank Dr. John Rubenstein for the suggestion of prox-1 as a granule cell marker and for comments on the manuscript, Dr. David Anderson for the in situ hybridization protocol, and Dr. Peter Gruss for the probe to prox-1. We also thank M. J. Chu, B. Antalffy, and J. Xu for their technical expertise. This work was supported by grants from the National Institutes of Health (to D.H.L., M.-J.T., and J.L.N) and the March of Dimes (to D.H.L.). S.J.P. was supported by a Howard Hughes Medical Institute postdoctoral fellowship for physicians. M.L. was supported by a National Research Service Award fellowship from the National Institute of Diabetes and Digestive and Kidney Diseases. A.E.C. was supported by a Howard Hughes Medical Institute medical student fellowship.
Abbreviations
- β-gal
β-galactosidase
- E
embryonic day
- P
postnatal day
References
- 1.Kageyama R, Ishibashi M, Takebayashi K, Tomita K. Int J Biochem Cell Biol. 1997;29:1389–1399. doi: 10.1016/s1357-2725(97)89968-2. [DOI] [PubMed] [Google Scholar]
- 2.Lee J E. Curr Opinion Neurobiol. 1997;7:13–20. doi: 10.1016/s0959-4388(97)80115-8. [DOI] [PubMed] [Google Scholar]
- 3.Akazawa C, Ishibashi M, Shimizu C, Nakanishi S, Kageyama R. J Biol Chem. 1995;270:8730–8738. doi: 10.1074/jbc.270.15.8730. [DOI] [PubMed] [Google Scholar]
- 4.Gradwohl G, Fode C, Guillemot F. Dev Biol. 1996;180:227–241. doi: 10.1006/dbio.1996.0297. [DOI] [PubMed] [Google Scholar]
- 5.Ma Q, Kintner C, Anderson D J. Cell. 1996;87:43–52. doi: 10.1016/s0092-8674(00)81321-5. [DOI] [PubMed] [Google Scholar]
- 6.Sommer L, Ma Q, Anderson D J. Mol Cell Neurosci. 1996;8:221–241. doi: 10.1006/mcne.1996.0060. [DOI] [PubMed] [Google Scholar]
- 7.Cau E, Gradwohl G, Fode C, Guillemot F. Development. 1997;124:1611–1621. doi: 10.1242/dev.124.8.1611. [DOI] [PubMed] [Google Scholar]
- 8.Ma Q, Sommer L, Cserjesi P, Anderson D J. J Neurosci. 1997;17:3644–3652. doi: 10.1523/JNEUROSCI.17-10-03644.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Arie N, Bellen H J, Armstrong D L, McCall A E, Gordadze P R, Guo Q, Matzuk M M, Zoghbi H Y. Nature (London) 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 10.Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. Neuron. 1998;20:483–494. doi: 10.1016/s0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- 11.Ma Q, Chen Z, del Barco Barrantes I, de la Pompa J L, Anderson D J. Neuron. 1998;20:469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- 12.Bartholomea A, Nave K A. Mech Dev. 1994;48:217–228. doi: 10.1016/0925-4773(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 13.Lee J E, Hollenberg S M, Snider L, Turner D L, Lipnick N, Weintraub H. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 14.Naya F J, Stellrecht C M, Tsai M-J. Genes Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu C, Akazawa C, Nakanishi S, Kageyama R. Eur J Biochem. 1995;229:239–248. doi: 10.1111/j.1432-1033.1995.tb20461.x. [DOI] [PubMed] [Google Scholar]
- 16.McCormick M B, Tamimi R M, Snider L, Asakura A, Bergstrom D, Tapscott S J. Mol Cell Biol. 1996;16:5792–5800. doi: 10.1128/mcb.16.10.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naya F J, Huang H P, Qiu Y, Mutoh H, DeMayo F J, Leiter A B, Tsai M-J. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwab M H, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner M J, Nave K A. J Neurosci. 1998;18:1408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrow E M, Furukawa T, Lee J E, Cepko C L. Development. 1999;126:23–36. doi: 10.1242/dev.126.1.23. [DOI] [PubMed] [Google Scholar]
- 20.Miyata T, Maeda T, Lee J E. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliver G, Sosa-Pineda B, Geisendorf S, Spana E P, Doe C Q, Gruss P. Mech Dev. 1993;44:3–16. doi: 10.1016/0925-4773(93)90012-m. [DOI] [PubMed] [Google Scholar]
- 22.Rickmann M, Amaral D G, Cowan W M. J Comp Neurol. 1987;264:449–479. doi: 10.1002/cne.902640403. [DOI] [PubMed] [Google Scholar]
- 23.Frotscher M. Curr Opinion Neurobiol. 1998;8:570–575. doi: 10.1016/s0959-4388(98)80082-2. [DOI] [PubMed] [Google Scholar]
- 24.Altman J, Bayer S A. J Comp Neurol. 1990;301:325–342. doi: 10.1002/cne.903010302. [DOI] [PubMed] [Google Scholar]
- 25.Altman J, Bayer S A. J Comp Neurol. 1990;301:365–381. doi: 10.1002/cne.903010304. [DOI] [PubMed] [Google Scholar]
- 26.Ferrer I, Serrano T, Soriano E. Neurosci Res. 1990;8:60–66. doi: 10.1016/0168-0102(90)90058-m. [DOI] [PubMed] [Google Scholar]
- 27.Pellegrini M, Mansouri A, Simeone A, Boncinelli E, Gruss P. Development. 1996;122:3893–3898. doi: 10.1242/dev.122.12.3893. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida M, Suda Y, Matsuo I, Miyamoto N, Takeda N, Kuratani S, Aizawa S. Development. 1997;124:101–111. doi: 10.1242/dev.124.1.101. [DOI] [PubMed] [Google Scholar]
- 29.Porter F D, Drago J, Xu Y, Cheema S S, Wassif C, Huang S P, Lee E, Grinberg A, Massalas J S, Bodine D, et al. Development. 1997;124:2935–2944. doi: 10.1242/dev.124.15.2935. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Y, Sheng H Z, Amini R, Grinberg A, Lee E, Huang S, Taira M, Westphal H. Science. 1999;284:1155–1158. doi: 10.1126/science.284.5417.1155. [DOI] [PubMed] [Google Scholar]
- 31.Houser C R. Brain Res. 1990;535:195–204. doi: 10.1016/0006-8993(90)91601-c. [DOI] [PubMed] [Google Scholar]
- 32.Houser C R. Epilepsy Res Suppl. 1992;7:223–234. [PubMed] [Google Scholar]
- 33.Fernandez G, Effenberger O, Vinz B, Steinlein O, Elger C E, Döhring W, Heinze H J. Neurology. 1998;50:909–917. doi: 10.1212/wnl.50.4.909. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell L A, Jackson G D, Kalnins R M, Saling M M, Fitt G J, Ashpole R D, Berkovic S F. Neurology. 1999;52:327–336. doi: 10.1212/wnl.52.2.327. [DOI] [PubMed] [Google Scholar]
- 35.Vertes R P, Kocsis B. Neuroscience. 1997;81:893–926. doi: 10.1016/s0306-4522(97)00239-x. [DOI] [PubMed] [Google Scholar]
- 36.Whishaw I Q, Bland B H, Bayer S A. Brain Res. 1978;146:249–268. doi: 10.1016/0006-8993(78)90972-1. [DOI] [PubMed] [Google Scholar]