

Abstract
The MADS-box transcription factor Mcm1p and forkhead (FKH) transcription factor Fkh2p act in a DNA-bound complex to regulate cell-cycle dependent expression of the CLB2 cluster in Saccharomyces cerevisiae. Binding of Fkh2p requires prior binding by Mcm1p. Here we have investigated the molecular determinants governing the formation of the Mcm1p– Fkh2p complex. Fkh2p exhibits cooperativity in complex formation with Mcm1p and we have mapped a small region of Fkh2p located immediately upstream of the FKH DNA binding domain that is required for this cooperativity. This region is lacking in the related protein Fkh1p that cannot form ternary complexes with Mcm1p. A second region is identified that inhibits Mcm1p-independent DNA binding by Fkh2p. The spacing between the Mcm1p and Fkh2p binding sites is also a critical determinant for complex formation. We also show that Fkh2p can form ternary complexes with the human counterpart of Mcm1p, serum response factor (SRF). Mutations at analogous positions in Mcm1p, which are known to affect SRF interaction with its partner protein Elk-1, abrogate complex formation with Fkh2p, demonstrating evolutionary conservation of coregulatory protein binding surfaces. Our data therefore provide molecular insights into the mechanisms of Mcm1p– Fkh2p complex formation and more generally aid our understanding of MADS-box protein function.
INTRODUCTION
The transcription factor Mcm1p regulates several diverse processes including mating type determination, arginine metabolism and cell-cycle control in Saccharomyces cerevisiae (reviewed in 1,2). In each case, Mcm1p recruits distinct coregulatory proteins to promoters to enable specific responses to be elicited. During the cell cycle, Mcm1p controls the expression of early cell-cycle box-regulated genes at the M/G1 phase boundary (3) and the expression of the CLB2 cluster in the G2 and M phases (4), where it functions in a complex with the forkhead (FKH) transcription factor, Fkh2p (reviewed in 5) (6–9). Fkh2p cooperatively binds with Mcm1p to form swi five factor (SFF) complexes on the promoters of genes in the CLB2 cluster including SWI5 (8,10). In contrast, the related transcription factor Fkh1p does not appear to form part of the SFF complex that forms on the SWI5 promoter or exhibit binding cooperativity with Mcm1p (8,10), suggesting a specific role for Fkh2p in regulating gene expression through these complexes. The Mcm1p–Fkh2p complex activates transcription in the G2 and M phases of the cell cycle in conjunction with the coactivator Ndd1p (6) and, along with Fkh1p, appears to play a repressive role on target genes during the rest of the cell cycle (8).
The Mcm1p–Fkh2p complex exhibits several similarities with the mammalian Elk-1–serum response factor (SRF) complex. The Elk-1–SRF complex is also thought to play a role in cell-cycle control but acts to regulate cell-cycle entry in response to mitogenic signalling (reviewed in 11,12). Together with SAP-1 and SAP-2, Elk-1 constitutes part of the ternary complex factor (TCF) subfamily of ETS-domain proteins. Like FKH transcription factors, ETS-domain proteins contain a winged helix–turn–helix DNA binding domain. SRF and Mcm1p are both members of the MADS-box family of transcription factors and their core DNA binding domains are 70% identical (reviewed in 2). This similarity is emphasised by their related DNA binding specificities and the abilities of Mcm1p and SRF to interact with several of their respective binding partners (13–15). In addition to protein– DNA contacts, protein–protein interactions are essential for the formation of the Elk-1–SRF complex. Structural and mutagenic studies on the related Elk-1–SRF and SAP-1–SRF complexes have identified the reciprocal interaction surfaces in these transcription factors (14,16–18). A key feature of this interaction is the insertion of an aromatic residue in the TCF component into a hydrophobic pocket on the surface of SRF. The analogous pocket on the surface of Mcm1p is used to bind to the coregulatory protein MATα2 (19).
In this study, we have investigated the molecular determinants required for the formation of the Mcm1p–Fkh2p complex. We show that the spacing of the Fkh2p and Mcm1p binding sites is important for permitting complex function. Moreover, Fkh2p contains an autoinhibitory domain that stops autonomous DNA binding in the absence of Mcm1p and a second motif that is required for cooperative DNA binding with Mcm1p. Upon disruption of the reciprocal binding pocket on Mcm1p, Fkh2p binding is abrogated. Mcm1p is essential for viability and versions containing mutations of the Fkh2p binding pocket cannot rescue a deletion of the MCM1 gene. Similarly, a mutation in the Mcm1p binding motif of Fkh2p affects Fkh2p activity in vivo. The molecular determinants of Mcm1p–Fkh2p complex formation therefore show several parallels with the Elk-1–SRF complex although important differences also exist.
MATERIALS AND METHODS
Plasmid constructions and mutagenesis
The pBluescript-KS+-derived plasmids, pAS1 (encoding coreSRF; amino acids 132–222), pAS37 (encoding METcoreSRF; amino acids 142–222) (20), and pAS704 (encoding coreMcm1; amino acids 1–98) (21) and pAS798 (22) have been described previously.
The following plasmids were created for in vitro transcription/translation. CoreMcm1 mutants: plasmids encoding single point mutants, pAS1611 (T71A), pAS1612 (T82E), pAS1613 (V69E) and pAS1128 (T71E), were constructed using fragments obtained from a two-step PCR protocol (23) with two flanking primers FOR and REVL, the template pAS704 and the mutagenic primers ADS941, ADS944, ADS943 and ADS983, respectively. PCR products were cleaved with XbaI and either NcoI or HindIII and ligated into pAS37 derivatives cleaved with the same enzymes. pAS1607 and pAS1608 (encoding Fkh1p amino acids 227–421 and 287–421, respectively) were constructed by inserting NcoI/XhoI-cleaved PCR fragments (generated using primers ADS906/908 and ADS907/908, respectively, and yeast genomic DNA as a template) into the same sites in pAS798. pAS1241 [encoding Fkh2(458–862)], pAS1243 [encoding Fkh2(1–458)], pAS1244 [encoding Fkh2(180–458)], pAS1246 [encoding Fkh2(216–458)], pAS1247 [encoding Fkh2(254–458)], pAS1248 [encoding Fkh2(292–458)], pAS1249 [encoding Fkh2(325–458)], pAS1830 [encoding Fkh2(302–458)] and pAS1829 [encoding Fkh2(312–458)] were constructed by inserting NcoI/XhoI-cleaved PCR fragments (generated using primers ADS863/862, ADS857/871, ADS870/871, ADS877/871, ADS878/871, ADS879/871, ADS880/871, ADS1003/871 and ADS1004/871, respectively) into the same sites in pAS798. pAS1242 [encoding Fkh2(1–862)], was constructed by inserting a NcoI-cleaved PCR fragment (generated using primers ADS857/858, on yeast genomic DNA) into NcoI-cleaved pAS1241. pAS1764 encoding Fkh2(292–458)(Y315E) was constructed using fragments obtained from a two-step PCR protocol (23) with two flanking primers ADS879 and REVL, the template pAS1247 and the mutagenic primer ADS1905. The PCR product was cleaved with NcoI and XhoI and ligated into pGBKT7 (Clontech) cleaved with NcoI and SalI.
The following bacterial expression vectors were created: pAS1752 (encoding Fkh2p amino acids 254–458 fused to GST) was constructed by inserting a EcoRI/XhoI-cleaved PCR fragment into the same sites in pGEXKG (24). pAS799 [encoding Mcm1p amino acids 1–96 fused to maltose binding protein (MBP)] was kindly provided by Cynthia Wolberger.
The following yeast expression vectors were created: pMW20-NDD1 (URA3 CEN/ARS) was created by ligating a BamHI-digested PCR fragment carrying the full-length NDD1 gene into the same site in pMW20 (kindly provided by M. Walberg). pAS797 [Mcm1 amino acids 1–98; aka pSL2190 (25); Mcm1(1–98) under the control of the MCM1 upstream promoter region in vector pRS314, a CEN/ARS-TRP1 yeast shuttle vector]. pAS1768, encoding the V69E mutant Mcm1p clone was created by replacing the EcoRI/BglII fragment from pAS797 with an identically cleaved PCR fragment from pAS1613. pAS1762 (encoding a GAL4 DNA binding domain fusion to Fkh2p amino acids 1–458) was created by inserting a NcoI/EcoRI-cleaved PCR fragment into the same sites in pAS1241. pAS1767 (encoding a GAL4 DNA binding domain fusion to the Y315E mutant of Fkh2p amino acids 1–458) was created using a two-step PCR protocol (23) on the template pAS1242 using the mutagenic primer ADS1905. The PCR product was cleaved with NcoI and XhoI and ligated into pGBKT7 (Clontech) cleaved with NcoI and SalI.
Details of PCR primers can be supplied upon request. The sequences of all plasmids encoding mutant proteins and PCR-derived sequences were confirmed by automated dideoxy sequencing.
Protein production and pulldown assays
Wild-type and mutant Mcm1p proteins (using wheat germ lysates, Promega), SRF (using rabbit reticulocyte lysates, Promega) and Fkh2p derivatives (using wheat germ or rabbit reticulocyte lysates, Promega) were produced by coupled or sequential in vitro transcription and translation and subsequently analysed and quantified as described previously (26). MBP–Mcm1(1–96) fusion proteins were purified as described previously (27) and the Mcm1p moiety released by factor Xa cleavage. GST fusion proteins were prepared and pulldown assays were carried out as described previously (28) using GST–Fkh2(254–458) and in vitro translated Mcm1p derivatives.
Gel retardation analysis
Gel retardation assays on the SWI5 site were carried out essentially as described previously (8) or the indicated derivatives (see Fig. 5A). All sites had CTAG overhangs at their 5′ ends for labelling with Klenow. The following sources of protein were used: bacterially-expressed Mcm1p (Figs 3 and 5), in vitro translated Mcm1p (wheat germ lysate, Figs 1, 2, 4 and 6), in vitro translated Fkh2p (wheat germ lysate, Figs 3 and 5; rabbit reticulocyte lysate, Figs 1, 2, 4 and 6).
Figure 5.

Influence of binding site spacing on Mcm1p–Fkh2p complex formation. (A) Schematic of the Mcm1p–Fkh2p ternary complex bound to the P(SWI5) site. The sequence of the wild-type site and ‘spacer’ mutants are shown below. The fkh binding motif is shown in bold and underlined and the CArG box Mcm1p binding motif is boxed. (B and C) Gel retardation analysis of Fkh2(1–862) (B) and Fkh2(325–458) (C) in the presence (B) or absence (C) of Mcm1(1–96) bound to the indicated wild-type (WT) and ‘spacer’ mutant binding sites. (D) Schematic of the Mcm1p–Fkh2p complex and the P(SW15)Rev binding site. The arrows represent the relative orientation of the Mcm1p and Fkh2p binding sites. (E and F) Gel retardation analysis of Fkh2(1–862) (E) and Fkh2(325–458) (F) in the presence (E) and absence (F) of Mcm1p on the WT and Rev versions of the P(SWI5) site. The locations of Mcm1p, Fkh2p and Mcm1p–Fkh2p complexes are indicated in each panel.
Figure 3.

Fine mapping of the Fkh2p ‘cooperativity domain’. (A) A schematic of the truncated Fkh2(292–458) protein. The amino acid sequence of the sequence immediately preceding the FKH domain of Fkh2p and the analogous region in Fkh1p are shown. The amino acids at the truncation endpoints are highlighted. (B) Gel retardation analysis of the indicated Fkh2p truncated proteins in the absence (lanes 1–4) or presence (lanes 5–8) of Mcm1(1–96) on the P(SWI5) site. (C) Gel retardation analysis of increasing amounts of the truncated Fkh2p truncated proteins in the presence (lanes 1–10) or absence (lanes 11–20) of Mcm1(1–96). Relative molar amounts of Fkh2p were 1 (lanes 1, 6, 11 and 16); 3 (lanes 2, 7, 12, 17); 5 (lanes 3, 8, 13, 18); 10 (lanes 4, 9, 14, 19); 20 (lanes 5, 10, 15, 20). (D) Quantification of the data in (C). The binding observed with the highest concentrations of Fkh2p used is not shown as the binding in lane 5 is beyond the linear range. (E) Alignment of amino acids 312–333 of Fkh2p with the B-box region (amino acids 150–168) of Elk-1. Amino acids in Elk-1 that affect interactions with SRF by >50% (18) are shaded. These are conserved in Fkh2p. (F) Gel retardation analysis of the indicated Fkh2p mutant proteins in the absence (lanes 1–3) or presence (lanes 4–6) of Mcm1(1–96) on the P(SWI5) site.
Figure 1.
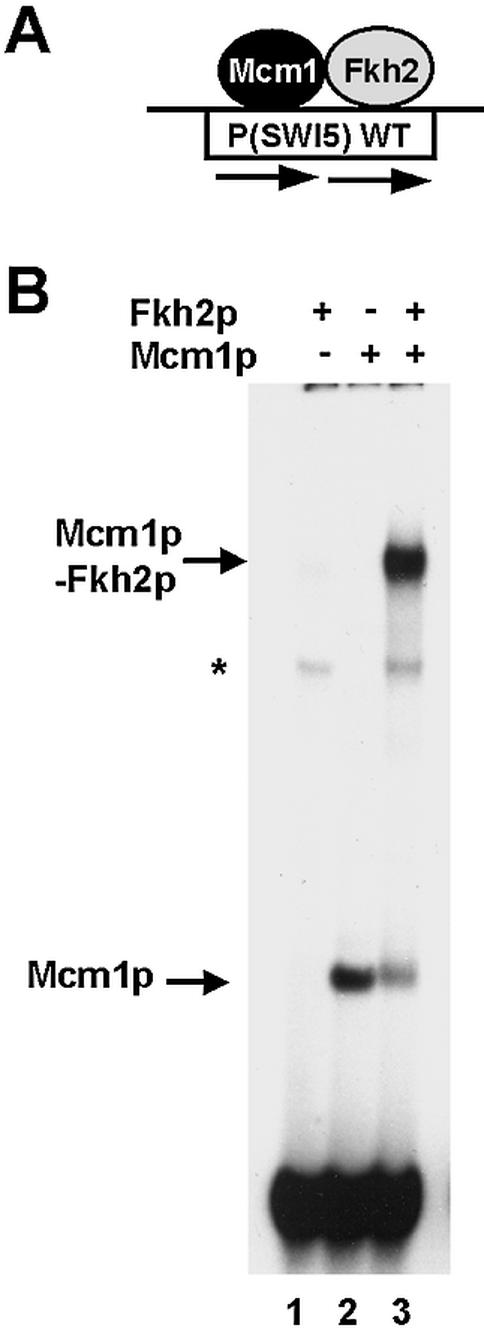
Fkh2p requires Mcm1p to bind the SWI5 promoter. (A) Schematic of the ternary Mcm1p–Fkh2p complex that forms on the composite CArG–Fkh binding motif in the SWI5 promoter. (B) Gel retardation analysis of in vitro translated Mcm1(1–98) and full-length Fkh2p on the CArG–fkh motif from the SWI5 promoter. The addition of Fkh2p and Mcm1p, and the locations of the DNA-bound Mcm1p and Mcm1p–Fkh2p complexes are indicated. The asterisk represents a non-specific band arising from the reticulocyte lysates.
Figure 2.

Mapping the ternary complex determinants in Fkh2p. (A) A schematic of the truncated Fkh2p constructs generated for the mapping analysis. The locations of the FHA and FKH domains are indicated. The minimal region required for ternary complex formation and the location of the autoinhibitory domain are indicated. The dotted line indicates that the C-terminal end of the autoinhibitory motif is unknown. The ability of each Fkh2p construct to form ternary complexes and exhibit autoinhibitory DNA binding properties is indicated. (B and C) Gel retardation analysis of the indicated Fkh2p proteins in the presence or absence of Mcm1(1–98) (B) or in the presence of SRF(132–222) on the P(SWI5) site. Asterisks indicate Fkh2p truncations that bind DNA in the absence of Mcm1p.
Figure 4.

Fkh1p does not bind cooperatively with Mcm1p. (A) Schematic of full-length and truncated versions of Fkh1p and Fkh2p. (B) Gel retardation analysis of the indicated Fkh1p and Fkh2p truncated proteins in the absence (lanes 1–4) or presence (lanes 5–8) of Mcm1(1–98) on the P(SW15) site. The locations of complexes corresponding to Mcm1p alone, Fkh1p/Fkh2p alone or Mcm1p–Fkh2p complexes are indicated. The asterisk indicates the location of a band corresponding to a weak ternary complex.
Figure 6.

Identification of the Fkh2p binding surface on Mcm1p. (A) Alignment of the Elk-1 binding regions in SRF with the equivalent region in Mcm1p. Residues mutated in this study are highlighted. Dots between the two sequences indicate identical amino acids. Residues comprising secondary structural elements are bracketed. (B) A structural representation of Mcm1p bound to DNA (monomers coloured in yellow and orange) (19). The residues mutated in this study are shown in blue. (C and D) Gel retardation analysis of the indicated Mcm1(1–98) mutants in the absence (C and D, lanes 1–5) or the presence of either Fkh2(1–862) (C, lanes 6–10) or Fkh2(254–458) (D, lanes 6–10) on the P(SWI5) site. The locations of complexes corresponding to Mcm1p alone or Mcm1p–Fkh2p are indicated. The amount of binary Mcm1p complex was normalised to give equivalent binding. (E) A GST pulldown experiment of GST–Fkh2(254–458) and the indicated in vitro translated Mcm1(1–98) mutants. Ten percent of input protein is shown. Quantification of the data normalised to input protein is shown below and compared with wild-type Mcm1p (taken as ‘100’). The average of three independent experiments is shown.
Yeast strains, growth conditions and 5-fluoroorotic acid (5-FOA) selection
Yeast strains used were: YY2052 [MATa P(PAL)-lacZ::fus1/pSL1574], where pSL1574 is a plasmid that contains MCM1 on a CEN/ARS-URA3 vector (25), AP173 (a/α ade– leu2 trp1 his3 ura3 FKH2/fkh2::URA3 NDD1/ndd1::HIS3); AP175 (a/α ade– leu2 trp1 his3 ura3 FKH2/fkh2::LEU2 NDD1/ndd1:: HIS3) and AP183 (a ade– leu2 trp1 his3 ura3 fkh2::LEU2 ndd1::HIS3). To construct a fkh2Δndd1Δ haploid strain, one copy of the NDD1 gene was replaced by the HIS3 gene, and the URA3 selectable marker disrupting one copy of the FKH2 gene in AP173, was replaced by the LEU2 gene, to create AP175. The HIS3 and LEU2 cassettes were amplified from YDp-HIS and YDp-LEU (29) by PCR using the Ndd1delfow/Ndd1delrev and F2-1/F2-2 oligonucleotide primers, respectively. Gene replacements were confirmed by PCR. The fkh2Δndd1Δ haploid strain, AP183, was isolated following sporulation of the AP175 diploid containing pMW20-NDD1. Details of primers can be provided on request.
Yeast strains were grown at 30°C in rich (YPD) or minimal (SD) media (28). Sporulation medium was 1% potassium acetate, 0.1% yeast extract, 0.05% dextrose and 2% agar (27). Yeast cells were transformed using the lithium acetate method (30).
FOA selection was used to select for cells that could lose pMW20-NDD1 (in a fkh2Δ background) and pSL1574 (in a mcm1Δ background). The fkh2Δndd1Δ haploid strain, AP183, was co-transformed with pMW20-NDD1 and either pGBKT7 (vector), pGBKT7-Fkh2(1–458) or pGBKT7-Fkh2(1–458) (Y315E). YY2052 was transformed with pAS797 or pAS1768. Equal numbers of cells from 20 colonies of each transformation were re-suspended in 1 ml of water. Ten-fold serial dilutions (neat, 1/10, 1/100, 1/1000) were then spotted onto SD plates or plates containing 5-FOA and the appropriate supplements. Plates were incubated at 30°C for 2–5 days to kill cells containing the URA3 gene (31).
Figure generation
All figures were generated electronically from either phosphorimager files (Bio-Rad Quantity One software) or scanned autoradiographic images using Adobe PhotoDeluxe (Adobe) and PowerPoint (Microsoft) software. Final images are representative of the original autoradiographic images.
RESULTS
Identification of regions of Fkh2p involved in cooperative Mcm1p–Fkh2p complex formation
Fkh2p forms ternary complexes with Mcm1p on the promoters of genes in the CLB2 gene cluster such as SWI5 (Fig. 1A) (6–9). To establish whether Fkh2p requires Mcm1p to bind to the SWI5 promoter, we carried out gel retardation analysis with a fragment of the SWI5 promoter and in vitro translated Mcm1(1–98) and full-length Fkh2(1–862) proteins (Fig. 1B). Whilst Mcm1p binds in the absence of Fkh2p, Fkh2p binding is absolutely dependent on the presence of Mcm1p (Fig. 1B, lanes 1 and 3). This is indicative of cooperative binding, and is consistent with other recent observations (10).
A series of truncated Fkh2p proteins was created to map the regions required for ternary complex formation with Mcm1p (Fig. 2A). These proteins were tested for DNA binding in the presence (Fig. 2B, lanes 1–8) or absence (Fig. 2B, lanes 9–16) of Mcm1p. Ternary complex formation was still observed upon deletion of the C-terminus (amino acids 458–862) of Fkh2p (Fig. 2B, lane 3), and was retained upon further N‐terminal deletion to amino acid 292 (Fig. 2B, lane 7). However, ternary complex formation was lost upon further N‐terminal truncation to amino acid 325 (Fig. 2B, lane 8). Unexpectedly, the formation of stable ternary complexes was lost in the Fkh2(180–458) and Fkh2(216–458) constructs (Fig. 2B, lanes 4 and 5) although the band smearing in these lanes is indicative of complex breakdown during electrophoresis (see Discussion). We did not expect the Fkh2p truncations to bind to the SWI5 site in the absence of Mcm1p and indeed this was the case upon N‐terminal deletion to amino acid 254 (Fig. 2B, lanes 9–14). However, upon N‐terminal truncation to amino acid 292, autonomous DNA binding by Fkh2p was revealed (Fig. 2B, lane 15). Thus, sequences located between amino acids 254 and 292 act to inhibit autonomous DNA binding by Fkh2p.
Previously we proposed that the Mcm1p–Fkh2p complex might be functionally analogous to the human Elk-1–SRF complex (8). The Mcm1p and SRF core DNA binding domains exhibit a high degree of sequence similarity and related DNA binding specificities (70% identity) (15), hence we next tested whether coreSRF (amino acids 132–222) could interact with Fkh2p on the SWI5 site (Fig. 2C). As observed with Mcm1p, ternary complex formation with SRF was identifiable upon C- and N‐terminal deletion of Fkh2p to amino acids 292–458. However, unlike Mcm1p–Fkh2p ternary complexes, ternary complexes between either Fkh2(216–458) or Fkh2(254–458) and SRF did not undergo dissociation during electrophoresis, suggesting a more stable interaction. However, the binding of longer Fkh2 derivatives was reduced compared with Fkh2(254–458), suggesting a possible role for additional inhibitory regions in this context.
To further delineate the minimal part of Fkh2p that could form ternary complexes with Mcm1p, an additional series of N‐terminal truncations was made (Fig. 3A). In this series of experiments, Mcm1(1–96) was used. DNA binding by the Fkh2p derivatives was similar in the absence of Mcm1p (Fig. 3B, lanes 1–4). However, ternary complex formation was reduced upon truncation beyond amino acid 312 (Fig. 3B, lanes 5–8). To further investigate this cooperativity, DNA binding was tested using titrations of Fkh2(312–458) and Fkh2(325–458) and DNA binding tested in the presence or absence of Mcm1p. Cooperativity in ternary complex formation was observed using Fkh2(312–458) but greatly reduced upon N‐terminal truncation to amino acid 325 (Fig. 3C and D). Thus these data demonstrate that sequences located between amino acids 312 and 325 are essential for cooperative ternary complex formation with Mcm1p.
To identify important residues within this region, we carried out site-directed mutagenesis experiments (Fig. 3E and F). We specifically targeted aromatic residues in this region as these had previously been shown to be important for the binding of coregulatory proteins to other MADS-box proteins (16,17,19). Indeed alignment of amino acids 213–333 from Fkh2p with the B-box SRF binding region of Elk-1 shows a potential conservation of several such residues (Fig. 3E). Mutation of amino acid Y315 in Fkh2p had little effect on its Mcm1p-independent DNA binding capacity (Fig. 3F, lane 2), but abolished cooperative binding with Mcm1p (Fig. 3F, lane 5). In contrast, mutation of either Y321 or F324 had little effect on ternary complex formation with Mcm1p (data not shown).
Collectively, our truncation studies have identified Mcm1(1–96) and Fkh2(312–458) as minimal determinants for the cooperative formation of Mcm1p–Fkh2p ternary DNA-bound complexes. Y325 has been shown to be a key amino acid in Fkh2 that is required for the formation of these complexes.
Cooperative ternary complex formation is not observed with Fkh1p
Previously we demonstrated that Fkh1p does not constitute a major part of the ternary complex that forms on the SWI5 promoter in yeast cell extracts (8). However, the regions encompassing the FKH DNA binding domains of Fkh1p and Fkh2p are very similar in sequence (72% identity through amino acids 329–454 of Fkh2p). However, this sequence similarity diverges in the region immediately upstream from the FKH domain that we have shown to be important for the formation of Mcm1p–Fkh2p complexes (Fig. 3A). We therefore compared analogously truncated Fkh1p and Fkh2p proteins for their ability to form ternary complexes with Mcm1p on the SWI5 promoter (Fig. 4).
Fkh2(254–458) efficiently formed ternary complexes with Mcm1p (Fig. 4B, lane 7) but the analogous Fkh1p construct, Fkh1(227–421) was unable to efficiently form ternary complexes (Fig. 4B, lane 5). The FKH domains of both proteins bind autonomously to the SWI5 site, albeit at a lower efficiency with Fkh1p (Fig. 4B, lanes 2 and 4). Thus, Fkh1p exhibits a lower intrinsic binding propensity for the SWI5 promoter and moreover, unlike Fkh2p, is unable to cooperatively form ternary complexes with Mcm1p.
Binding site spacing requirements for the formation of Mcm1p–Fkh2p complexes
The DNA binding sites in the Mcm1p–Fkh2p complex are closely juxtaposed (Fig. 5A) (8). Furthermore, our mapping data have identified a motif that is required for cooperative interactions with Mcm1p that is located close to the Fkh2p DNA binding domain (Fig. 3). This motif is likely to represent the Mcm1p interacting region of Fkh2p. In the SRF–Elk-1 complex, the spacing between the DNA binding sites can be highly variable (32). We therefore tested the spacing requirements of the Fkh2p and Mcm1p binding sites, which permit efficient complex formation. A series of binding sites was created in which nucleotides were either added or removed from the spacer region located between the core Fkh2p and Mcm1p binding sites (Fig. 5A).
The insertion of 2 or 5 bp between Fkh2p and Mcm1p binding sites led to a large decrease in ternary complex formation (Fig. 5B, lanes 2 and 3). However, the addition of 10 bp (corresponding to one helical turn) restored ternary complex formation, albeit to a reduced level in comparison to the wild-type spacing (Fig. 5B, lane 1). The deletion of either 1 or 3 bp between the two binding sites also caused a large decrease in the efficiency of Mcm1p–Fkh2p complex formation (Fig. 5B, lanes 5 and 6). To ensure that the changes we had introduced did not affect protein–DNA interactions mediated by the FKH domain of Fkh2p, we tested the ability of Fkh2(325–458) to bind to each of the sites (Fig. 5C). Binding of Fkh2(325–458) to all the sites was readily detectable, in contrast to the large decreases observed in the context of ternary complexes. We also tested the requirement for binding site orientation by inverting the asymmetric Fkh2p binding site (Fig. 5D). Inversion of the binding site resulted in abrogation of complex formation (Fig. 5E, lane 4) but still allowed autonomous binding of the Fkh2p FKH DNA binding domain (Fig. 5F, lanes 1 and 2).
Collectively these data demonstrate the critical importance of binding site spacing and orientation for the efficient formation of DNA-bound Mcm1p–Fkh2p ternary complexes.
Identification of Fkh2p interaction determinants on Mcm1p
The observation that Fkh2p can form ternary complexes with both Mcm1p and its mammalian counterpart SRF (Fig. 2B and C) indicates that the binding surface on Mcm1p and SRF is likely to be conserved. To explore this further, a series of mutant SRF derivatives was examined for binding ability to Fkh2p on the SWI5 promoter. One of these mutants, V194E, abolished ternary complex formation (data not shown). Previously, this mutation was shown to also abolish Elk-1 binding to SRF (18). Predictions from structural studies suggest that this mutation would block the insertion of aromatic residues into a binding pocket on the surface of Mcm1p (16). Indeed, we have identified Y315 as a residue that has a critical role in mediating ternary complex formation between Fkh2p and Mcm1p (Fig. 3F). To establish whether the equivalent residue in Mcm1p was also important for Fkh2p interactions, a Mcm1(1–98) mutant protein was constructed containing the V69E mutation. In addition, several additional mutants were obtained at T71 (T71A, T71E), which are known to be important for SRF interactions with its coregulatory partner, Elk-1 (Fig. 6A) (18). The T82E mutant Mcm1p protein was used as a negative control that is predicted not to effect ternary complex formation based on the SRF–Elk-1 paradigm (18). The locations of these residues on the surface of Mcm1p are shown in Figure 6B.
Ternary complex formation between Mcm1(1–98)(V69E) and full-length Fkh2p was virtually abolished (Fig. 6C, lane 7). In contrast, the introduction of the T71E mutation only had a moderate effect on ternary complex formation, while T71A actually exhibited enhanced binding (Fig. 6C, lanes 8 and 9; see Discussion). The T82E mutation did not affect ternary complex formation as predicted. Similar results were obtained in the presence of the truncated Fkh2(254–458) (Fig. 6D), demonstrating the importance of this region of Mcm1p in binding to the interaction surface we have mapped in Fkh2p.
To examine whether the abrogation of ternary complex formation between Mcm1(1–98)(V69E) and Fkh2p was due to disruption of protein–protein interactions, we carried out GST pulldown assays with GST–Fkh2(254–458) and wild-type and mutant Mcm1p derivatives (Fig. 6E). The L68E mutant version of Mcm1p was previously shown to disrupt DNA bending and hence Mcm1p–Fkh2p complex formation but does not disrupt protein–protein interactions with Fkh2p (33). In comparison to wild-type and L68E mutant versions, the V69E mutant exhibited much reduced binding to Fkh2p, demonstrating that this mutation directly affects protein– protein interactions between these two transcription factors.
Together, these results identify the pocket surrounding V69 in Mcm1p (see Fig. 6B) as a critical component of the Mcm1p–Fkh2p interface. This same pocket is also important for SRF–Fkh2p and SRF–Elk-1 interactions, demonstrating an evolutionary conservation of this pocket for driving cooperative ternary complex formation.
The effect of ternary complex disrupting mutations in vivo
We have identified mutations in both Mcm1p (V69E) and Fkh2p (Y315E) that disrupt ternary complex formation. To assess the importance of these residues, we next examined whether the corresponding mutant alleles of MCM1 and FKH2 affected the cellular function of these proteins.
Previous studies have demonstrated that the inviability of an mcm1Δ mutant can be rescued by a truncated version of Mcm1p containing only the minimal DNA binding domain [Mcm1(1–98)] (34). Indeed, we were able to rescue the lethality of an mcm1Δ mutant with a plasmid encoding Mcm1(1–98) (Fig. 7A). However, in contrast, a plasmid encoding Mcm1(1–98)(V69E) was unable to support growth of the mcm1Δ mutant (Fig. 7A).
Figure 7.
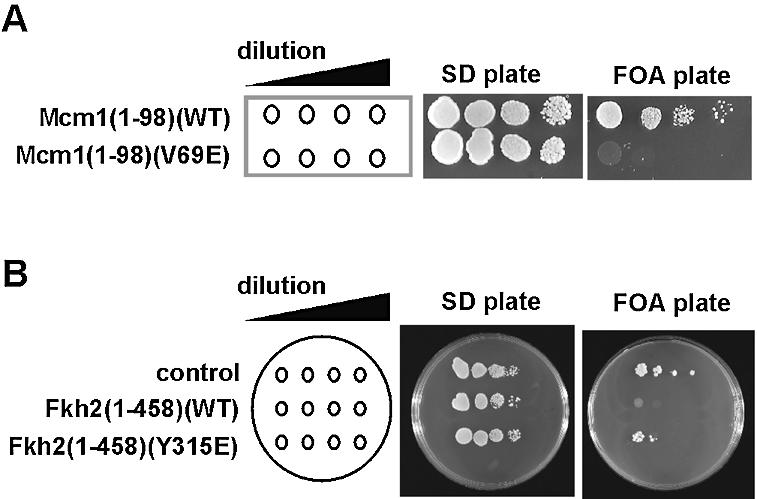
The role of Mcm1p–Fkh2p interactions in vivo. (A) The mcm1Δ mutant strain, YY2052, was transformed with plasmids encoding wild-type or V69E version of Mcm1(1–98) and plated on SD or FOA plates (to remove the plasmid-borne mcm1 allele). (B) The fkh2Δndd1Δ double mutant (AP183) was transformed with pMW20-NDD1 and either the pGBKT7 vector (control), pGBKT7–Fkh2(1–458) or pGBKT7–Fkh2(1–458)(Y315E) plasmids. Neat and 10-fold serial dilutions of transformants were spotted onto SD and FOA plates. Plates were incubated at 30°C for 2–5 days.
Previously, it has been shown that the lethality associated with a deletion of the NDD1 gene (35) can be rescued by deletion of the FKH2 gene (6). Hence, to examine the function of mutations of Fkh2p we constructed a fkh2Δndd1Δ double mutant strain (AP183) and co-transformed this strain with pMW20-NDD1, a URA3 plasmid expressing the wild type NDD1 gene, and either vector (pGBKT7) or a plasmid expressing amino acids 1–458 of Fkh2p [pGBKT7 Fkh2(1–458)]. Transformants were then plated onto 5-FOA medium to kill cells that were unable to lose the URA3 plasmid pMW20-NDD1. As expected cells co-transformed with pKT7 were able to lose pMW20-NDD1 (Fig. 7B). In contrast, cells co-transformed with pGBKT7 Fkh2(1–458) were unable to grow on 5-FOA medium (Fig. 7B) suggesting that the inviability of ndd1Δ cells cannot be reversed by removing the C-terminal region of Fkh2p (amino acids 459–862). The N‐terminal region (amino acids 1–458) of Fkh2p contains the FKH DNA binding domain and also contains the region of Fkh2p required for ternary complex formation with Mcm1p. Hence, to test whether ternary complex formation was linked to the inviability of the ndd1Δ, fkh2Δndd1Δ cells containing pGBKT7 Fkh2(1–458)(Y315E) and pMW20-NDD1 were plated on to 5-FOA medium. In contrast to wild-type protein, the Y315E substitution reversed the lethality associated with the Fkh2(1–458) construct in this background (Fig. 7B). The Y315E substitution blocks interactions with Mcm1p in vitro suggesting that the inviability of ndd1Δ cells is linked to the inability of Mcm1p–Fkh2p ternary complexes to activate transcription of the CLB2 gene cluster in the absence of Ndd1p. Importantly, these data demonstrate that Y315 of Fkh2p is required for the function of this protein in vivo.
Taken together, our data reveal that mutations of Mcm1p and Fkh2p that affect the activities of these proteins in vitro also affect their in vivo function. Hence, our in vitro studies of the Mcm1p–Fkh2p ternary complex provide important insights into the function and activity of this complex in vivo.
DISCUSSION
The Mcm1p–Fkh2p complex plays a pivotal role in regulating genes in the late G2 and M phases of the cell cycle in S.cerevisiae (reviewed in 5; 6–9). Here we provide molecular details of how this complex forms. Importantly, we identify regions and critical residues in Mcm1p and Fkh2p required for cooperative binding of Fkh2p to Mcm1p-occupied sites and demonstrate the importance of optimal DNA binding site spacing for complex formation to occur. The validity of these conclusions is supported by the demonstration that mutations of the regions identified as important for the formation of Mcm1p–Fkh2p complexes disrupt the activities of these proteins in vivo (Fig. 7).
The mammalian TCF–SRF complex represents a paradigm for understanding the basis for cooperative transcription factor complex formation (reviewed in 11,12). In this complex, a short region of the TCFs, the B-box, is presented on a flexible tether and binds to a surface exposed groove on the surface of SRF (16–18). These protein–protein interactions are essential for the recruitment of the TCF component into this complex. Due to the presence of the flexible tether, the spacing between the SRF and the TCF DNA binding sites can vary by over one helical turn with no discernable effect on ternary complex formation (32). The Mcm1p–Fkh2p complex shows many similarities with this complex. The sequences and structures of the MADS-box DNA binding domains of Mcm1p and SRF are highly conserved (19,36). Similarly, both the TCFs and Fkh2p contain a winged helix–turn–helix DNA-binding domain. Furthermore, both the TCF and Fkh2p binding sites are asymmetric. Here we demonstrate that Mcm1p and SRF use the same binding pocket on their surfaces to interact with Fkh2p and TCF, respectively (defined by the V69E mutation) (Fig. 6). The importance of this mutation is further underscored by the inability to rescue mcm1 deleted strains with Mcm1(1–98)(V69E), although this lethality probably reflects a loss of interaction with other coregulatory partners in addition to Fkh2p. Sequences located outside the DNA binding domain are required on both TCFs and Fkh2p to promote cooperativity in complex formation (Figs 1–4). How ever, the Mcm1p–Fkh2p complex differs in several key respects. Unlike in the TCFs, a long flexible linker does not exist in Fkh2p and the motif required for Mcm1p binding is closely juxtaposed to its DNA binding domain. This suggests that the DNA binding sites must be closely positioned. Indeed, this view is supported by our observation that alteration of the distance between the Mcm1p and Fkh2p binding sites in the SWI5 promoter strongly reduces recruitment of Fkh2p by Mcm1p (Fig. 5). In agreement with these data, inspection of the potential binding sites for Mcm1p–Fkh2p complexes in the promoters of genes that are known to be regulated by this complex, in conjunction with Ndd1p (37), shows little variability of this spacing (Fig. 8). Interestingly, some binding is regained upon addition of a full helical turn between the DNA binding sites. This suggests that contacts might be regained through bending of the intervening DNA when the proteins are located on the same side of the helix. Mcm1p is known to induce significant DNA bending and this might facilitate complex formation between Mcm1p and Fkh2p (19,38). Indeed, mutant Mcm1p proteins that are compromised in their DNA bending abilities exhibit reduced recruitment of Fkh2p (33).
Figure 8.

Binding sites for the Mcm1p–Fkh2p complex. Potential binding motifs are shown for the Mcm1p–Fkh2p complex located within the promoters of genes revealed to be direct targets of the Mcm1p–Fkh2p–Ndd1p complex by chromatin IP analysis (37). The Mcm1p and Fkh2p binding sites (boxed and shaded) and spacer region are shown. A conserved dinucleotide motif in the spacer is underlined.
The sequence of the region in Fkh2p that is required for cooperative binding of Fkh2p to Mcm1p shares some similarity with the B-box motif of the Elk-1 (Fig. 3F). In particular, the three critical aromatic residues in Elk-1 are conserved. The first two of these show identical spacing and encompass a conserved serine residue (Ser318 in Fkh2p) that also plays an important role in Elk-1 interactions with SRF. Apart from these amino acids, no other residues were identified as critical for complex formation between Elk-1 and SRF (17). Of the amino acids tested, only Y315 appears critical in Fkh2p, suggesting a difference in how the interactions mediated by Fkh2p and Elk-1 take place. Presumably, in Fkh2p, other interactions compensate for the lack of effect seen by mutation of other positions, possibly due to direct contacts between the MADS-box and the FKH domains themselves due to their close juxtapositioning. By extrapolating from the SRF–SAP-1 structure, the most likely role of Y315 is in making contacts with Mcm1p but also in determining and stabilising the trajectory of the Fkh2p motif across the surface of Mcm1p (16). In contrast, the increased binding of Fkh2p observed with the T71A mutant of Mcm1p is reminiscent of the binding of Fli-1 to the analogous SRF mutant (T196A) (18), suggesting that the interaction surface on Mcm1p used by Fkh2p retains characteristics from both TCF– and Fli-1–SRF interactions. Thus while Fkh2p uses the binding surface on Mcm1p that is analogous to that used by TCF and Fli-1 to bind to SRF, the exact interactions are likely to differ. Structural studies are required to define this interface more precisely.
Our DNA binding studies also uncovered additional regulatory activities acting on Fkh2p. In particular, deletion analysis identified an autoinhibitory domain that prevents the binding of Fkh2p to DNA in the absence of Mcm1p (Fig. 2). Similar domains are found in many transcription factors (including mammalian TCFs) that act to block spurious DNA binding in the absence of coregulatory partner proteins (39). In addition, our data revealed that deletion of the first 180 amino acids of Fkh2p destabilises Mcm1p–Fkh2p ternary complexes until further deletions of residues 180–254 are made. This is not observed in complexes of Fkh2p with SRF, indicating that SRF–Fkh2p complexes are likely to be stronger (Fig. 2). This observation points to a potential role for the N‐terminal part of Fkh2p [that includes the FKH-associated (FHA) domain] in stabilising Mcm1p–Fkh2p ternary complexes in combination with amino acids 180–254. As FHA domains are often sites of protein–protein interactions, this might have regulatory implications for the complex. Finally, it should be noted that the region of Fkh2p required for DNA binding cooperativity is not conserved with Fkh1p (Fig. 4) and Fkh1p is incapable of cooperatively forming ternary complexes with Mcm1p (Fig. 4) (10). Thus, our results clearly define in molecular terms why the highly related transcription factors Fkh1p and Fkh2p exhibit different propensities in forming complexes with Mcm1p and hence divergent functions in vivo.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Anne Clancy for excellent technical assistance. We are grateful to members of our laboratories for helpful discussions and to George Sprague and Cynthia Wolberger for reagents. This work was supported by the BBSRC, Cancer Research UK and the Wellcome Trust. J.B. was an ERASMUS programme exchange student. A.D.S. is a Research Fellow of the Lister Institute of Preventive Medicine.
REFERENCES
- 1.Dolan J.W. and Fields,S. (1991) Cell-type-specific transcription in yeast. Biochim. Biophys. Acta, 1088, 155–169. [DOI] [PubMed]
- 2.Shore P. and Sharrocks,A.D. (1995) The MADS-box family of transcription factors. Eur. J. Biochem., 229, 1–13. [DOI] [PubMed]
- 3.McInerny C.J., Partridge,J.F., Mikesell,G.E., Creemer,D.P. and Breeden,L.L. (1997) A novel Mcm1-dependent element in the SWI4, CLN3, CDC6, and CDC47 promoters activates M/G1-specific transcription. Genes Dev., 11, 1277–1288. [DOI] [PubMed]
- 4.Althoefer H., Schleiffer,A., Wassmann,K., Nordheim,A. and Ammerer,G. (1995) Mcm1 is required to coordinate G2-specific transcription in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 5917–5928. [DOI] [PMC free article] [PubMed]
- 5.Breeden L.L. (2000) Cyclin transcription: timing is everything. Curr. Biol., 10, R586–588. [DOI] [PubMed]
- 6.Koranda M., Schleiffer,A., Endler,L. and Ammerer,G. (2000) Forkhead-like transcription factors recruit Ndd1 to the chromatin of G2/M-specific promoters. Nature, 406, 94–98. [DOI] [PubMed]
- 7.Kumar R., Reynolds,D.M., Shevchenko,A., Shevchenko,A., Goldstone,S.D. and Dalton,S. (2000) Forkhead transcription factors, Fkh1p and Fkh2p, collaborate with Mcm1p to control transcription required for M-phase. Curr. Biol., 10, 896–906. [DOI] [PubMed]
- 8.Pic A., Lim,F.-L., Ross,S.J., Johnson,A.L., Sultan,R.A., West,A.G., Johnston,L.H., Sharrocks,A.D. and Morgan,B.A. (2000) The forkhead protein Fkh2 is a component of the yeast cell cycle transcription factor SFF. EMBO J., 19, 3750–3761. [DOI] [PMC free article] [PubMed]
- 9.Zhu G., Spellman,P.T., Volpe,T., Brown,P.O., Botstein,D., Davis,T.N. and Futcher,B. (2000) Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature, 406, 90–94. [DOI] [PubMed]
- 10.Hollenhorst P.C., Pietz,G. and Fox,C.A. (2001) Mechanisms controlling differential promoter-occupancy by the yeast forkhead proteins Fkh1p and Fkh2p: implications for regulating the cell cycle and differentiation. Genes Dev., 15, 2445–2456. [DOI] [PMC free article] [PubMed]
- 11.Sharrocks A.D. (2002) Complexities in ETS-domain transcription factor function and regulation; lessons from the TCF subfamily. Biochem. Soc. Trans, 30, 1–9. [DOI] [PubMed]
- 12.Treisman R. (1994) Ternary complex factors: growth regulated transcriptional activators. Curr. Opin. Genet. Dev., 4, 96–101 [DOI] [PubMed]
- 13.Mueller C.G. and Nordheim,A. (1991) A protein domain conserved between yeast MCM1 and human SRF directs ternary complex formation. EMBO J., 10, 4219–4229. [DOI] [PMC free article] [PubMed]
- 14.Shaw P.E. (1992) Ternary complex formation over the c-fos serum response element: p62TCF exhibits dual component specificity with contacts to DNA and an extended structure in the DNA-binding domain of p67SRF. EMBO J., 11, 3011–3019. [DOI] [PMC free article] [PubMed]
- 15.Wynne J. and Treisman,R. (1992) SRF and MCM1 have related but distinct DNA binding specificities. Nucleic Acids Res., 20, 3297–3303 [DOI] [PMC free article] [PubMed]
- 16.Hassler M. and Richmond,T.J. (2001) The B-box dominates SAP-1–SRF interactions in the structure of the ternary complex. EMBO J., 20, 3018–3028. [DOI] [PMC free article] [PubMed]
- 17.Ling Y., Lakey,J., Roberts,E.C. and Sharrocks,A.D. (1997) Molecular characterisation of the B-box protein–protein interaction motif of the ETS-domain transcription factors Elk-1. EMBO J., 16, 2431–2440. [DOI] [PMC free article] [PubMed]
- 18.Ling Y., West,A.G., Roberts,E.C., Lakey,J.H. and Sharrocks,A.D. (1998) Interaction of transcription factors with SRF: identification of the Elk-1 binding surface. J. Biol. Chem., 273, 10506–10514. [DOI] [PubMed]
- 19.Tan S. and Richmond,T.J. (1998) Crystal structure of the yeast MATalpha2/MCM1/DNA ternary complex. Nature, 391, 660–666. [DOI] [PubMed]
- 20.Sharrocks A.D., Von Hesler,F. and Shaw,P.E. (1993) The identification of elements determining the different DNA binding specificities of the MADS-box proteins p67SRF and RSRFC4. Nucleic Acids Res., 21, 215–221. [DOI] [PMC free article] [PubMed]
- 21.West A.G., Causier,B.E., Davies,B. and Sharrocks,A.D. (1998) DNA binding and dimerisation determinants of the Antirrhinum majus MADS-box transcription factors. Nucleic Acids Res., 26, 5277–5287. [DOI] [PMC free article] [PubMed]
- 22.West A.G. and Sharrocks,A.D. (1999) MADS-box transcription factors adopt different mechanisms to bend DNA. J. Mol. Biol., 286, 1311–1323. [DOI] [PubMed]
- 23.Shore P. and Sharrocks,A.D. (1994) The transcription factors Elk-1 and serum response factor interact by direct protein–protein contacts mediated by a short region of Elk-1. Mol. Cell. Biol., 14, 3283–3291. [DOI] [PMC free article] [PubMed]
- 24.Guan K. and Dixon,J.E. (1991) Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem., 192, 262–267. [DOI] [PubMed]
- 25.Bruhn L. and Sprague,G.F.S.,Jr (1994) MCM1 point mutants deficient in expression of α-specific genes: residues important for interaction with α1. Mol. Cell. Biol., 14, 2534–2544. [DOI] [PMC free article] [PubMed]
- 26.West A.G., Shore,P. and Sharrocks,A.D. (1997) DNA binding by MADS-box transcription factors: a molecular mechanism for differential DNA bending. Mol. Cell. Biol., 17, 2876–2887 [DOI] [PMC free article] [PubMed]
- 27.Galanis A., Yang,S.-H. and Sharrocks,A.D. (2001) Targeting of MAP kinase signal transduction pathways to SAP-1. J. Biol. Chem., 276, 965–973. [DOI] [PubMed]
- 28.Sherman F., Fink,G.R. and Hicks,J.B. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 29.Berben G., Dumont,J., Gilliquet,V., Bolle,P.A. and Hilger,F. (1991) The YDp plasmids: a uniform set of vectors bearing versatile gene disruption cassettes for Saccharomyces cerevisiae. Yeast, 7, 475–477. [DOI] [PubMed]
- 30.Schiestl R.H. and Gietz,R.D. (1989) High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier. Curr. Genet., 16, 339–346. [DOI] [PubMed]
- 31.Boeke J.D., La Croute,F. and Fink,G.R. (1984) A positive screen for mutants lacking 5′-phosphate decarboxylase activity in yeast: 5′-fluoroorotic acid resistance. Mol. Gen. Genet., 197, 345–346. [DOI] [PubMed]
- 32.Treisman R., Marais,R. and Wynne,J. (1992) Spatial flexibility in complexes between SRF and its accessory proteins. EMBO J., 11, 4631–4640. [DOI] [PMC free article] [PubMed]
- 33.Lim F.-L., Hayes,A., West,A.G., Pic-Taylor,A., Darieva,Z., Morgan,B.A., Oliver,S.G. and Sharrocks,A.D. (2003) Mcm1p-induced DNA bending regulates the formation of ternary transcription factor complexes. Mol. Cell. Biol., 23, 450–461. [DOI] [PMC free article] [PubMed]
- 34.Bruhn L., Hwang-Shum,J.J. and Sprague,G.F.,Jr (1992) The N‐terminal 96 residues of MCM1, a regulator of cell type-specific genes in Saccharomyces cerevisiae, are sufficient for DNA binding, transcription activation, and interaction with alpha 1. Mol. Cell. Biol., 12, 3563–3572. [DOI] [PMC free article] [PubMed]
- 35.Loy C.J., Lydall,D. and Surana,U. (1999) NDD1, a high-dosage suppressor of cdc28–1N, is essential for expression of a subset of late-S-phase-specific genes in Saccharomyces cerevisiae. Mol. Cell. Biol., 19, 3312–3327. [DOI] [PMC free article] [PubMed]
- 36.Pellegrini L., Tan,S. and Richmond,T.J. (1995) Structure of serum response factor core bound to DNA. Nature, 376, 490–498. [DOI] [PubMed]
- 37.Simon I., Barnett,J., Hannett,N., Harbison,C.T., Rinaldi,N.J., Volkert,T.L., Wyrick,J.J., Zeitlinger,J., Gifford,D.K., Jaakkola,T.S. and Young,R.A. (2001) Serial regulation of transcriptional regulators in the yeast cell cycle. Cell, 106, 697–708. [DOI] [PubMed]
- 38.Smith D.L., Desai,A.B. and Johnson,A.D. (1995) DNA-bending by the a1 and α2 homeodomain proteins from yeast. Nucleic Acids Res., 23, 1239–1243. [DOI] [PMC free article] [PubMed]
- 39.Graves B.J., Cowley,D.O., Goetz,T.L., Petersen,J.M., Jonsen,M.D. and Gillespie,M.E. (1998) Autoinhibition as a transcriptional regulatory mechanism. Cold Spring Harb. Symp. Quant. Biol., 63, 621–629. [DOI] [PubMed]