

Abstract
tRNA 3′ processing endoribonuclease (3′ tRNase) is an enzyme responsible for the removal of a 3′ trailer from precursor tRNA (pre-tRNA). We purified ∼85 kDa 3′ tRNase from pig liver and determined its partial sequences. BLAST search of them suggested that the enzyme was the product of a candidate human prostate cancer susceptibility gene, ELAC2, the biological function of which was totally unknown. We cloned a human ELAC2 cDNA and expressed the ELAC2 protein in Escherichia coli. The recombinant ELAC2 was able to cleave human pre-tRNAArg efficiently. The 3′ tRNase activity of the yeast ortholog YKR079C was also observed. The C-terminal half of human ELAC2 was able to remove a 3′ trailer from pre-tRNAArg, while the N‐terminal half failed to do so. In the human genome exists a gene, ELAC1, which seems to correspond to the C-terminal half of 3′ tRNase from ELAC2. We showed that human ELAC1 also has 3′-tRNase activity. Furthermore, we examined eight ELAC2 variants that seem to be associated with the occurrence of prostate cancer for 3′-tRNase activity. Seven ELAC2 variants which contain one to three amino acid substitutions showed efficient 3′-tRNase activities, while one truncated variant, which lacked a C-terminal half region, had no activity.
INTRODUCTION
Prostate cancer is a complex disorder that is caused by a multitude of genetic and environmental factors. Recently, a candidate prostate cancer susceptibility gene, ELAC2, has been identified at chromosome 17p by positional cloning and mutation screening (1). An insertion/frameshift mutation and a non-conservative missense mutation in ELAC2 segregate with prostate cancer in two kindreds (1). Several studies report that two common missense variants in the gene are also significantly associated with the occurrence of prostate cancer (1–3), although several groups argue against this association (4–6). Sequence analysis suggested that ELAC2 encodes an evolutionally conserved, metal-dependent hydrolase (1,7), which could partly explain environmental effects on human prostate cells by imagining differential interaction of the enzyme variants with some environmental exposure (1). In addition, the gene product showed similarity to a family of DNA interstrand crosslink repair proteins (8), which seemed consistent with a potential role in cancer susceptibility. Despite the fundamental biological interest, however, the genuine function of the ELAC2 product was totally unknown. Here we demonstrate experimentally that the prostate cancer susceptibility gene encodes an unfamiliar but important enzyme which is essential for tRNA biosynthesis, that is, tRNA 3′ processing endoribonuclease (3′ tRNase).
3′ tRNase is an enzyme responsible for the removal of a 3′ trailer from precursor tRNA (pre-tRNA) which is transcribed as a larger form (9). The enzyme cleaves pre-tRNA immediately downstream of a discriminator nucleotide (10,11), onto which the CCA residues are added to produce mature tRNA. We have shown that mammalian 3′ tRNase can remove 3′ trailers from various pre-tRNAs with various efficiencies (11–14). Pre-tRNA 5′ leaders affect the efficiency of tRNA 3′ processing reaction in vitro; leaders of 9 nt or longer severely inhibit the tRNA 3′ processing reaction, and even small 5′ leaders, when stably base-paired with 3′ trailers, hinder removal of the 3′ trailers by 3′ tRNase (14). Besides the 5′ leaders, the 3′ trailers also affect the cleavage efficiency of pre-tRNAs, which varies depending on both the length and the 5′ end nucleotide of the 3′ trailer (11).
Mammalian 3′ tRNase is unique in that a relatively stable complex between the enzyme and a 3′-truncated tRNA can function as a four-base-recognizing RNA cutter (RNase 65) (15–17). Although little is known about the physiological role and substrate of RNase 65, it has been demonstrated that the 3′-truncated tRNA directs substrate specificity via four base-pairings (10). Another unique point is that mammalian 3′ tRNase possesses a property of being able to cleave any RNA at any site under the direction of small guide RNA (18–20) like an RNA-induced silencing complex (RISC) underlying RNA interference (21).
In addition to these studies focusing on 3′-tRNase interactions with various pre-tRNAs and pre-tRNA-like complexes, we had also been making efforts towards cloning a 3′-tRNase cDNA, and finally came across the prostate cancer susceptibility gene.
MATERIALS AND METHODS
Enzyme purification and microsequencing
Pig 3′ tRNase was intensively fractionated from liver basically as described before (10), and further purified by glycerol gradient ultracentrifugation. We layered the 3′-tRNase fractions after Mono Q column chromatography onto a 10 ml 15–30% glycerol gradient in a buffer (20 mM Tris–HCl, pH 7.5, 0.2 mM EDTA), and centrifuged them at 100 000 g for 48 h at 4°C. The final active fraction was separated on an SDS–10% polyacrylamide gel. A protein band was visualized by staining the gel with Coomassie brilliant blue R-250.
We excised a gel strip containing the protein band, and subjected the protein to trypsin digestion and analyzed resulting peptides with a mass spectrometer. The microsequencing was carried out by Protana (Odense, Denmark).
Cloning of 3′-tRNase cDNAs
The complete protein coding region (2478 bp) of a human ELAC2 cDNA was PCR-amplified with LA Taq DNA polymerase (Takara Shuzo) from a human testis cDNA library (Clontech), and cloned between SapI and XhoI sites of the Escherichia coli expression vector pTYB11 (New England BioLabs). The two primer pairs 5′-GACAAGCTCTTCCAACATGTGGGCGCTTTGCTCG-3′/5′-ACACAGTTCTTCAGCCAA-3′ and 5′-TTGGCTGAAGAACTGTGT-3′/5′-CCGCTCGAGTTACTGGGCTCTGACCTTCT-3′ were used. Because the obtained cDNA contained several base substitutions that change amino acids, we repaired them by PCR using appropriate primer sets with Pyrobest DNA polymerase (Takara Shuzo) that has a proof-reading activity (22,23). We confirmed that the sequence of the ELAC2 coding region of the resultant plasmid pTYB11/ELAC2 is the same as the sequence previously published (GenBank accession no. AF304370), using a CEQ Dye Terminator Cycle Sequencing Kit (Bechman Coulter).
In a similar fashion, the full-length coding regions of a human ELAC1 cDNA (1089 bp) and the yeast YKR079C gene (2514 bp) were PCR-amplified from the human testis cDNA library and from the Saccharomyces cerevisiae genome, respectively. The primer pairs 5′-CGGGATCCATGTCTATGGATGTGACA-3′/5′-CCGCTCGAGTTATTTCTTGATTGGAATGC-3′ and 5′-GGAATTCCATGTTCACATT TATACCCATC-3′/5′-GGAATTCTTAATTTTTCTTGTGTTTCTTAA-3′ were used for the amplification of ELAC1 and YKR079C, respectively. The amplified ELAC1 cDNA was cloned between the BamHI and XhoI sites of pGEX-4T-3 (Amersham Pharmacia Biotech), and the amplified YKR079C gene was cloned into the EcoRI site of pGEX-4T-3. After corrections of PCR errors as above, we confirmed that the insert regions of pGEX-4T-3/ELAC1 and pGEX-4T-3/YKR079C are the same as the sequences previously published (GenBank accession nos AF308695 and Z28304, respectively).
Construction of expression plasmids for 3′-tRNase variants
Based upon pTYB11/ELAC2, we constructed its ten derivatives to express in E.coli 10 different 3′-tRNase variants: pTYB11/ELAC2-ΔC for the N‐terminal half ELAC2 (residues 1–480), pTYB11/ELAC2-ΔN for the C-terminal half ELAC2 (residues 482–826), pTYB11/ELAC2Ser217Leu for ELAC2 con taining a Ser217Leu substitution, pTYB11/ELAC2Ala541Thr for ELAC2 containing an Ala541Thr substitution, pTYB11/ELAC2Arg781His for ELAC2 containing an Arg781His substitution, pTYB11/ELAC2Ser217Leu/Ala541Thr for ELAC2 containing Ser217Leu and Ala541Thr substitutions, pTYB11/ELAC2Ala541Thr/Arg781His for ELAC2 containing Ala541Thr and Arg781His substitutions, pTYB11/ELAC2Arg781His/Ser217Leu for ELAC2 containing Arg781His and Ser217Leu substitutions, pTYB11/ELAC2Ser217Leu/Ala541Thr/Arg781His for ELAC2 containing Ser217Leu, Ala541Thr and Arg781His substitutions, and pTYB11/ELAC21641insG for ELAC2 containing the C-terminal truncation due to the G-insersion/frameshift change at nucleotide 1641. These pTYB11/ELAC2 derivatives were generated by site-directed mutagenesis by overlap extension using PCR (22,23) and/or by the conventional DNA recombination technique with DNA restriction enzymes and DNA ligase. We confirmed that the insert sequences are changed correctly by DNA sequencing as above.
Expression and purification of recombinant proteins encoded in the pTYB11 constructs
The recombinant ELAC2 and its variants were over-expressed in E.coli and purified with chitin beads as described previously (22). The expressed proteins were identified by liquid chromatographic mass spectrometry (LC/MS/MS) analyses with the ion trap mass spectrometer LCQ Advantage (Thermo Finnigan) equipped with an electrospray ionization source and the high performance liquid chromatography system MAGIC 2002 (Michrom BioResources).
Expression and purification of GST fusion proteins
Escherichia coli strain DH5α that harbors the expression plasmid for GST–YKR079C or GST–ELAC1 was incubated at 37°C in 250 ml of LB medium containing 50 µg/ml ampicillin until the A600 of the culture reached 0.6. At this point, the fusion protein was induced by adding IPTG (500 µM). After further incubation at 37°C for 4 h, the cells were harvested by centrifugation. Cell pellets were resuspended in 10 ml of lysis buffer (50 mM Tris–HCl, pH 7.6, 500 mM NaCl, 10% glycerol, 1 mM dithiothreitol (DTT), 23 mM AEBSF, 100 mM EDTA, 2 mM Bestatin, 0.3 mM E-64, 0.3 mM pepstatin A). The cells were sonicated and centrifuged at 100 000 g for 1 h. The cleared lysate was incubated with 0.5 ml of glutathione–Sepharose beads at 4°C for 12 h. After exhaustive washing, the retained proteins were eluted from the beads with 0.1 ml of buffer (50 mM Tris–HCl, pH 7.6, 10% glycerol) containing 20 mM glutathione. All of the purification steps were carried out at 4°C.
Pre-tRNA synthesis
The pre-tRNAsArg R-11TUUU and R-6L6TUUU were synthesized with T7 RNA polymerase (Takara Shuzo) from the synthetic DNA templates (22). The transcription reactions were carried out in the presence or absence of [α-32P]UTP (Amersham Pharmacia Biotech) under the conditions recommended by the manufacturer (Takara Shuzo), and the transcribed pre-tRNAs were gel-purified.
The unlabeled pre-tRNA transcript R-6L6TUUU was subsequently labeled with fluorescein according to the manufacturer’s protocol (Amersham Pharmacia Biotech). Briefly, after the removal of the 5′ phosphates of the transcript with bacterial alkaline phosphatase (Takara Shuzo), the transcript was phosphorylated with T4 polynucleotide kinase (Takara Shuzo) and ATPγS. Then a single fluorescein moiety was appended onto the 5′-phosphorothioate site. The resulting fluorescein-labeled R-6L6TUUU was gel-purified before assays.
In vitro tRNA 3′ processing assay
The 3′ processing reactions for 32P-labeled or fluorescein-labeled pre-tRNA (0.1 pmol) were performed with 3′ tRNases of various origins in a mixture (10 µl) containing 10 mM Tris–HCl (pH 7.5), 1.5 mM DTT and 3.2 mM MgCl2 at 37°C for 10 min. After resolution of the reaction products on a 10% polyacrylamide–8 M urea gel, the gel was analyzed with a Typhoon 9210 (Amersham Pharmacia Biotech).
RNA sequencing
The unlabeled pre-tRNAArg R-11TUUU (2 pmol) was reacted with pig liver 3′ tRNase (20 ng), ELAC2 (50 ng), YKR079C (50 ng) or ELAC1 (50 ng) under the standard assay conditions at 37°C for 10 min, extracted with phenol/chloroform, and precipitated with ethanol. The reaction products dissolved in water were 3′-end-labeled with T4 ligase (Takara Shuzo) and [5′-32P]pCp at 4°C for 10 h. The 5′ cleavage products were gel-purified, and their 3′-terminal sequences were determined by the chemical RNA sequencing method (23). The gel was analyzed with the Typhoon 9210.
RESULTS
Microsequencing of 3′ tRNase purified from pig liver
We previously reported that pig 3′ tRNase appeared to be a protein of ∼45 kDa (10), but further enzyme purification indicated that an apparent mass of the pig liver enzyme is ∼85 kDa on an SDS–10% polyacrylamide gel (Fig. 1A). Although we were not able to identify the ∼45 kDa protein, it could have been a proteolytic cleavage product of the ∼85 kDa protein. To determine an amino acid sequence of this further purified protein, we subjected the protein in an excised gel strip to trypsin digestion and analyzed resulting peptides with a mass spectrometer. Four internal peptide sequences, AEFLLLNHFSQR, LPLFSPDFNEK, VGLAFDHMK and ALFAGDLEEMEER, were derived and searched using BLAST against GenBank. The search suggested that the protein is the ELAC2 gene product based on the fact that the human ELAC2 amino acid sequence includes all these peptide sequences with a few amino acid mismatches probably due to species difference (Fig. 1B). The calculated molecular mass of human ELAC2 is 92 kDa, which is comparable to that of the pig enzyme estimated by the SDS–polyacrylamide gel electrophoresis (Fig. 1A).
Figure 1.

Purification and partial sequencing of pig 3′ tRNase. (A) The enzyme was intensively fractionated from pig liver basically as described before (10), and further purified by glycerol gradient ultracentrifugation. The final active fraction was separated on an SDS–10% polyacrylamide gel. (B) The four sequences derived from mass spectrometric analysis of the ∼85 kDa band are aligned with a C-terminal portion of the human ELAC2 sequence.
Human ELAC2 has the 3′-tRNase activity
To confirm that the ELAC2 product really has the 3′-tRNase activity, the complete protein coding region of an ELAC2 cDNA was PCR-amplified from a human testis cDNA library, and cloned into the E.coli expression vector pTYB11. The ELAC2 product was over-expressed as an intein-containing precursor in E.coli, and purified with chitin beads by removing its intein portion. The protein sample was checked for purity by SDS–polyacrylamide gel electrophoresis. The protein profile showed two protein bands, one of ∼60 kDa and the other of ∼100 kDa (Fig. 2). We analyzed both proteins with a mass spectrometer, and elucidated that the large and small proteins correspond to human ELAC2 and E.coli GroEL, respectively. An in vitro tRNA 3′ processing assay was performed with the recombinant ELAC2 and human pre-tRNAArg R-11TUUU synthesized in the presence of [α-32P]UTP by using an in vitro T7 RNA polymerase transcription system (Fig. 3A), and the reaction products were separated on a 10% polyacrylamide–8 M urea gel. As expected, this protein was able to cleave pre-tRNAArg at a specific site as pig liver 3′ tRNase was (Fig. 3B). A control reaction with the protein sample from the E.coli cells transformed with the vector pTYB11 generated no cleavage products (Figs 2 and 3B). We also examined commercially obtained E.coli GroEL for the 3′-tRNase activity, and confirmed that the protein has no such activity (data not shown).
Figure 2.
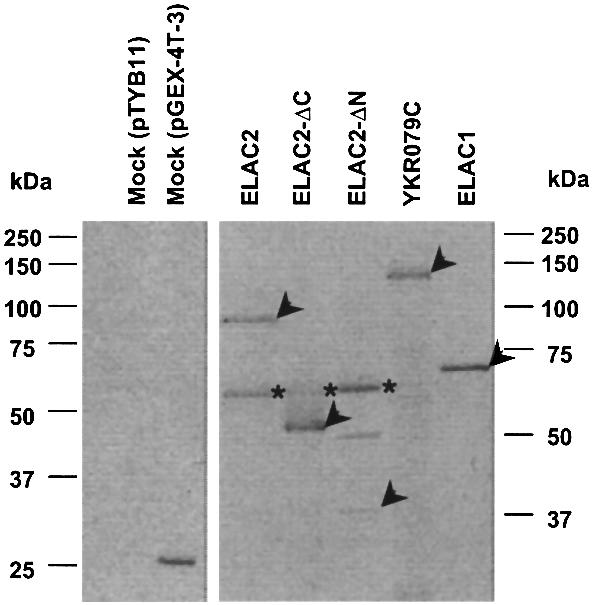
Recombinant protein profiles. The full, 5′-half and 3′-half coding regions of a human ELAC2 cDNA were cloned into the E.coli expression vector pTYB11 to produce the recombinant proteins ELAC2, ELAC2-ΔC (residues 1–480) and ELAC2-ΔN (residues 482–826), respectively. The YKR079C and ELAC1 coding regions were cloned into the E.coli expression vector pGEX-4T-3 to generate the recombinant proteins YKR079C and ELAC1, respectively. These recombinant proteins were over-expressed in E.coli, and purified with chitin or glutathione beads by a batch method. The vectors pTYB11 and pGEX-4T-3 were also used for mock transformations. The purified proteins (1 µg) were analyzed on an SDS–10% polyacrylamide gel, and stained with Coomassie brilliant blue R-250. By mass spectrometric analysis, we confirmed that each protein band designated by an arrowhead corresponds to each recombinant protein and that a band designated by an asterisk corresponds to E.coli GroEL. An ∼50 kDa protein in the ELAC2-ΔN sample was identified as E.coli 2-oxoglutarate dehydrogenase.
Figure 3.
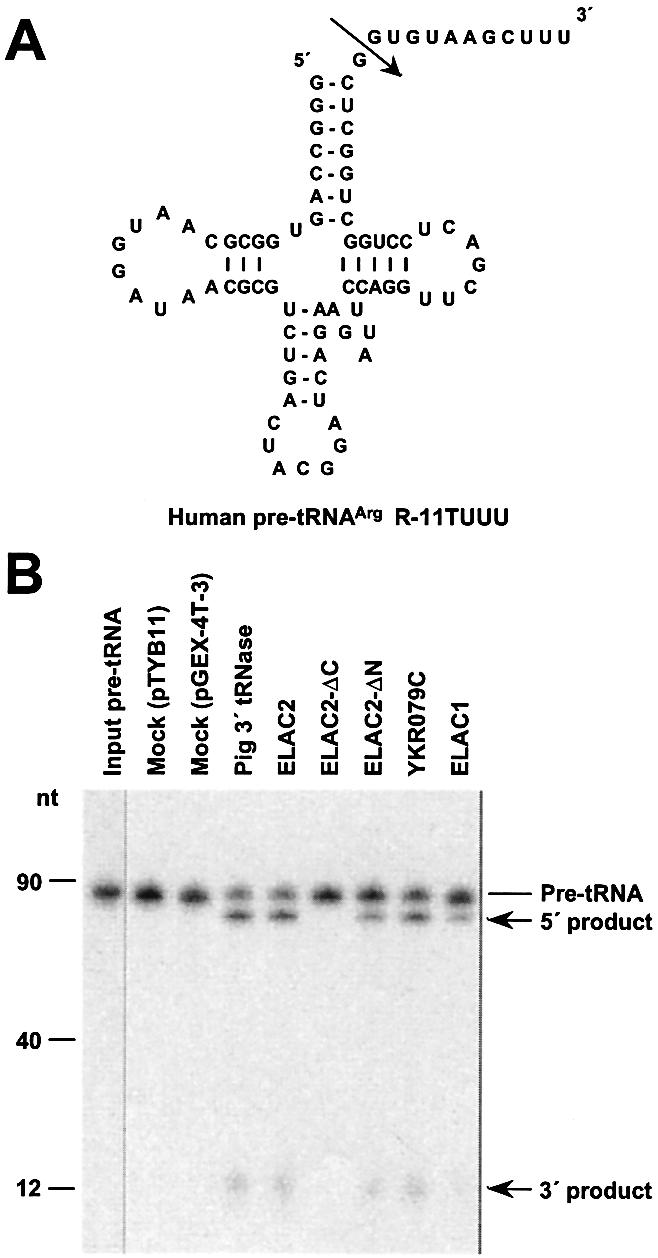
Tests of various ELAC2-related recombinant proteins for in vitro tRNA 3′ processing activity. (A) A secondary structure of human pre-tRNAArg R-11TUUU. An arrow denotes the cleavage site by 3′ tRNase. (B) The in vitro tRNA 3′ processing assays. Each recombinant protein (50 ng) or pig liver 3′ tRNase (20 ng) was incubated with uniformly 32P- labeled human pre-tRNAArg R-11TUUU (0.1 pmol) at 37°C for 10 min under the standard assay conditions (13). The cleavage reactions were analyzed on a 10% polyacrylamide–8 M urea gel.
To determine the exact cleavage site, we incubated cold human pre-tRNAArg with the recombinant ELAC2, gel-purified the 5′ cleavage product and 3′-end-labeled it with [5′-32P]pCp. Subsequently, the 32P-end-labeled product was subjected to chemical RNA sequencing (24). Figure 4 shows that the 3′ sequence reads ‘GCUCGGUCGG… (3′ to 5′)’, indicating that the cleavage occurs after the discriminator nucleotide G as the cleavage by the pig liver enzyme does (11).
Figure 4.
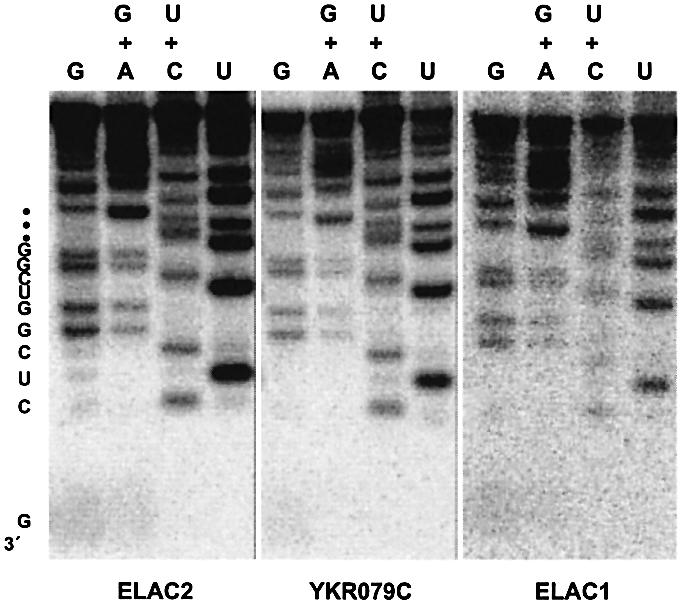
Determination of the exact cleavage site by recombinant 3′ tRNases, ELAC2, YKR079C and ELAC1. The unlabeled 5′ cleavage products generated in the in vitro 3′-tRNase assays were 3′-end-labeled with [5′-32P]pCp and T4 RNA ligase, gel-purified and subjected to chemical RNA sequencing (24). The sequencing reactions were analyzed on a 20% polyacrylamide–8 M urea gel. The 3′-terminal nucleotide G corresponds to the discriminator.
Identification of the yeast 3′-tRNase gene
BLAST searches of the ELAC2 amino acid sequence against GenBank revealed a single ortholog in S.cerevisiae (YKR079C) (1). ELAC2 and YKR079C share 21% identical residues and show 49% sequence similarity. To corroborate that the biological function of ELAC2 is to remove 3′ trailers endoribonucleolytically from pre-tRNAs, we tested the YKR079C product for 3′-tRNase activity. A PCR-amplified protein coding region of YKR079C was cloned into the E.coli expression plasmid pGEX-4T-3, and a glutathione-S-transferase (GST)-fused protein was expressed in E.coli and purified with glutathione beads. The GST–YKR079C fusion protein appeared as a band of ∼130 kDa on an SDS–10% polyacrylamide gel (Fig. 2). The size is consistent with the calculated molecular mass of 123 kDa, which consists of 26 kDa of GST and 97 kDa of YKR079C. The identity of the protein was confirmed by LC/MS/MS analysis.
The purified GST–YKR079C fusion protein was incubated with the human pre-tRNAArg R-11TUUU under the standard conditions. As shown for human ELAC2, the two species of cleavage products were generated by the fusion protein, while the cleavage was not observed in the presence of the ∼27 kDa GST protein alone (Figs 2 and 3B). This result shows that the cleavage of pre-tRNAArg is mediated directly by YKR079C and not by GST or by contaminating E.coli proteins. The 3′-terminal sequence of the long cleavage product was determined as above, and the obtained sequence indicated that the cleavage occurs after the discriminator as in the case of human ELAC2 (Fig. 4). Therefore, we concluded that the YKR079C product is yeast 3′ tRNase. Taken together, these results clearly demonstrate that ELAC2 indeed encodes tRNA 3′ processing endoribonuclease.
The C-terminal half of ELAC2 is essential for the 3′-tRNase activity
The multiple protein alignment of ELAC2 orthologs revealed that the C-terminal half regions have much higher similarity than the N‐terminal half regions (1). Thus, we assumed that the essential domains including the catalytic domain should exist in the C-terminal half of 3′ tRNase. To test this possibility, we constructed two pTYB11 expression plasmids lacking either N‐terminal (residues 1–481) or C-terminal (residues 481–826) half-coding region, expressed and purified half proteins as described for the full-length ELAC2. The LC/MS/MS analysis showed that the recombinant C-terminal half (ELAC2-ΔN) and N‐terminal half (ELAC2-ΔC) 3′ tRNases had apparent molecular weights of ∼53 and ∼38 kDa, respectively, on an SDS–10% polyacrylamide gel (Fig. 2). Consistently, the calculated masses were 54 and 38 kDa, respectively. Both protein samples were contaminated with E.coli GroEL, and another unrelated E.coli protein of ∼50 kDa, 2-oxoglutarate dehydrogenase, was detected in the ELAC2-ΔN sample. In the tRNA 3′ processing assay, ELAC2-ΔN was able to cleave the pre-tRNAArg, while ELAC2-ΔC was not at all (Fig. 3B). These results suggest that the C-terminal half of 3′ tRNase is essential for its activity.
Human ELAC1 has also the tRNA 3′ processing activity
Interestingly, in the human genome exists a gene, ELAC1, which is very similar to, but about half the size of ELAC2 (1), and seems to correspond to the C-terminal half of 3′ tRNase from ELAC2. To examine whether ELAC1 has also the tRNA 3′ processing activity as ELAC2-ΔN, we cloned a human cDNA encoding ELAC1 into the plasmid pGEX-4T-3, and expressed and purified the GST–ELAC1 fusion protein as described for GST–YKR079C (Fig. 2). On an SDS–10% polyacrylamide gel, GST–ELAC1 appeared as a protein of ∼70 kDa (Fig. 2), which is consistent with the calculated molecular mass of 66 kDa composed of 26 kDa from GST and 40 kDa from ELAC1. The protein identity was confirmed by LC/MS/MS analysis. From the 3′-tRNase assay for GST– ELAC1 and the RNA sequencing of the cleavage product, we demonstrated that the ELAC1 enzyme can also cleave pre-tRNAArg at the expected site (Figs 3B and 4). This result emphasizes the importance of the C-terminal half domain of 3′ tRNase from ELAC2.
Pre-tRNAArg containing a 5′ leader is also a good substrate for ELAC2 and ELAC1
Because pig liver 3′ tRNase can remove 3′ trailers efficiently even from pre-tRNAs containing 5′ leaders if the length of leaders is smaller than 9 nt, we examined whether the human pre-tRNAArg containing a 5′ leader R-6L6TUUU is also a substrate for human ELAC2. R-6L6TUUU was synthesized in vitro with T7 RNA polymerase, and subsequently 5′-end-labeled with fluorescein (Fig. 5A). In the 3′-tRNase assay under the standard conditions, human recombinant ELAC2 cleaved R-6L6TUUU as efficiently as the pig liver enzyme (Fig. 5B). We also examined the ELAC2-ΔC, ELAC2-ΔN, YKR079C and ELAC1 enzymes, and showed that these enzymes can cleave R-6L6TUUU with the exception of ELAC2-ΔC (Fig. 5B).
Figure 5.
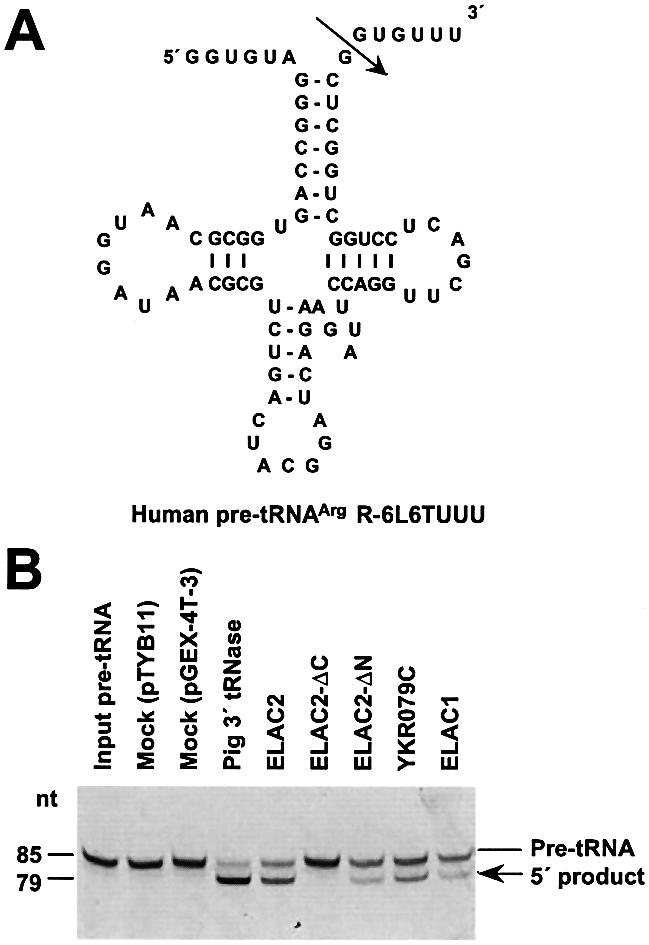
Assays of ELAC2-related recombinant proteins for the 3′-tRNase activity. (A) A secondary structure of the human pre-tRNAArg R-6L6TUUU. An arrow indicates the cleavage site by 3′ tRNase. (B) The in vitro 3′-tRNase assays. Each recombinant protein (50 ng) or pig liver 3′ tRNase (20 ng) was incubated with fluorescein-labeled R-6L6TUUU (0.1 pmol) at 37°C for 10 min under the standard assay conditions (13). The cleavage reactions were analyzed on a 10% polyacrylamide–8 M urea gel.
tRNA 3′ processing activities of enzyme variants associated with the occurrence of prostate cancer
It has been reported that two mutations in ELAC2, an insertion/frameshift (1641 ins G) and a missense change (Arg781His), segregate with prostate cancer in two pedigrees (1). The frameshift mutation results in premature termination after the miscoding of 67 residues. In addition, two common missense changes, Ser217Leu and Ala541Thr, seem to be associated with the occurrence of prostate cancer (1–3). In order to investigate the effects of these mutations on the 3′-tRNase activity, we constructed eight expression plasmids which can produce one frameshift (ELAC21641insG), three single missense (ELAC2Ser217Leu, ELAC2Ala541Thr and ELAC2Arg781His), three double missense (ELAC2Ser217Leu/Ala541Thr, ELAC2Ala541Thr/Arg781His and ELAC2Arg781His/ Ser217Leu) or one triple missense (ELAC2Ser217Leu/Ala541Thr/Arg781His) variant enzyme, based upon pTYB11/ELAC2. These variants were expressed in E.coli, purified and assayed for 3′-tRNase activity. Seven ELAC2 variants which contain one to three amino acid substitutions showed efficient 3′-tRNase activities, while one truncated variant, which lacked a C-terminal half region, had no activity, emphasizing again the importance of the C-terminal domain (Fig. 6).
Figure 6.

3′-tRNase activities of enzyme variants associated with the occurrence of prostate cancer. (A) Positions of the mutations in human ELAC2. Regions of the Walker A motif and the histidine motif are denoted by shaded rectangles. The regions corresponding to ELAC2-ΔC and ELAC2-ΔN are also indicated by thick bars. (B) The 3′-tRNase assays were performed with each variant enzyme (25 and 50 ng) and fluorescein-labeled R-6L6TUUU (0.1 pmol) at 37°C for 10 min under the standard conditions (13). The cleavage reactions were analyzed on a 10% polyacrylamide–8 M urea gel. Values of percent cleavage for low and high enzyme dosages were represented by open and filled bars, respectively. Data are the mean ± SD of three independent experiments.
DISCUSSION
We purified 3′ tRNase from pig liver and determined its partial sequences by mass spectrometry. The BLAST search of them against GenBank suggested that the human ELAC2 gene is the gene that encodes 3′ tRNase. To demonstrate this directly, we cloned the cDNA from human testis into the expression plasmid pTYB11, expressed it in E.coli and purified the ELAC2 product. The obtained ELAC2 sample clearly showed the 3′-tRNase activity, although it was contaminated with the bacterial GroEL. The possibility that GroEL has the 3′-tRNase activity was excluded by the experiment using the purchased pure GroEL. Another possibility that the observed activity is due to a trace amount of other contaminated bacterial proteins was also ruled out by the assay using the sample from mock transformed bacteria. Furthermore, we confirmed that a single yeast ortholog, YKR079C, has the 3′-tRNase activity by testing the recombinant yeast enzyme obtained through a different expression system with the vector pGEX-4T-3. We also showed that both recombinant enzymes, ELAC2 and YKR079C, can cleave pre-tRNAArg precisely after the discriminator nucleotide and that they can process not only pre-tRNA without a 5′ leader but also pre-tRNA with the leader like the pig liver 3′ tRNase. Therefore, we conclude that the gene ELAC2 indeed encodes 3′ tRNase. In addition, our data suggest that no other protein factors and no eukaryote-specific modifications are essential for the 3′-tRNase activity of ELAC2.
The YKR079C gene has been shown to be indispensable for yeast viability (1), suggesting that the yeast genome may have only one gene that encodes an enzyme responsible for correctly removing 3′ trailers from pre-tRNAs. Although a yeast 3′ tRNase with a molecular weight of 45/60 kDa has been identified, which can catalyze precise cleavage of a pre-tRNA at the discriminator site (25), the gene of this enzyme was unidentified. It is possible that the 45/60 kDa proteins detected on the polyacrylamide gel are degradation products of the 97 kDa intact YKR079C protein. Even these about half-size degradation products, if they contain the majority of the C-terminal domain, should show the 3′-tRNase activity like ELAC2-ΔN. Although several studies suggested the existence in yeast of another pathway to remove 3′ trailers by utilizing only exoribonucleases (26), the necessity of the YKR079C gene may argue against such pathways. Even in the situations where exoribonucleases can be involved in the pre-tRNA 3′ processing, the final cut to generate the discriminator termini for the CCA-adding enzyme may be carried out by 3′ tRNase. An alternative interpretation is also possible. The reason that the YKR079C gene is indispensable may be because the gene has another unknown essential function. Several unidentified yeast genes that are not related to YKR079C might also encode 3′ processing endoribonucleases.
We showed that human ELAC1, which is very similar to the C-terminal half domain of human ELAC2, has the 3′-tRNase activity. An ELAC1 ortholog of Arabidopsis thaliana has been also reported to show 3′-tRNase activity (27). These results are consistent with the observation that the C-terminal half of human ELAC2 is sufficient for the 3′-tRNase activity. Human ELAC2, human ELAC1 and their orthologs contain the histidine motif hhh[S/T]HXHXDHXXG (where h can be any bulky hydrophobic residue), which is highly conserved among them and forms a potential divalent metal ion binding site (1,7). It has been demonstrated experimentally that several histidine motifs are crucial for catalysis by enzymes such as β-lactamases and cyclic nucleotide phosphodiesterases (7,28). Curiously, ELAC2-ΔC did not show the 3′-tRNase activity at all, even though the N‐terminal half sequence is similar to the C-terminal one (1). This may be because of the lack of an intact histidine motif. The above consideration implies that the histidine motif is important for the 3′-tRNase catalytic activity. In contrast, the Walker A motif (29) that exists in the N‐terminal half region of human ELAC2 (1) is not essential for the activity. This ATP/GTP-binding motif may be involved in regulation of the catalysis by means of ATP or GTP.
BLAST searches revealed that the human genome contains both ELAC1 and ELAC2 genes, while the genomes of Caenorhabditis elegans and S.cerevisiae have only ELAC2 (1). This raises an interesting question whether the two distinct 3′ tRNases from ELAC1 and ELAC2 play differential roles in tRNA processing in the cells. It is possible that one is for nuclear tRNA processing, and that the other is for mitochondrial use. Although the iPSORT analysis for human ELAC2 predicted a mitochondrial localization, it has been reported recently that human ELAC2 exists both in cytoplasm and nuclei and interacts with the γ-tubulin complex (30). The ELAC2/γ-tubulin interaction suggests an additional function of ELAC2 such as cell cycle regulation.
As expected, ELAC21641insG, which lacks most of the C-terminal domain, did not show the enzymatic activity at all. Thus, 1641insG homozygotes must be lethal, unless the cells have another equivalent backup enzyme. Our data suggests that ELAC1 might be such an enzyme. Alternatively, some compensatory frameshift mechanism of the human translation system might generate an active enzyme. How could 1641insG heterozygotes confer increased risk of prostate cancer? The aberrant enzyme could interact with some unidentified regulatory factors, sequester them and interfere with the intact enzyme’s function, resulting in perturbation of the network of cellular gene regulation. In contrast, from the present assays, we did not observe meaningful differences in the 3′-tRNase activity between the wild-type ELAC2 and three single missense, three double missense and one triple missense variant enzymes. It is possible, however, that some differences could be detected when different pre-tRNA species are tested or kinetic parameters are measured. In any case, these missense variants all had sufficiently strong 3′-tRNase activities, although the substitution of Ser217 with the bulky hydrophobic residue Leu could affect the protein structure, and Ala541 which lies at the amino border of the histidine motif could be important for the enzymatic activity. Whether there are causalities between prostate cancer and the above missense mutations in ELAC2 is still controversial (1–6). From our results, it seems that the causalities could be fairly complicated, if they exist. The variant enzymes could differentially interact with some unknown regulatory proteins, or could be degraded more easily than the wild type, resulting in perturbation of cellular normal metabolism. Although it would not be straightforward to elucidate how such mutations could lead the cells to a malignant state, the present study in which we found out the cellular function of the ELAC2 product would be a great leap.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. Takeda and members of the NUPALS Undergraduate Molecular Biology Club for technical assistance.
REFERENCES
- 1.Tavtigian S.V., Simard,J., Teng,D.H., Abtin,V., Baumgard,M., Beck,A., Camp,N.J., Carillo,A.R., Chen,Y., Dayananth,P. et al. (2001) A candidate prostate cancer susceptibility gene at chromosome 17p. Nature Genet., 27, 172–180. [DOI] [PubMed]
- 2.Rebbeck T.R., Walker,A.H., Zeigler-Johnson,C., Weisburg,S., Martin,A.M., Nathanson,K.L., Wein,A.J., and Malkowicz,S.B. (2000) Association of HPC2/ELAC2 genotypes and prostate cancer. Am. J. Hum. Genet., 67, 1014–1019. [DOI] [PMC free article] [PubMed]
- 3.Fujiwara H., Emi,M., Nagai,H., Nishimura,T., Konishi,N., Kubota,Y., Ichikawa,T., Takahashi,S., Shuin,T., Habuchi,T., et al. (2002) Association of common missense changes in ELAC2 (HPC2) with prostate cancer in a Japanese case-control series. Hum. Genet., 47, 641–648. [DOI] [PubMed]
- 4.Wang L., McDonnell,S.K., Elkins,D.A., Slager,S.L., Christensen,E., Marks,A.F., Cunningham,J.M., Peterson,B.J., Jacobsen,S.J., Cerhan,J.R., et al. (2001) Role of HPC2/ELAC2 in hereditary prostate cancer. Cancer Res., 61, 6494–6499. [PubMed]
- 5.Xu J., Zheng,S.L., Carpten,J.D., Nupponen,N.N., Robbins,C.M., Mestre,J., Moses,T.Y., Faith,D.A., Kelly,B.D., Isaacs,S.D. et al. (2001) Evaluation of linkage and association of HPC2/ELAC2 in patients with familial or sporadic prostate cancer. Am. J. Hum. Genet., 68, 901–911. [DOI] [PMC free article] [PubMed]
- 6.Meitz J.C., Edwards,S.M., Easton,D.F., Murkin,A., Ardern-Jones,A., Jackson,R.A., Williams,S., Dearnaley,D.P., Stratton,M.R., Houlston,R.S. and Eeles,R.A. (2002) HPC2/ELAC2 polymorphisms and prostate cancer risk: analysis by age of onset of disease. Br. J. Cancer, 87, 905–908. [DOI] [PMC free article] [PubMed]
- 7.Melino S., Capo,C., Dragani,B., Aceto,A. and Petruzzelli,R. (1998) A zinc-binding motif conserved in glyoxalase II, beta-lactamase and arylsulfatases. Trends Biol. Sci., 23, 381–382. [DOI] [PubMed]
- 8.Niegemann E. and Brendel,M. (1994) A single amino acid change in SNM1-encoded protein leads to thermoconditional deficiency for DNA cross-link repair in Saccharomyces cerevisiae. Mutat. Res., 315, 275–279. [DOI] [PubMed]
- 9.Schurer H., Schiffer,S., Marchfelder,A. and Morl,M. (2001) This is the end: processing, editing and repair at the tRNA 3′-terminus. Biol. Chem., 382, 1147–1156. [DOI] [PubMed]
- 10.Nashimoto M. (1995) Conversion of mammalian tRNA 3′ processing endoribonuclease to four-base-recognizing RNA cutters. Nucleic Acids Res., 23, 3642–3647. [DOI] [PMC free article] [PubMed]
- 11.Nashimoto M. (1997) Distribution of both lengths and 5′ terminal nucleotides of mammalian pre-tRNA 3′ trailers reflects properties of 3′ processing endoribonuclease. Nucleic Acids Res., 25, 1148–1155. [DOI] [PMC free article] [PubMed]
- 12.Nashimoto M., Tamura,M. and Kaspar,R.L. (1999) Selection of cleavage site by mammalian tRNA 3′ processing endoribonuclease. J. Mol. Biol., 287, 727–740. [DOI] [PubMed]
- 13.Nashimoto M., Tamura,M. and Kaspar,R.L. (1999) Minimum requirements for substrates of mammalian tRNA 3′ processing endoribonuclease. Biochemistry, 38, 12089–12096. [DOI] [PubMed]
- 14.Nashimoto M., Wesemann,D.R., Geary,S., Tamura,M. and Kaspar,R.L. (1999) Long 5′ leaders inhibit removal of a 3′ trailer from a precursor tRNA by mammalian tRNA 3′ processing endoribonuclease. Nucleic Acids Res., 27, 2770–2776. [DOI] [PMC free article] [PubMed]
- 15.Nashimoto M., Kominami,R., Nishi,S. and Mishima,Y. (1991) A novel spermidine-dependent endoribonuclease activity caused by RNA–protein complex in mouse FM3A cell extracts. Biochem. Biophys. Res. Commun., 176, 1163–1169. [DOI] [PubMed]
- 16.Nashimoto M. (1992) Characterization of the spermidine-dependent, sequence-specific endoribonuclease that requires transfer RNA for its activity. Nucleic Acids Res., 20, 3737–3742. [DOI] [PMC free article] [PubMed]
- 17.Nashimoto M. (1993) 3′ truncated tRNAArg is essential for in vitro specific cleavage of partially synthesized mouse 18S rRNA. Nucleic Acids Res., 21, 4696–4702. [DOI] [PMC free article] [PubMed]
- 18.Nashimoto M. (1996) Specific cleavage of target RNAs from HIV-1 with 5′ half tRNA by mammalian tRNA 3′ processing endoribonuclease. RNA, 2, 523–534. [PMC free article] [PubMed]
- 19.Nashimoto M., Geary,S., Tamura,M. and Kaspar,R. (1998) RNA heptamers that direct RNA cleavage by mammalian tRNA 3′ processing endoribonuclease. Nucleic Acids Res., 26, 2565–2571. [DOI] [PMC free article] [PubMed]
- 20.Nashimoto M. (2000) Anomalous RNA substrates for mammalian tRNA 3′ processing endoribonuclease. FEBS Lett., 472, 179–186. [DOI] [PubMed]
- 21.McManus M.T. and Sharp,P.A. (2002) Gene silencing in mammals by small interfering RNAs. Nature Rev. Genet., 3, 737–747. [DOI] [PubMed]
- 22.Nashimoto M., Nashimoto,C., Tamura,M., Kaspar,R.L. and Ochi,K. (2001) The inhibitory effect of the autoantigen La on in vitro 3′ processing of mammalian precursor tRNAs. J. Mol. Biol., 312, 975–984. [DOI] [PubMed]
- 23.Ho S.N., Hunt,H.D., Horton,R.M., Pullen,J.K. and Pease,L.R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene, 77, 51–59. [DOI] [PubMed]
- 24.Peattie D.A. (1979) Direct chemical method for sequencing RNA. Proc. Natl Acad. Sci. USA, 76, 1760–1764. [DOI] [PMC free article] [PubMed]
- 25.Papadimitriou A. and Gross,H.J. (1996) Pre-tRNA 3′-processing in Saccharomyces cerevisiae. Purification and characterization of exo- and endoribonucleases. Eur. J. Biochem., 242, 747–759. [DOI] [PubMed]
- 26.Yoo C.J. and Wolin,S.L. (1997) The yeast La protein is required for the 3′ endonucleolytic cleavage that matures tRNA precursors. Cell, 89, 393–402. [DOI] [PubMed]
- 27.Schiffer S., Rosch,S. and Marchfelder,A. (2002) Assigning a function to a conserved group of proteins: the tRNA 3′-processing enzymes. EMBO J., 21, 2769–2777. [DOI] [PMC free article] [PubMed]
- 28.Francis S.H., Turko,I.V. and Corbin,J.D. (2001) Cyclic nucleotide phosphodiesterases: relating structure and function. Prog. Nucleic Acid Res. Mol. Biol., 65, 1–52. [DOI] [PubMed]
- 29.Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed]
- 30.Korver W., Guevara,C., Chen,Y., Neuteboom,S., Bookstein,R., Tavtigian,S. and Lees,E. (2003) The product of the candidate prostate cancer susceptibility gene ELAC2 interacts with the γ-tubulin complex. Int. J. Cancer, 104, 283–288. [DOI] [PubMed]