

Abstract
The availability of almost the complete human genome as cloned BAC libraries represents a valuable resource for functional genomic analysis, which, however, has been somewhat limited by the ability to modify and transfer this DNA into mammalian cells intact. Here we report a novel comprehensive Escherichia coli-based vector system for the modification, propagation and delivery of large human genomic BAC clones into mammalian cells. The GET recombination inducible homologous recombination system was used in the BAC host strain E.coli DH10B to precisely insert an EGFPneo cassette into the vector portion of a ∼200 kb human BAC clone, providing a relatively simple method to directly convert available BAC clones into suitable vectors for mammalian cells. GET recombination was also used for the targeted deletion of the asd gene from the E.coli chromosome, resulting in defective cell wall synthesis and diaminopimelic acid auxotrophy. Transfer of the Yersinia pseudotuberculosis invasin gene into E.coli DH10B asd– rendered it competent to invade HeLa cells and deliver DNA, as judged by transient expression of green fluorescent protein and stable neomycin-resistant colonies. The efficiency of DNA transfer and survival of HeLa cells has been optimized for incubation time and multiplicity of infection of invasive E.coli with HeLa cells. This combination of E.coli-based homologous recombination and invasion technologies using BAC host strain E.coli DH10B will greatly improve the utility of the available BAC libraries from the human and other genomes for gene expression and functional genomic studies.
INTRODUCTION
The availability of a complete array of fully sequenced cloned BACs that span most of the human genome is a remarkable development in biology, and represents a valuable resource for studying gene expression and other chromosomal functions (1–3). However, the utility of these BACs is limited by the difficulty in manipulating and transferring them into mammalian cells. BACs can contain as much as 350 kb of genomic DNA, and will therefore carry the complete coding sequence of most human genes (average size 30–60 kb) as well as the surrounding regulatory DNA for correct spatio-temporal gene expression. Furthermore, functional chromosome structures such as centromeres are formed on DNA that is several hundred kilobases in size, which has proven difficult to transfer intact without significant rearrangements.
The propagation of BAC libraries in recombination- deficient Escherichia coli DH10B imparts greater stability to the large BAC clones but has made their further manipulation using direct homologous recombination very difficult. A relatively simple and straightforward method has recently been demonstrated that provides transiently inducible recombination proficiency to E.coli DH10B by expression of the bacteriophage lambda gam gene and E.coli recE and recT from an arabinose promoter on plasmid pGETrec (4). The GET recombination system has been used to insert reporter constructs and disease causing mutations into BAC inserts containing the beta-globin locus (5,6). Additional strategies for the homologous recombination of BAC clones have also been reported (7,8).
The delivery of large DNA into mammalian cells also presents technical difficulties, particularly the ‘naked DNA’ stage where purified large DNA may undergo mechanical degradation, and considerable efforts have been directed to develop efficient delivery methods. Escherichia coli BM2710 has been demonstrated to invade and deliver plasmid DNA into HeLa, COS-1 and CHO cells when they are expressing the Yersinia pseudotuberculosis invasin gene, which binds to mammalian integrin receptors and results in internalization in primary vesicles, and are auxotrophic for diaminopimelic acid (DAP), resulting in a cell wall deficiency (9,10). Other attenuated bacterial pathogens have also been used as gene delivery vectors (11–15). Recently, the transfer of plasmids up to 15 kb into CHO-K1 cells has been achieved by donor controlled conjugation from E.coli DH5α containing the broad host range RK2 plasmid (16). Additionally, the HSV family of viruses have been used for the infectious delivery of genomic BACs up to the ∼150 kb viral packaging limit with efficiencies ranging from 25 to 100% into the human MRC-5V2 and mouse primary hepatocytes (17–19).
Here we present a novel combination of GET recombination and bacterial invasion of mammalian cells, providing a comprehensive E.coli-based vector system designed for the modification, propagation and delivery of genomic BAC clones using their host E.coli DH10B. GET recombination has been used here to delete the asd gene in E.coli DH10B, resulting in DAP auxotrophy, and to precisely insert the EGFPneo cassette from plasmid pEGFP-N2 into the vector portion of a 200 kb BAC clone. After transfer of a plasmid coding for the Y.pseudotuberculosis invasin gene into this E.coli DH10B asd– host, invasion and transfer of both plasmid and BAC DNA into HeLa cells was demonstrated by green fluorescent protein (GFP) and neomycin expression.
MATERIALS AND METHODS
Plasmids and bacterial strains
Escherichia coli DH10B is F– mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 deoR recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ– rpsL nupG (20). Plasmid pEGFP-N2 (4.7 kb) (BD Biosciences Clontech, Palo Alto, CA) carries the EGFP reporter gene driven by the CMV promoter and a kanamycin/neomycin (kan/neo) gene that confers resistance to kanamycin (Km) and neomycin analogs in prokaryotes and eukaryotes, respectively. Plasmid pGB2Ωinv (7.6 kb) contains the Y.pseudotuberculosis invasin (inv) gene and carries resistance to both spectinomycin and streptomycin on the non-autotransferable low-copy plasmid pGB2 (9). Plasmid pGETrec contains the arabinose inducible recombination genes, recE, recT and gam, as described previously (4). The 3 kb pZeoSV2(+) plasmid carrying the prokaryotic EM7 promoter-zeor cassette was obtained from Invitrogen Corporation, Carlsbad, CA.
Preparation and purification of PCR products
The EM7 promoter-zeor cassette from pZeoSV2(+) was amplified using primers asdZF (5′-GAG ACC GGC ACA TTT ATA CAG CAC ACA TCT TTG CAG GAA AAA AAC GCT TAc ggt gca ttg gaa cgg acc g-3′) and asdZR (5′-CTC CTG TAT TAC GCA CTA ACA GGG GCG GCA TCG CGC CCC AGA TTT AAT GAt gca ata aac aag ttt cga g-3′). The resultant 590 bp PCR product was designated zeo590. The zeo590 fragment was designed to delete the E.coli DH10B asd gene after the A from the ATG start codon to seven bases downstream after the TAA stop codon.
The EGFP-kanamycin/neomycin (EGFP-kmr-neor) cassette from pEGFP-N2 was amplified using primers GK-F (5′- TTC CGG TCA CAC CAC ATA CGT TCC GCC ATT CCT ATG CGA TGC ACA TGC TGT ATG CCG GTA taa ccg tat tac cgc cat gc-3′) and GK-R (5′-AGA CTT CCG TTG AAC TGA TGG ACT TAT GTC CCA TCA GGC TTT GCA GAA CTT TCA GCG GTA acg acc caa cac cgt gcg tt-3′). The resultant 3.8 kb PCR product was designated EGFPneo. Upper-case characters indicate homologous nucleotide sequences to either the 5′ or 3′ end of the targeted locus. Lower-case characters indicate nucleotides complementary to the template that is used for the amplification of the PCR product. The PCR products were gel purified using the QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA) and resuspended in 10 mM Tris–HCl, pH 8.5 at a concentration of ∼300 ng/µl.
Construction of the E.coli DH10B asd–
GET recombination in E.coli DH10B was performed as described (4). Approximately 600 ng of purified PCR product zeo590 was electroporated into l-arabinose-induced electrocompetent DH10B (pGETrec) cells using a Gene Pulser II unit set at 2.5 kV, 200 Ω and 25 µF (Bio-Rad Laboratories, Hercules, CA). After recovery for 90 min at 37°C in 1 ml of LB medium containing 0.5 mM DAP, cells were plated on LB agar containing 0.5 mM DAP and 25 µg/ml Zeo. Zeor colonies were genotyped by plating onto LB, LB containing DAP, and LB containing DAP and 25 µg/ml Zeo. Zeor DAP auxotrophs (asd–) were verified by PCR across the asd locus using primers asd1 (5′-TGGTGAAAGATGTGCCAAGA-3′) and asd2 (5′-TCCCGGTAAATCATGAAACA-3′). Plasmid pGETrec was eliminated by streaking DH10B asd– colonies onto LB plates containing DAP, Zeo and the inducer l-arabinose, and omitting ampicillin (Ac) selection. Single colonies that require DAP, were resistant to Zeo and sensitive to Ac were identified as DH10B asd– with plasmid pGETrec eliminated.
Retrofitting of the EGFPneo cassette on the 200 kb hgUroS-1 BAC
The 200 kb hgUroS-1 BAC clone containing the complete human Uroporphyrinogen-III synthase gene was kindly provided by Drs Gerardo Aizencang and Robert Desnick (21). Purified PCR product EGFPneo was electroporated into l-arabinose-induced electrocompetent DH10B (pGETrec, hgUroS-1). The subsequent electroporation and selection steps were carried out with the following brief modifications. After recovery in 1 ml of LB medium at 37°C for 90 min, cells were plated on LB agar containing 12.5 µg/ml chloramphenicol (Cm) and 30 µg/ml Km, and resistant colonies verified by PCR amplification across the recombination junction using primers KF (5′-ATCTGGGAAGTGACGGACAG-3′) and KR (5′-CAGCATCGCAACCGCATCAG-3′) (4). The modified hgUroS-1-EGFPneo was subsequently re-electroporated into DH10B cells that did not contain pGETrec. BAC DNA from this E.coli was digested with SalI and NotI following manufacturers protocols and subjected to pulsed field gel electrophoresis (PFGE) [Bio-Rad CHEF-DRII, 1% (w/v) agarose gel, 150 V, switch time of 0.1–30 s, 12°C, 0.5× TBE, for 21 h].
Invasion of HeLa cells with E.coli DH10B
Saturated overnight or late-log cultures of E.coli DH10B asd– (pGB2Ωinv) (invasive) or DH10B asd– (non-invasive) containing the pEGFP-N2 or BAC hgUroS-1-EGFPneo were grown at 37°C in brain heart infusion (BHI) broth with 0.5 mM DAP and appropriate antibiotics. Overnight, bacteria (in 10 ml) at a concentration of ∼1 × 109 c.f.u./ml (OD600 ∼1.2) were pelleted and resuspended in 1 ml of RPMI 1640 medium (Cellgro by Mediatech, Inc., Herndon, VA). Aliquots of bacteria at the desired multiplicity of infection (MOI) were then diluted in RPMI 1640 to give a final volume of 2 ml, vortexed and overlaid onto monolayer cultures of 1 × 105 HeLa cells per well (in a 6-well plate) that had been washed twice with PBS. The plates were centrifuged at 1000 r.p.m. for 10 min before being incubated at 37°C in 5% CO2 for the desired incubation time. Subsequently, the cells were washed twice with PBS to remove non-adherent bacteria, fresh medium containing 20 µg/ml gentamicin (Gm) added, and further incubated for 48 h.
GFP-expressing cells were viewed directly in 6-well plates using an Axiovert 135 fluorescent microscope (Carl Zeiss, Inc., Germany) equipped with a Sony DKC-5000 CCD camera. Subsequently, cells were trypsinized, resuspended in PBS, and 4 × 104 cells were quantified for GFP expression cells using a FACScan flow cytometer with CellQuest software (Beckton-Dickinson, Mountain View, CA). Stable G418-resistant colonies were obtained by replating HeLa cells 48 h after invasion and addition of G418 (1 mg/ml) 24 h later. Entry of bacteria into HeLa cells was determined using the Gm protection assay (22). After each incubation time point (10, 30, 60 or 120 min), the cells were washed twice with PBS to remove non-adherent bacteria before adding fresh RPMI 1640 medium containing 80 µg/ml Gm and incubated for a further 30 min at 37°C in a 5% CO2 atmosphere incubator. Cells were washed twice with PBS and internalized bacteria released from the monolayer by a 5 min incubation with 1% (w/v) Triton X-100 and titered on BHI plates containing 0.5 mM DAP and the appropriate antibiotics.
RESULTS
Modification of E.coli DH10B using GET recombination
In order to increase the utility of the available genomic BAC resources, the GET recombination system was developed to permit the targeted modification of BAC inserts and vectors in host strain E.coli DH10B (4). We have used a novel application of the GET recombination system for the targeted deletion of the aspartate β-semialdehyde (asd) gene from the E.coli chromosome, converting E.coli DH10B into a DAP auxotroph that lyses upon cell division in the absence of DAP supplementation (12,23). A 590 bp PCR fragment (zeo590) containing the complete zeocin-resistance (zeor) gene was amplified from pZeoSV2(+) using PCR primers asdZF and asdZR, which each contain 50 bp of DNA that is homologous to the E.coli asd locus. Electroporation of zeo590 into arabinose-induced E.coli DH10B (pGETrec) resulted in homologous recombination of the 50 bp flanking DNA with the asd locus, and replacement of the asd coding sequences with the zeor fragment (Fig. 1A). The correct targeting of this homologous recombination was verified by PCR amplification across the asd locus using primers asd1 and asd2 which flank the chromosomal region replaced by zeo590, which amplify a 691 bp PCR fragment from correctly targeted cells (Fig. 1C, lanes 2 and 3, two independent isolates) and a 1.3 kb PCR fragment (Fig. 1C, lane 3) from untargeted cells. The pGETrec plasmid continued to persist in the asd– DH10B cells in the absence of Ac selection, in contrast to that reported by Muyrers et al. (24). Therefore, l-arabinose induction of the strong PBAD promoter on the pGETrec plasmid in the absence of selection was used to eliminate the plasmid.
Figure 1.

GET recombination in E.coli DH10B. (A) Modification of the E.coli chromosome. The zeo590 PCR product contains the zeo-resistance gene flanked at both ends with 50 bp sequences (stippled boxes) that are homologous to regions of the asd gene. Introduction of zeo590 into arabinose-induced E.coli (pGETrec) results in precise replacement of the asd gene with the zeo-resistance gene. After elimination of pGETrec, introduction of pGB2Ωinv encoding the Y.pseudotuberculosis inv gene confers the ability to invade mammalian cells and deliver either pEGFP-N2 or a modified BAC clone. (B) Modification of a 200 kb BAC clone. PCR product, EGFPneo, which contains the EGFP-kmr-neor cassette flanked on either side by 60 bp of DNA homologous to pBeloBAC11 vector centered on the Bst11701 site. Homologous recombination results in insertion of DNA encoding for EGFP and neomycin resistance. Arrows represent primers KF and KR which flank the recombination junction. B, Bst11071; N, NotI; S, SalI. (C) PCR analysis of E.coli DH10B asd–. Primers asd1 and asd2 amplify across the asd locus in E.coli DH10B. Lanes 1 and 2, the predicted 691 bp PCR product confirming correct targeted replacement of the asd locus in two independent zeor colonies. Lane 3, the 1.3 kb fragment predicted for an intact asd locus. Lane M, marker X (Roche Diagnostics Corporation, IN). (D) Restriction analysis of the modified BAC clone. Lane 1, unmodified BAC digested with NotI, showing 6.9 kb NotI fragment. Lanes 2 and 3, independent isolates of modified BAC digested with NotI showing 7.8 and 2.7 kb NotI fragments, which confirm the correct targeting of EGFPneo into the vector portion of the BAC clone. Lane 4, unmodified BAC digested with SalI showing a 6.7 kb SalI fragment. Lanes 5 and 6, independent isolates of modified BAC digested with SalI, showing 8.2 and <2 kb SalI fragments, which confirm the correct targeting of EGFPneo into the vector portion of the BAC clone. The 200 kb NotI and 175 and 24 kb SalI fragments representing the BAC insert have not changed during the homologous recombination. Higher contrast was used on the gel photograph below the 6.55 kb marker for clarity of the lower molecular weight ethidium bromide stained bands.
Modification of a 200 kb genomic BAC clone
In order to assay the functional delivery of BAC DNA into mammalian cells, we have used the GET recombination system to insert DNA encoding the GFP and G418 resistance into the pBeloBAC11 vector of a 200 kb human BAC that contains the human UroS-1 gene (hgUROS-1) (21). A 3.8 kb PCR fragment (EGFPneo) containing the genes for GFP and neomycin was amplified from pEGFP-N2 using PCR primers GK-F and GK-R, which each contain 60 bp of flanking DNA that is homologous to the 120 bp of DNA centered on the unique Bst1107I site on pBeloBAC11 (Fig. 1B). Insertion of DNA into this Bst1107I site has been shown to not disrupt BAC replication and maintenance (4). After electroporation of EGFPneo DNA into arabinose-induced DH10B (pGETrec, hgUroS-1), DNA from Kmr colonies were screened by PCR amplification across the recombination junction using primers KF and KR (Fig. 1B). Amplification of the predicted 4.0 kb fragment confirmed the correct targeting of EGFPneo within the vector portion of the hgUroS-1 BAC (data not shown). BAC DNA isolated from two Kmr colonies were analyzed by NotI and SalI restriction digestions using PFGE, which further confirmed the correct targeting of the BAC vector (Fig. 1D). Furthermore, the stability of the BAC insert during this recombination procedure is demonstrated by the consistent 200 kb NotI and 175 and 24 kb SalI insert bands in both independent recombinant BAC isolates (Fig. 1D). This modified BAC clone was designated hgUroS-1-EGFPneo.
Plasmid delivery and expression in human cells
The Y.pseudotuberculosis invasin gene on pGB2Ωinv (9) was introduced into E.coli DH10B asd– carrying pEGFP-N2 (Fig. 1A). F-derived BACs and pUC-based high copy plasmids are compatible with pGB2Ωinv and can therefore co-exist in the DH10B host. Although the optimal growth temperature for invasion of mammalian cells by Y.pseudotuberculosis is at 28°C, invasin expression in E.coli is regulated in a temperature-independent manner (25). Therefore, the invading E.coli were grown overnight at 37°C, which simplifies the invasion procedure, and were observed to improve the efficiency in delivering pEGFP-N2 into HeLa cells (data not shown).
The E.coli DH10B asd– (pEGFP-N2, pGB2Ωinv) grown at 37°C was overlaid onto target HeLa and HT1080 cells, and the transfection efficiency monitored by the expression of GFP using both fluorescence microscopy and fluorescence- activated cell sorting (FACS). Figure 2 shows an increase in GFP expression in response to increasing MOI of bacteria incubated with HeLa cells for 2 h. MOIs of 2000 (Fig. 2B) and 4000 (Fig. 2C) produced GFP expression in ∼12% of the HeLa cells. The DH10B asd– (pEGFP-N2) that did not contain the Y.pseudotuberculosis invasin (inv) gene resulted in minimal GFP expression even at an MOI of 4000 (Fig. 2D). Invasion of HT1080 cells with DH10B asd– (pGB2Ωinv, pEGFP-N2) with MOIs ranging from 500 to 4000 using identical conditions to HeLa transfection resulted in 2.66% of the cells expressing GFP at an MOI of 1500 (data not shown).
Figure 2.

Invasion of HeLa cells by E.coli DH10B asd– (pGB2Ωinv, pEGFPN2). Overnight culture of invasive DH10B asd– bacteria or non- invasive DH10B asd– control were overlaid onto monolayer cultures of HeLa cells at the desired MOI for 120 min. HeLa cells were washed to remove extracellular bacteria before being incubated at 37°C for 48 h prior to detection of GFP expression. (A–E) Left, photomicrograph showing GFP fluorescence; center, phase contrast microscopy (red filter) showing same field of cells; right, FACS analysis of cells sorted for GFP (x-axis) and orange auto-fluorescence (y-axis); cells in lower right quadrant represent GFP-positive cells (F, % GFP-expressing cells). (A) MOI = 1000, F = 3.17. (B) MOI = 2000, F = 12.08. (C) MOI = 4000, F = 12.12. (D) Non-invasive E.coli, MOI = 2000, F = 1.67. (E) HeLa colony stably expressing GFP. F = 62.5% after G418 selection for 32 days.
In order to assess the ability of this invading E.coli to stably transfect HeLa cells, HeLa colonies resistant to 1 mg/ml G418 were selected from cells invaded with DH10B asd– (pGB2Ωinv, pEGFP-N2). A representative stable transfected colony is shown in Figure 2E, with 62.5% of the cells positive for GFP expression, as visualized by fluorescence microscopy after 32 days of growth and quantified by FACS. Thus, this E.coli DH10B-based delivery system can both transiently and stably transfect HeLa cells.
Rapid internalization of invasive E.coli DH10B into HeLa cells
The co-incubation of E.coli with HeLa cells for 2 h, as shown above (Fig. 2A–D), resulted in a significant loss of viability of the HeLa cells, especially at the higher MOIs. Therefore, in order to maximize HeLa cell viability, the minimal co-incubation time required for efficient invasion and plasmid delivery into HeLa cells was determined using a Gm plating assay (22). HeLa cells were incubated with E.coli at an MOI of 2000 for varying time points and cells washed to remove uninvaded E.coli. The bacteriocidal antibiotic Gm, which does not penetrate HeLa cell membranes, was added to kill any remaining adherent E.coli (22). HeLa cells were harvested and lysed, and viable E.coli in the cells measured by plating on selective media. Surprisingly, invasion of E.coli reached a maximum at the shortest time point tested (10 min), and remained approximately constant for up to 120 min (Fig. 3A). Uninvasive bacteria [DH10B asd– (pEGFP-N2)] showed only minimal background levels (please note the log scale used on the histogram in Fig. 3A). This result shows that E.coli invasion is efficient at short exposure times, which greatly increases the overall viability of the HeLa cells. Based on these results, an incubation time of 30 min with late-log invasive E.coli was chosen to optimize invasion efficiency (Fig. 3A) yet maintain good HeLa cell viability (>75%).
Figure 3.
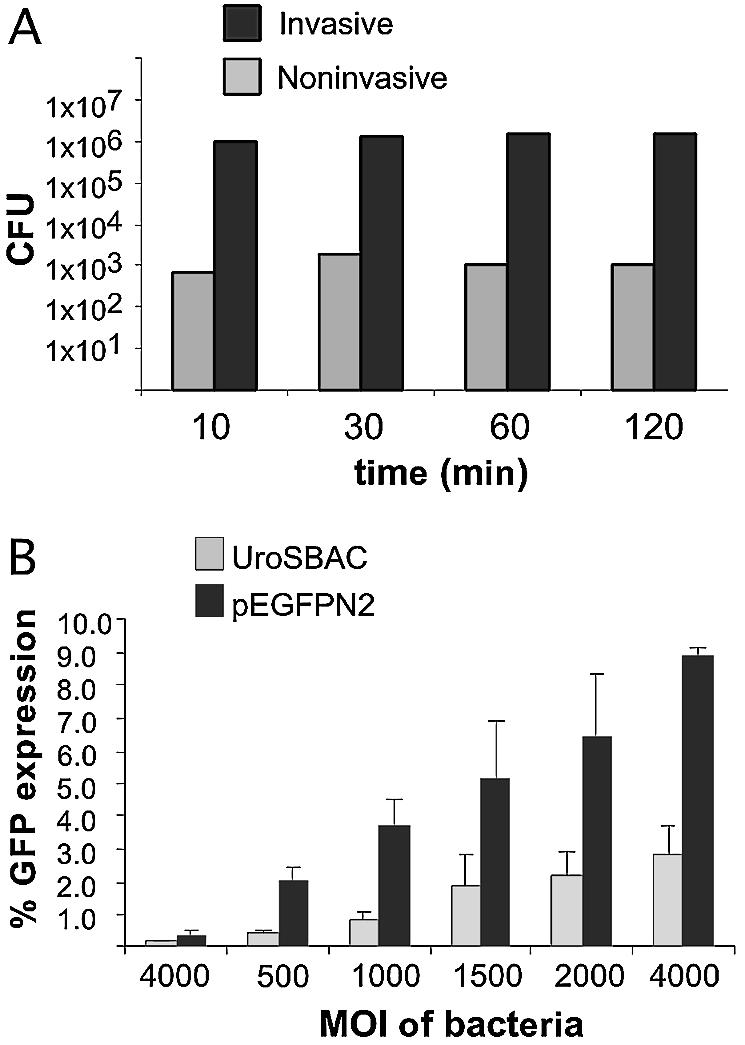
Invasive E.coli is internalized and delivers plasmid and a 200 kb BAC clone into HeLa cells. (A) Internalization of the E.coli DH10B asd– upon exposure to HeLa cells for various time points. Mean values are calculated from two independent experiments. (B) Percentage of GFP expression in HeLa cells by invading E.coli harboring either pEGFP-N2 or hgUroS-1 BAC. Mean values are calculated from three independent experiments. An MOI of 4000 using uninvasive bacteria is shown in leftmost histograms.
Invasive E.coli delivers plasmid and BAC DNA into HeLa cells
The transfection efficiency of HeLa cells using invasive E.coli DH10B asd– (pGB2Ωinv, pEGFP-N2) was performed for a co-incubation time of 30 min, which greatly increases HeLa cell viability. Less than 0.4% of HeLa cells expressed GFP when incubated with non-invasive E.coli DH10B asd– (pEGFP-N2) at an MOI of 4000. At increasing MOIs of invasive E.coli and HeLa cells, the percentage of HeLa cells expressing GFP increases, from ∼2% at an m.o.i of 500 to 8.9% at an MOI of 4000, 23-fold over background (Fig. 3B). Using similar conditions, the transfection efficiency of the invasive E.coli was investigated for the genomic BAC hgUroS-1-EGFPneo, which we modified using GET recombination (Fig. 1B and D). This BAC has a relatively large 200 kb insert containing the entire human Uroporphyrinogen-III synthase gene and surrounding regulatory sequences (21). This BAC was transferred into the invasive E.coli to create DH10B asd– (pGB2Ωinv, hgUroS-1-GFPneo), which was co-incubated with HeLa cells at a range of MOIs for 30 min (Fig. 3B). The background level of GFP expression using non-invasive E.coli asd– DH10B (hgUroS-1-GFPneo) was lower (<0.2%) than that observed for E.coli DH10B asd– (pGB2Ωinv, pEGFP-N2), likely due to the much lower copy number per E.coli of the BAC relative to pEGFP-N2. FACS analysis 48 h after transfection showed that the percentage of HeLa cells expressing GFP from BAC hgUroS-1-GFPneo increased with increasing MOI, reaching a maximum of 2.8% at an MOI of 4000, 15-fold over background (Fig. 3B).
DISCUSSION
We have shown that E.coli DH10B can be engineered to (i) permit targeted modification of plasmid and BAC DNA and (ii) deliver this modified DNA to human cells. Escherichia coli presents several advantages for use as a gene manipulation and delivery vector. Escherichia coli is widely used for the cloning and propagation of DNA, with protocols for growth, transformation, selection and DNA purification standard in most molecular biology laboratories. Genomic BAC libraries, which represent the building blocks of current genome assemblies, are propagated in E.coli DH10B, which has a natural potential to carry extrachromosomal DNA of over 1 Mb in size (26). However, the recombination deficiency of E.coli DH10B has limited the manipulation of these large BAC clones (>100 kb) using conventional recombinant DNA techniques. Here we have demonstrated the use of the GET recombination inducible system (4) to insert selectable markers and reporter genes (from pEGFP-N2) directly into the vector portion of an ∼200 kb BAC clone, providing a relatively simple and widely applicable method for the conversion of available genomic BAC clones directly into vectors suitable for transfection into human cells.
The utility of these modified BACs has also been limited by lack of an efficient delivery system into mammalian cells. Here we have developed a strategy using the E.coli DH10B BAC host strain directly as a delivery vector. Using a novel application of the GET recombination system, we have created a targeted deletion of the asd gene from the E.coli DH10B chromosome, resulting in DAP auxotrophy and defective cell wall synthesis. Transformation of plasmid pGB2Ωinv expressing the Y.pseudotuberculosis invasin gene renders these E.coli competent to invade mammalian cells and deliver plasmid and BAC DNA. pGETrec can be used to directly replace the asd gene with the invasin gene in the E.coli chromosome in one step, making this second transformation with pGB2Ωinv unnecessary and eliminating any biohazard risk from inadvertent plasmid transfer to non-auxotrophic bacteria.
Incubation of the E.coli DH10B asd– (pGB2Ωinv) carrying pEGFP-N2 with both HeLa and HT1080 cells resulted in the delivery and transient expression of GFP in as high as 12% of cells, and isolation of stable G418-resistant colonies. Using the Gm protection assay (22), we found that incubation for as little as 10 min allowed for optimal internalization of bacteria while greatly increasing the viability of recipient cells. Incubation of E.coli DH10B asd– (pGB2Ωinv) carrying the ∼200 kb modified BAC clone resulted in ∼2.8% of HeLa cells expressing GFP. This E.coli-based DNA vector represents the first comprehensive system capable of modification, propagation and delivery of full-sized BACs on the order of several hundred kilobases in size, and will greatly increase the utility of available BAC libraries for functional genomics and gene expression studies. These studies will provide important information regarding the regulation of temporal and tissue-specific gene expression for the development of improved gene therapy vectors (27).
Acknowledgments
ACKNOWLEDGEMENTS
We are very grateful to C. Grillot-Courvalin and S. Goussard (Institut Pasteur, France) for plasmid pGB2Ωinv and for helpful advice. We would like to thank A. Radu and L. F. P. Cunha (both from the Mount Sinai School of Medicine, New York), R. R. Isberg (Tufts University School of Medicine, Boston) and G. Ismail (Malaysia University of Science and Technology, Kuala Lumpur) for helpful discussion and assistance. K.N. would like to dedicate this work to S. Shantini for her support, encouragement and warmth. This work was supported by NIH grant R21DK58672.
REFERENCES
- 1.Morley M., Arcaro,M., Burdick,J., Yonescu,R., Reid,T., Kirsch,I.R. and Cheung,V.G. (2001) GenMapDB: a database of mapped human BAC clones. Nucleic Acids Res., 29, 144–147. [DOI] [PMC free article] [PubMed]
- 2.Osoegawa K., Mammoser,A.G., Wu,C., Frengen,E., Zeng,C., Catanese,J.J. and de Jong,P.J. (2001) A bacterial artificial chromosome library for sequencing the complete human genome. Genome Res., 11, 483–496. [DOI] [PMC free article] [PubMed]
- 3.Zhao S. (2001) A comprehensive BAC resource. Nucleic Acids Res., 29, 141–143. [DOI] [PMC free article] [PubMed]
- 4.Narayanan K., Williamson,R., Zhang,Y., Stewart,A.F. and Ioannou,P.A. (1999) Efficient and precise engineering of a 200 kb β-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther., 6, 442–447. [DOI] [PubMed]
- 5.Orford M., Nefedov,M., Vadolas,J., Zaibak,F., Williamson,R. and Ioannou,P.A. (2000) Engineering EGFP reporter constructs into a 200 kb human beta-globin BAC clone using GET recombination. Nucleic Acids Res., 28, e84. [DOI] [PMC free article] [PubMed]
- 6.Nefedov M., Williamson,R. and Ioannou,P.A. (2000) Insertion of disease-causing mutations in BACs by homologous recombination in Escherichia coli. Nucleic Acids Res., 28, e79. [DOI] [PMC free article] [PubMed]
- 7.Lee E.C., Yu,D., Martinez de Velasco,J., Tessarollo,L., Swing,D.A., Court,D.L., Jenkins,N.A. and Copeland,N.G. (2001) A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics, 73, 56–65. [DOI] [PubMed]
- 8.Mahony T.J., McCarthy,F.M., Gravel,J.L., West,L. and Young,P.L. (2002) Construction and manipulation of an infectious clone of the bovine herpesvirus 1 genome maintained as a bacterial artificial chromosome. J. Virol., 76, 6660–6668. [DOI] [PMC free article] [PubMed]
- 9.Grillot-Courvalin C., Goussard,S., Huetz,F., Ojcius,D.M. and Courvalin,P. (1998) Functional gene transfer from intracellular bacteria to mammalian cells. Nat. Biotechnol., 16, 862–866. [DOI] [PubMed]
- 10.Grillot-Courvalin C., Goussard,S. and Courvalin,P. (1999) Bacteria as gene delivery vectors for mammalian cells. Curr. Opin. Biotechnol., 10, 477–481. [DOI] [PubMed]
- 11.Courvalin P., Goussard,S. and Grillot-Courvalin,C. (1995) Gene transfer from bacteria to mammalian cells. C. R. Acad. Sci. III, 318, 1207–1212. [PubMed]
- 12.Sizemore D.R., Branstrom,A.A. and Sadoff,J.C. (1995) Attenuated Shigella as a DNA delivery vehicle for DNA-mediated immunization. Science, 270, 299–302. [DOI] [PubMed]
- 13.Sizemore D.R., Branstrom,A.A. and Sadoff,J.C. (1997) Attenuated bacteria as a DNA delivery vehicle for DNA-mediated immunization. Vaccine, 15, 804–807. [DOI] [PubMed]
- 14.Paglia P., Terrazzini,N., Schulze,K., Guzman,C.A. and Colombo,M.P. (2000) In vivo correction of genetic defects of monocyte/macrophages using attenuated Salmonella as oral vectors for targeted gene delivery. Gene Ther., 7, 1725–1730. [DOI] [PubMed]
- 15.Krusch S., Domann,E., Frings,M., Zelmer,A., Diener,M., Chakraborty,T. and Weiss,S. (2002) Listeria monocytogenes mediated CFTR transgene transfer to mammalian cells. J. Gene Med., 4, 655–667. [DOI] [PubMed]
- 16.Waters V.L. (2001) Conjugation between bacterial and mammalian cells. Nature Genet., 29, 375–376. [DOI] [PubMed]
- 17.Wade-Martins R., White,R.E., Kimura,H., Cook,P.R. and James,M.R. (2000) Stable correction of a genetic deficiency in human cells by an episome carrying a 115 kb genomic transgene. Nat. Biotechnol., 18, 1311–1314. [DOI] [PubMed]
- 18.Wade-Martins R., Smith,E.R., Tyminski,E. and Saeki,Y. (2001) An infectious transfer and expression system for genomic DNA loci in human and mouse cells. Nat. Biotechnol., 19, 1067–1070. [DOI] [PubMed]
- 19.White R.E., Wade-Martins,R. and James,M.R. (2002) Infectious delivery of 120-kilobase genomic DNA by an epstein-barr virus amplicon vector. Mol. Ther., 5, 427–435. [DOI] [PubMed]
- 20.Grant S.G., Jessee,J., Bloom,F.R. and Hanahan,D. (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl Acad. Sci. USA, 87, 4645–4649. [DOI] [PMC free article] [PubMed]
- 21.Aizencang G., Solis,C., Bishop,D.F., Warner,C. and Desnick,R.J. (2000) Human uroporphyrinogen-III synthase: genomic organization, alternative promoters and erythroid-specific expression. Genomics, 70, 223–231. [DOI] [PubMed]
- 22.Isberg R. and Falkow,S. (1985) A single genetic locus encoded by Yersinia pseudotuberculosis permits invasion of cultured animal cells by Escherichia coli K-12. Nature, 317, 262–264. [DOI] [PubMed]
- 23.Haziza C., Stragier,P. and Patte,J.-C. (1982) Nucleotide sequence of the asd gene of Escherichia coli: absence of a typical attenuation signal. EMBO J., 1, 379–384. [DOI] [PMC free article] [PubMed]
- 24.Muyrers J.P., Zhang,Y., Testa,G. and Stewart,A.F. (1999) Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res., 27, 1555–1557. [DOI] [PMC free article] [PubMed]
- 25.Isberg R.R., Swain,A. and Falkow,S. (1988) Analysis of expression and thermoregulation of the Yersinia pseudotuberculosis inv gene with hybrid proteins. Infect. Immun., 56, 2133–2138. [DOI] [PMC free article] [PubMed]
- 26.Low K.B. (1972) Escherichia coli K-12 F-prime factors, old and new. Bacteriol. Rev., 36, 587–607. [DOI] [PMC free article] [PubMed]
- 27.Verma I.M. and Somia,N. (1997) Gene therapy: promises, problems and prospects. Nature, 398, 239–242. [DOI] [PubMed]