

Abstract
Mitochondria constitute a major source of reactive oxygen species and have been proposed to integrate the cellular responses to stress. In animals, it was shown that mitochondria can trigger apoptosis from diverse stimuli through the opening of MTP, which allows the release of the apoptosis-inducing factor and translocation of cytochrome c into the cytosol. Here, we analyzed the role of the mitochondria in the generation of oxidative burst and induction of programmed cell death in response to brief or continuous oxidative stress in Arabidopsis cells. Oxidative stress increased mitochondrial electron transport, resulting in amplification of H2O2 production, depletion of ATP, and cell death. The increased generation of H2O2 also caused the opening of the MTP and the release of cytochrome c from mitochondria. The release of cytochrome c and cell death were prevented by a serine/cysteine protease inhibitor, Pefablock. However, addition of inhibitor only partially inhibited the H2O2 amplification and the MTP opening, suggesting that protease activation is a necessary step in the cell death pathway after mitochondrial damage.
Generation of reactive oxygen species (ROS) in plants has been implicated in biotic and abiotic stresses. A biphasic oxidative burst is triggered in the plant cells following recognition of invading avirulent pathogens. Despite extensive research on the source of ROS, the subcellular location and the mechanism of ROS generation has not been unequivocally clarified (Bolwell, 1999). Many different mechanisms have been shown to be involved in pathogen-induced ROS production, including peroxidases and diverse oxidases (oxalate oxidase, amine oxidase, and NADPH oxidase; Pugin et al., 1997; Wojtaszek, 1997; Rea et al., 1998). An intracellular and an apoplastic source of ROS production were detected during elicitation of tobacco (Nicotiana tabacum) epidermal cells with cryptogein from Phytophthora cryptogea (Allan and Fluhr, 1997). The connection between pathogen response and respiration is further substantiated by the recent finding that salicylic acid, an important component of the pathogen response mechanism, inhibits ATP formation and uptake of respiratory O2 in nonphotosynthetic tobacco cells (Xie and Chen, 1999). Mitochondria is a major source of ROS formation, and it is possible that this organelle could participate in the oxidative burst in plants. During respiration, molecular oxygen may undergo a univalent reduction at the sites of ROS generation in complexes I and III of the respiratory chain, forming superoxide, which subsequently dismutates to hydrogen peroxide (Braidot et al., 1999).
We have shown previously that a brief treatment with H2O2 induced a programmed cell death (PCD) in suspension-cultured soybean (Glycine max) cells (Levine et al., 1994, 1996). Recently, Desikan and coworkers (1998) showed that a similar response is activated in suspension-cultured Arabidopsis cells. In those experiments, PCD induction by H2O2 in the soybean and Arabidopsis cell cultures required a rather high concentration of H2O2, presumably due to rapid decomposition of H2O2 by various antioxidant systems. It is important to note that in both plant systems, the cell death process induced by H2O2 depended on active cellular metabolism and could be blocked by protease inhibitors. Subsequent studies have shown that H2O2 caused activation of Cys proteases in soybean cells that were instrumental in the execution of the cell death program (Solomon et al., 1999).
Cys proteases play a crucial role in animal apoptosis and were shown to be involved in many forms of plant PCD (del Pozo and Lam, 1998). In animal systems, it was shown that mitochondria are directly involved in the activation of cytosolic Asp-specific Cys proteases (caspases) via release of cytochrome c (Green and Reed, 1998). Translocation of cytochrome c from mitochondria has been reported during heat stress in cucumber (Cucumis sativus; Balk et al., 1999) and in menadione-induced cell death of tobacco protoplasts (Sun et al., 1999). On the other hand, in petunia (Petunia hybrida), the release of cytochrome c was found to be dispensable for induction of PCD (Xu and Hanson, 2000).
In this work, we analyzed the role of mitochondria in the generation of oxidative burst triggered by a temporary or by a continuous oxidative stress. We show that exposure of Arabidopsis cells to a mild constant oxidative stress increased respiratory electron transport and oxygen uptake in isolated mitochondria, leading to increased production of ROS, effectively amplifying the oxidative stress. H2O2 caused the mitochondrial permeability transition and release of cytochrome c from the inner mitochondrial membranes, resulting in ATP depletion and cell death. Blocking of the MTP with cyclosporin A (CsA) prevented cell death. We also show that protease inhibitors blocked cytochrome c release and partially prevented the permeability transition and the depletion of ATP.
RESULTS
Induction of PCD by H2O2
Cell death induced by accumulating oxidative stress was studied in nonphotosynthetic Arabidopsis cells that were incubated with an H2O2-generating mixture of Glc oxidase (GO) and Glc (G). This treatment resulted in a rise of H2O2 in the medium from 90 μm in the control culture to 348 and 474 μm H2O2 after 6 and 12 h, respectively. This concentration of hydrogen peroxide was only slightly higher than the oxidative burst produced by harpin treatment, which was measured as 344 μm H2O2 after 3 h of treatment. However, these results are below the millimolar concentration that is generated after oligogalacturonide-induced oxidative burst in cultured soybean cells (Legendre et al., 1993). The addition of 10 mm G plus 10 units mL−1 GO caused death in 66.9% of cells within 18 h, whereas very little cell death occurred after addition of 2 units mL−1 GO (Fig. 1A). To test whether the continuous oxidative stress activated an active signaling mechanism, as implied by the relatively slow cell death process similar to the brief pulse of H2O2, cultures were preincubated with protease inhibitors that blocked H2O2-dependent cell death in soybean and Arabidopsis cultures (Levine et al., 1996; Solomon et al., 1999). Ser/Cys protease inhibitor Pefablock (AEBSF) reduced cell death by almost 70%, resulting in 24% of dead cells in the inhibitor-treated culture. Addition of a caspase-3 inhibitor, Z-YVAD (del Pozo and Lam, 1998), and of a Cys protease inhibitor, l-trans-epoxysuccinyl-leucylamido(4-guanidino) butane (Barrett et al., 1982), reduced the degree of cell death to 29% and 40%, respectively, indicating that oxidative stress induced an active hypersensitive response-like PCD pathway (Fig. 1B).
Figure 1.

Induction of PCD by oxidative stress. A, Cells were incubated with 10 mm G and 2, 5, or 10 units mL−1 GO. Cell death was measured 1, 3, 6, and 18 h later. Each point represents mean value of three independent experiments. B, Inhibition of oxidative stress-induced death by protease inhibitors. Arabidopsis cells were treated with 10 mm G plus 10 units mL−1 GO as in A in the presence of l-trans-epoxysuccinyl-leucylamido(4-guanidino) butane (50 μm), AEBSF (1 mm), or caspase-3 (Casp3) inhibitor (50 μm), and cell death was measured after 18 h. Results are mean values of three independent replica.
Depletion of ATP by Oxidative Stress
To study the mechanism of H2O2-induced PCD, we examined the effect of oxidative stress on level of ATP in the cells. H2O2 was continuously generated by addition of 10 units mL−1 GO with 10 mm G, and the ATP concentration was measured after 1, 3, and 6 h. A strong drop in ATP concentration was observed 1 h after treatment, and decreased further with time (Fig. 2A). Because the suspension-cultured cells used in this study do not possess functional chloroplasts, the major ATP-producing organelle in these cells is mitochondria. To directly assay the effect of a temporary oxidative stress on mitochondrial ATP production, cells were treated with 1 or 5 mm H2O2, and ATP production was measured after 3 h in isolated mitochondria. ATP generation was initiated by the addition of malate plus Glu, which constitutes the complex I substrates. ATP generation by the mitochondria was decreased with an increase in the concentrations of H2O2 (Fig. 2B). Because exogenously added H2O2 was decomposed before the isolation of mitochondria and the addition of respiratory substrates (Desikan et al., 1998), these results indicate sustained damage to the mitochondria.
Figure 2.

ATP depletion during oxidative stress. A, Analysis of ATP levels in whole cells treated with G/GO as in Figure 1B. ATP was extracted after 1, 3, or 6 h and was measured in a Bioluminometer. White bars represent the ATP level in control cells and black bars represent the ATP level in treated cells. Data presented are mean values of three replica ± se. B, Generation of ATP in isolated mitochondria. Cells were treated with 1 or 5 mm H2O2 for 3 h. Mitochondria were isolated as described in “Materials and Methods.” ATP production in isolated mitochondria was initiated by the addition of 10 mm malate plus Glu, 200 μm NAD, and 0.1 mm ADP. The ATP concentration was measured after 15 min. Data are the mean values of three replica ± se.
The Effect of Oxidative Stress on Mitochondrial Electron Transport
The effects of temporary and continuous oxidative stress on mitochondrial electron transport was examined by measuring oxygen consumption (Braidot et al., 1999). Comparison of oxygen consumption rates between control and treated cells showed faster oxidation of complex I or complex III substrate (malate plus Glu or succinate, respectively) by mitochondria from the oxidatively stressed cells (Fig. 3A). The faster oxygen consumption was also observed in cells that received a single pulse of H2O2 3 h prior to mitochondria isolation (Fig. 3C). In both cases, the mitochondria from stressed cells exhibited a partial uncoupling between the substrate oxidation and dependence on ADP availability, consistent with the low ATP levels that were detected in those cells (Fig. 2B). It is important to note that the differences in electron transport were not associated with the alternative oxidase activity because oxygen consumption was completely blocked by the addition of KCN (Fig. 3B). Oxygen consumption triggered by malate plus Glu was also fully stopped after the addition of rotenone, a complex I inhibitor (Fig. 3A), verifying that all measurements represent the mitochondrial respiratory pathway (Herz et al., 1994). In a similar manner, oxygen consumption that was initiated by succinate could be inhibited by a complex III inhibitor, antimycin A (data not shown).
Figure 3.

Oxygen consumption in oxidatively stressed mitochondria. A, Arabidopsis cells were treated with G/GO as in Figure 1B. Mitochondria were isolated 3 h later, and electron transport was measured polarographically with an oxygen electrode. Electron transport was initiated by the addition of complex I substrates, 10 mm malate plus 10 mm Glu, and 200 μm NAD plus (Mal plus Glu). Coupling between the electron transport and ATP production was estimated by the addition of 100 μm ADP. The role of complex I on measured oxygen consumption was examined by addition of 4 μm otenone (Rot). Numbers indicate the calculated rate of oxygen consumption normalized by standardized measurements of oxygen consumption of double-distilled water. Time between arrows corresponds to 60 s. B, Cells were spiked with 5 mm H2O2, and mitochondria were isolated 3 h later. Electron transport across complex I was measured as described in A. C, Cells were treated with G/GO as in A. Electron transport across complex III was measured with 10 mm succinate plus 100 μm ADP. The dependence of oxygen consumption on the cytochrome c pathway was examined by the addition of 50 μm KCN. glut, Glu.
Generation of ROS by Oxidatively Stressed Mitochondria
One of the major damaging consequences of enhanced electron flow is an increased generation of oxygen radicals such as superoxide and hydrogen peroxide (Kowaltowski and Vercesi, 1999). To assess the level of H2O2 production in mitochondria from control and stressed cells, the isolated mitochondria were incubated in the presence of a H2O2-sensitive mitochondrial dye, dihydrorhodamine123 (Royall and Ischiropoulos, 1993), and the endogenous H2O2 production was visualized under a fluorescent microscope. Virtually no H2O2 production was seen in mitochondria of the control samples, indicating that the isolation procedure did not generate H2O2. However, bright fluorescence was emitted from almost all of the mitochondria that were derived from the stressed cells (Fig. 4A).
Figure 4.

ROS production in isolated mitochondria. A, Mitochondria isolated from control (C) or cells treated for 3 h with G/GO as in Figure 1B were stained with dihydrorhodamine123 and were observed under a fluorescent microscope. B, Arabidopsis cells were pulsed with 1 or 5 mm H2O2, and mitochondria were isolated 3 h later. Electron flow was initiated by the addition of complex I substrates, and H2O2 concentration was measured after 15 min. C, Mitochondria from control (white bars) or cells treated with G/GO (black bars) for 3 h were incubated in the presence of complex I (mal) or complex III (succ) substrates, and H2O2 concentration was measured after 15 min. D, Antioxidant activity in isolated mitochondria from control (white bars) or G/GO-treated (black bars) cells. One millimole H2O2 was added to the mitochondria, and the H2O2 concentration was measured after 0, 5, 10, 15, and 30 min. E, Inhibition of ROS generation in mitochondria by inhibitors of electron transport. Mitochondria isolated from control (white bars) or from G/GO-treated (black bars) cells were incubated for 15 min with rotenone (4 μm) or antimycin A (1 μm) or catalase (5,000 units mL−1) in combination with complex I (mal plus glut) or complex III (suc) substrates. H2O2 concentration was measured after 15 min. t0 indicates the H2O2 concentration before the addition of the respiratory substrates. glut, Glu.
The production of O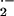 and H2O2 in isolated mitochondria was verified by using a chemical nonenzymatic assay (Snell and Snell, 1949). Arabidopsis cells were pulsed with 1 or 5 mm H2O2 and were incubated for 3 h prior to mitochondria isolation. Electron transport was initiated by the addition of malate plus Glu, and generation of H2O2 was measured for 30 min. The pulse of 5 mm H2O2 caused a 2.7-fold higher generation of H2O2 in mitochondria isolated from these cells than in mitochondria from control cells (Fig. 4B). An even higher, 4.3-fold increase in the H2O2 production was observed in the mitochondria from cultures that were continuously exposed to G/GO for the same period of time (Fig. 4C). Mitochondria provided with complex I substrate had a slightly higher activity than the complex III substrate. The addition of catalase together with the respiratory substrates strongly diminished the titanium sulfate oxidation, indicating that H2O2 constituted the major form of ROS. The increased production of H2O2 in the mitochondria from G/GO-treated cells was not a consequence of GO carryover during mitochondria preparation because addition of G did not increase H2O2 production, and omission of respiratory substrates stopped H2O2 production (data not shown). Oxidative stress-induced changes in the antioxidant capacity were tested by the addition of 1 mm H2O2 to the mitochondrial suspensions, and the decay was monitored for 30 min. A similar rate of H2O2 decomposition was observed in mitochondria from control and G/GO-treated cells, indicating that the increased H2O2 generation was not due to decreased antioxidant capacity (Fig. 4D). The direct involvement of electron transport in the generation of H2O2 was evaluated by the addition of complex I or complex III inhibitors (4 μm rotenone or 1 μm antimycin A) jointly with the complex I or III substrates. H2O2 was measured after 30 min of incubation. H2O2 production in the presence of the respiratory inhibitors ceased almost completely (Fig. 4E), in agreement with the earlier observed arrest in oxygen consumption (Fig. 3B). Production of H2O2 was verified by the addition of catalase at the time of the addition of complex I/III substrates.
and H2O2 in isolated mitochondria was verified by using a chemical nonenzymatic assay (Snell and Snell, 1949). Arabidopsis cells were pulsed with 1 or 5 mm H2O2 and were incubated for 3 h prior to mitochondria isolation. Electron transport was initiated by the addition of malate plus Glu, and generation of H2O2 was measured for 30 min. The pulse of 5 mm H2O2 caused a 2.7-fold higher generation of H2O2 in mitochondria isolated from these cells than in mitochondria from control cells (Fig. 4B). An even higher, 4.3-fold increase in the H2O2 production was observed in the mitochondria from cultures that were continuously exposed to G/GO for the same period of time (Fig. 4C). Mitochondria provided with complex I substrate had a slightly higher activity than the complex III substrate. The addition of catalase together with the respiratory substrates strongly diminished the titanium sulfate oxidation, indicating that H2O2 constituted the major form of ROS. The increased production of H2O2 in the mitochondria from G/GO-treated cells was not a consequence of GO carryover during mitochondria preparation because addition of G did not increase H2O2 production, and omission of respiratory substrates stopped H2O2 production (data not shown). Oxidative stress-induced changes in the antioxidant capacity were tested by the addition of 1 mm H2O2 to the mitochondrial suspensions, and the decay was monitored for 30 min. A similar rate of H2O2 decomposition was observed in mitochondria from control and G/GO-treated cells, indicating that the increased H2O2 generation was not due to decreased antioxidant capacity (Fig. 4D). The direct involvement of electron transport in the generation of H2O2 was evaluated by the addition of complex I or complex III inhibitors (4 μm rotenone or 1 μm antimycin A) jointly with the complex I or III substrates. H2O2 was measured after 30 min of incubation. H2O2 production in the presence of the respiratory inhibitors ceased almost completely (Fig. 4E), in agreement with the earlier observed arrest in oxygen consumption (Fig. 3B). Production of H2O2 was verified by the addition of catalase at the time of the addition of complex I/III substrates.
Oxygen Consumption and ROS Production in the Presence of AEBSF
Given the efficient inhibition of Arabidopsis cell death by treatment of cells with protease inhibitors (Fig. 1B), it was interesting to determine the mode of protection provided by the inhibitors. Arabidopsis culture was preincubated with AEBSF for 15 min and was treated with G/GO for 3 h. The accelerated oxygen consumption caused by the oxidative stress was reduced significantly in the presence of the inhibitor (Fig. 5A). One of the manifestations of damaged mitochondria is the permeability of the outer mitochondrial membrane to exogenous cytochrome c (Lemeshko, 2000, 2001). At early stages of apoptosis, the addition of exogenous cytochrome c can still restore respiratory functions (Mootha et al., 2001). The mitochondrial state of treated cells was examined by the addition of exogenous cytochrome c. No increase in O2 consumption was observed after cytochrome c supplementation of the mitochondria from control (healthy) cells, but oxygen consumption was strongly stimulated in the mitochondria of G/GO-treated cells (Fig. 5A). This response was not present when the cells were incubated with G/GO in the presence of AEBSF, suggesting improved preservation of the outer membrane.
Figure 5.

The effect of protease inhibitor on mitochondrial functioning. A, Cells were treated with G/GO in the presence or absence of protease inhibitor (PI) AEBSF (1 mm) for 3 h. Electron transport was measured in isolated mitochondria as in Figure 3A. Cytochrome c arrow indicates the effect of addition of exogenous cytochrome c (50 μg). B, The effect of protease inhibitor AEBSF (PI) on H2O2 generation in isolated mitochondria from control (C) or G/GO-treated cells. H2O2 concentration was measured after 15 min. C, The effect of protease inhibitor AEBSF (PI) on ATP production in isolated mitochondria from control (C) or G/GO-treated (GO) cells. ATP content was estimated and quantified as described in Figure 2B. glut, Glu.
The increase in the rate of ROS generation in the presence of the protease inhibitor (AEBSF) was reduced by 25% (Fig. 5B), and the amount of ATP production by the mitochondria was reduced by 33% (Fig. 5C), as compared with the culture treated with G/GO alone. The mitochondria from cells incubated in the presence of protease inhibitor also retained a better coupling between ATP synthesis and oxygen consumption, as measured by the drop in oxygen after addition of ADP (Fig. 5A).
Opening of Permeability Transition Pore and Release of Cytochrome c
One of the major molecular mechanisms that lead to profound changes in mitochondrial functioning is caused by an alteration in mitochondrial permeability transition pore (MTP), which protects the mitochondria from the loss of electrochemical potential for H+ by preventing nonspecific transfer of solutes of <1,500 D. In animal systems, permeability transition is sufficient and necessary for apoptosis (Kroemer, 1997). The state of MTP in the Arabidopsis cells treated with G/GO was assayed by measuring the mitochondrial swelling, which was initiated by addition of calcium (Pastorino et al., 1999). Although mitochondria from the untreated cells swelled considerably, little change in absorbance occurred in the mitochondria from oxidatively stressed cells, indicating that H2O2 caused sustained damage to the mitochondrial membrane (Fig. 6A). The MTP was still partially functional in mitochondria of cells treated with the protease inhibitor. In animal systems, among the critical proteins that are released from mitochondria through the MTP is cytochrome c, and its release into the cytosol triggers apoptosis (Green and Reed, 1998).
Figure 6.

The effect of oxidative stress on mitochondrial permeability transition and release of cytochrome c. A, Cells were treated with the G/GO in the presence or absence of protease inhibitor (PI) AEBSF (1 mm) for 3 h. Mitochondrial permeability transition was measured as the difference in the change in the A546. Electron flow in isolated mitochondria was generated by the addition of complex I substrates. Permeability transition was initiated by the addition of 16.5 nm CaCl2, and decrease in absorbance was measured at 546 nm. In a parallel experiment, 1 μm CsA was added to block mitochondrial swelling and this was used as a reference to calculate the change in absorbance. B, Cells were treated with G/GO in the presence or absence of protease inhibitor (PI) AEBSF (1 mm) for 3 h. Proteins were isolated from mitochondrial and cytosolic fractions, separated on 12.5% (w/v) SDS-PAGE, and analyzed by western blotting with antibodies against cytochrome c. Labeling was detected by chemiluminescence.
The localization of cytochrome c in oxidatively stressed Arabidopsis cells was studied by western blotting. Mitochondrial and cytosolic proteins from cells treated with G/GO in the presence or absence of AEBSF for 3 h were separated on 12.5% (w/v) SDS-PAGE and were probed with antibody against cytochrome c (Sun et al., 1999). In control cells, cytochrome c was detected only in the mitochondria, whereas in the induced cells, it was observed just in the cytosol, suggesting release of cytochrome c from mitochondria during oxidative stress (Fig. 6B). The loss of cytochrome c from the mitochondria was blocked in the cells treated with G/GO in the presence of AEBSF. However, AEBSF did not completely block the release of cytochrome c into the cytosol, and a small amount of cytochrome c was found also in the cytosol. The relatively weak signal of the cytochrome c in the G/GO-treated cells could be the result of its proteolytic degradation in the cytosol (Bobba et al., 1999).
CsA Protects Cells from Oxidative Damage
To test the role of MTP in the oxidative stress-induced PCD, we took advantage of the demonstrated inhibition of MTP by CsA (Fortes et al., 2001). Cells were preincubated with 1 μm CsA for 15 min prior to the introduction of the cell death-inducing concentration of G/GO. Staining the cells with Evan's blue after 20 h showed that CsA effectively reduced the amount of cell death from 64% to 33% (Fig. 7A). A similar conclusion was also reached by staining the cells with a viability dye, fluorescein diacetate, which resulted in a 7-fold increase in the fluorescence of the CsA-treated cultures (data not shown).
Figure 7.

Effect of CsA on cellular functioning. A, Cells were treated with G/GO in presence and absence of CsA (1 μm), which was added 15 min prior to the oxidative treatment. Cell death was measured after18 h. Each point represents the mean value of three replica. B, Cells were treated with G/GO in presence or absence of CsA. Mitochondria were isolated after 3 h and were incubated with complex I substrates. The H2O2 concentration was measured after 15 min. The results are the mean value of three replica ± se. C, Cells were treated with G/GO in presence and absence of CsA (1 μm). ATP was extracted after 3 h in isolated mitochondria and was measured as described in Figure 2B. The results are the mean value of three replica ± se.
The addition of CsA also had a positive effect on the production of H2O2 by the mitochondria isolated from the G/GO-treated cells (Fig. 7B). CsA addition reduced the increase in the rate of H2O2 generation resulting from the G/GO treatment by close to 50%. Analysis of the ATP contents indicated that the treatment of cells with CsA also improved the mitochondrial functioning and restored the mitochondrial ATP production to 89% as compared with the unstressed control cells (Fig. 7C). These measurements were conducted 3 h after addition of G/GO, which is before the considerable loss of membrane integrity, indicating that inhibition of MTP provided protection against oxidative stress damage to the mitochondrial functioning.
DISCUSSION
Generation of ROS by mitochondrial respiratory chain is a physiological and continuous process that ensues in a single electron reduction of up to 2% of the consumed oxygen in unstressed cells (Braidot et al., 1999; Kowaltowski and Vercesi, 1999). Under physiological conditions, toxic effects of ROS are removed by antioxidant systems, but during biotic or abiotic stresses, the concentration of ROS in the cells can rise significantly, reaching the threshold that can trigger PCD (Lamb and Dixon, 1997).
Our results show that even mild oxidative stress can lead to enhanced H2O2 generation by the mitochondria. H2O2 produced by various internal or external sources can be amplified within the mitochondria (Fig. 4), resulting in accumulation of oxidative stress damage to a level sufficient for activation of PCD (Fig. 1). A brief pulse of high concentration of H2O2 or a continuous generation of low amounts of H2O2 damaged the mitochondrial respiratory chain, resulting in increased leakage of electrons to O2 (Fig. 3). These findings can explain how a mild but prolonged oxidative stress that occurs during various adverse environmental conditions that are associated with oxidative stress such as chilling (Prasad et al., 1994; Van Breusegem et al., 1999), excessive light (Chamnongpol et al., 1996), salinity (Hernandez et al., 1993), and many other environmental stresses (Smirnoff, 1998) can produce necrotic lesions.
It has recently been suggested that plant mitochondria may act as an integrator of cellular conditions from different stimuli and, beyond a certain threshold, activate PCD (Lam et al., 1999; Jones, 2000). Our results support the crucial role of mitochondria in the regulation of stress responses. There are a number of checkpoints at which mitochondria can trigger PCD: at the level of substrate import into the mitochondria (Hautecler et al., 1994), at the level of substrate oxidation and electron transport (Moore et al., 1991), by the control over oxidative phosphorylation (Kesseler et al., 1992), as well as by regulation of MTP (Kroemer, 1997; Green and Reed, 1998). The electron flow can also be diverted by the upregulation of alternative oxidase (Day and Wiskich, 1995). Some of the above steps can be directly regulated by the cytosol, such as transport of malate into mitochondria. In addition, the mitochondrial responses can be regulated by calcium influx from the cytosol. Thus, various conditions that influence mitochondrial function as well as H2O2 production by other compartments such as plasma membrane NADPH oxidase system can be integrated to trigger PCD. Mitochondria can function independently as the source of H2O2 and can amplify the O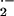 and H2O2 levels inside the cell. The production of H2O2 directly depended on the mitochondrial respiration, as was demonstrated by the addition of complex I or complex III inhibitors, which diminished the accumulation of H2O2 (Fig. 4E). The similar rate of H2O2 decomposition in mitochondria from control and oxidatively stressed cells exclude a decrease in antioxidant capacity as a possible mechanism of H2O2 accumulation (Fig. 4D).
and H2O2 levels inside the cell. The production of H2O2 directly depended on the mitochondrial respiration, as was demonstrated by the addition of complex I or complex III inhibitors, which diminished the accumulation of H2O2 (Fig. 4E). The similar rate of H2O2 decomposition in mitochondria from control and oxidatively stressed cells exclude a decrease in antioxidant capacity as a possible mechanism of H2O2 accumulation (Fig. 4D).
In mammalian mitochondria, it was shown that oxidants cause damage to membrane protein thiols, leading to crosslinking and opening of the permeability transition pore (Castilho et al., 1995; Fortes et al., 2001). MPT begins as a permeabilization of mitochondrial membrane to small sugars and osmotic support, and is later followed by permeabilization to low-molecular mass proteins, and finally resulting in irreversible mitochondrial dysfunction (Zoratti and Szabo, 1995; Castilho et al., 1996). In animal systems, permeability transition is necessary and sufficient for apoptosis to occur (Kroemer, 1997). The precise molecular site of oxidative damage to the plant mitochondrial components and the factors released by mitochondria have not been identified yet, although our results implicate the involvement of MPT and the electron transport systems in the plant PCD as well. The decreased oxygen consumption after the addition of ADP indicates that the oxidative stress caused uncoupling between respiration and generation of ATP (Fig. 3A). Such depletion of ATP can be sufficient to induce apoptosis in plants when coupled with calcium influx (Jones, 2000). H2O2 was shown to cause calcium influx in tobacco and in soybean cells (Price et al., 1994; Levine et al., 1996). Although those studies analyzed fluctuations in the cytosolic calcium, the regulation of calcium levels in the different subcellular compartments is closely linked (Trewavas and Malho, 1998). It is interesting that in potato (Solanum tuberosum) tubers, ROS induced MPT in a calcium-independent manner, suggesting that variations in the mechanism of PCD activation in plants may vary between species (Fortes et al., 2001). In some systems, it has also been found that oxidative burst was caused by the calcium entry (Piedras et al., 1998). Taken together, those results imply a tight regulatory network that links ROS generation and calcium fluxes, both of which are involved in control of PCD. Moreover, calcium influx into mitochondria is also regulated by different stimuli via changes in membrane potential (Silva et al., 1992).
The inhibition of G/GO-induced PCD by protease inhibitors, which were previously found to inhibit the H2O2-triggered death (Levine et al., 1996; Solomon et al., 1999), suggests that both stimuli activate the same cell death pathway. It is interesting that protease inhibitors blocked PCD without strongly reducing the mitochondrial generation of ROS or blocking the depletion of ATP. However, inhibition of protease activity prevented the loss of majority of cytochrome c from the inner mitochondria as evidenced by western blotting of mitochondrial proteins (Fig. 6B) and by measurements of oxygen consumption following addition of exogenous cytochrome c (Fig. 5A). The loss of cytochrome c from the inner mitochondrial membrane during apoptosis has been reported in many systems, and is considered as a crucial regulatory step in apoptosis (Kroemer, 1997). At the early phases of apoptosis, the readdition of exogenous cytochrome c can markedly restore respiratory functions (Mootha et al., 2001). The stimulation of oxygen consumption by added cytochrome c in the mitochondria of G/GO-treated cells indicates that these cells were still at the beginning, in line with the time course of PCD in these cells (Fig. 1A). The stimulatory effect of the exogenous cytochrome c was also not observed in the presence of the protease inhibitor (AEBSF), indicating an intact outer mitochondrial membrane (Fig. 5A). In the cytosol of G/GO-treated cells, the amount of cytochrome c detected by western analysis appeared as rather small, probably due to the proteolytic degradation of cytochrome c (Bobba et al., 1999), whereas in the presence of AEBSF, even small amounts of cytochrome c that were released into the cytosol were protected from degradation by the inhibitor. These observations are in agreement with the earlier results that showed activation of AEBSF-sensitive proteases by oxidative stress (Solomon et al., 1999).
In summary, our results show that a brief pulse of high concentration of H2O2 or a continuous generation of low concentrations of H2O2 caused amplification of H2O2 production by the mitochondria through fast uncoupled electron transport, leading to depletion of ATP, opening of MTP, and translocation of cytochrome c to the cytosol, terminating in PCD. Protease inhibition suppressed PCD of Arabidopsis cells, likely by preventing the damage to the outer mitochondrial membrane.
MATERIALS AND METHODS
Materials
Unless otherwise stated, the following concentrations of chemicals were used: antimycin A (1 μm), rotenone (4 μm), cytochrome c (50 μg), and catalase (5,000 units mL−1). The above materials were purchased from Sigma (St. Louis). GO was from Worthington Biochemicals (Freehold, NJ).
Cell Culture
Arabidopsis cell cultures isolated by May and Leaver (1993) were kept at 25°C under continuous light in Murashige and Skoog medium (pH 6.0) supplemented with 100 mg L−1 myoinositol, 0.4 mg L−1 thiamine, 0.5 mg L−1 naphthylacetic acid, 0.05 mg L−1 kinetin, and 3% (w/v) Suc. Cells were treated with H2O2 or with G/GO 3 d after subculture, which was performed weekly (1:10 dilution). Although the cultures appeared green after depletion of Suc several days after subculture, no functional activity of photosystem II was detected by measuring variable fluorescence with a pulse-amplitude-modulated fluorometer for at least 5 d after subculture.
Measurement of Electron Transport
Mitochondria were isolated according to the method of Day et al. (1985), and electron transport was measured polarographically using a Clark type oxygen electrode (Rank Brothers, Cambridge, UK). Mitochondrial suspension was adjusted to 80 μg of protein in the assay medium consisting of 0.3 m mannitol, 10 mm phosphate buffer (pH 7.5), 3 mm MgSO4, 10 mm NaCl, 5 mm KH2PO4, and 0.1% (w/v) bovine serum albumin. Complex I and complex III activities were initiated as described in Braidot et al. (1999). Coupling was assayed by the addition of 0.1 mm ADP. Complex IV activity was blocked by KCN (50 μm). Uncoupling between oxygen consumption and electron transport was verified by the addition of 5 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone, which drastically increased the oxygen consumption (data not shown).
Measurement of H2O2
H2O2 generation in isolated mitochondria were observed after staining for 5 min with dihydrorhodamine123 and were visualized under a fluorescent microscope (IX70; Olympus, Tokyo) using the excitation/emission filters (XF23; Omega Optical, Brattleboro, VT). Photographs were taken with a digital camera (Coolpix 900; Nikon, Tokyo). H2O2 production in isolated mitochondria or in cell culture medium was measured with a nonenzymatic assay according to Snell and Snell (1949). For the measurement of H2O2 production, 20 μL of mitochondrial suspension or culture medium was added to a cuvette containing 880 μL of double distilled water and 100 μL of titanium sulfate, and was incubated for 15 min at room temperature. Oxidation of titanium sulfate was recorded by reading A410. Readings were converted to corresponding concentration using a standard calibration plot. Endogenous antioxidant activity in mitochondria was assayed by measuring the decay of 1 mm H2O2 with time.
Measurement of ATP Pool
The amount of ATP was measured by the luciferin-luciferase method (St. John, 1970). ATP was dissolved in 5 mL of 25 mm HEPES buffer (pH 7.5), and luminescence from a 200-μL sample was assayed in a luminometer (Lumac 3M Biocounter M2010A; Lumitran Scientific, Jerusalem) together with 40 μL of luciferin-luciferase (Sigma) from 10 mg mL−1 stock for 30 s. The standard curve of ATP concentration was prepared from a known amount (1 pM–1 μm).
Measurement of Cell Death
Cell death was quantified by Evan's blue as described in Levine et al. (1994). For 100% cell death, the culture was heated at 100°C for 5 min. The percentage of cell death in selected treatments was also verified by counting several hundred cells under a microscope. Treatment of cells with 10 mm G plus 10 units mL−1 of GO routinely produced around 60% dead cells.
Western-Blot Analysis
Proteins from mitochondrial as well as cytosolic fraction (50 μg) were separated on 12.5% (w/v) SDS-PAGE. The proteins were transferred on to nitrocellulose, blocked in Tris-buffered saline, 0.3% (w/v) Tween 20, and 3% (w/v) dried skimmed milk, and they were labeled with antibody against cytochrome c (1/1,000; Zymed Laboratories, South San Francisco) at 4°C overnight. After washing (three times for 15 min) in block buffer, the membrane was incubated in rabbit anti-mouse horseradish peroxidase conjugate (1/5,000) in Tris-buffered saline/Tween 20 for 3 h at room temperature and washed again. Labeling was detected by chemiluminescence.
Measurement of MPT
MPT was assayed by measuring mitochondrial swelling as described by Pastorino et al. (1999). In brief, mitochondria were suspended in medium containing 400 mm mannitol, 10 mm phosphate buffer (pH 7.5), and 1 mm EDTA. One millimole Glu and 1 mm malate were added as respiratory substrates for complex I, and the MPT was initiated by the addition of 16.5 nm CaCl2. The mitochondrial swelling was measured by decreased A546. Swelling was inhibited by the addition of 1 μm of CsA for reference to calculate the change in absorbance.
Footnotes
This work was supported by the Israel Science Foundation.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.010999.
LITERATURE CITED
- Allan AC, Fluhr R. Two distinct sources of elicited reactive oxygen species in tobacco epidermal cells. Plant Cell. 1997;9:1559–1572. doi: 10.1105/tpc.9.9.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balk J, Leaver CJ, McCabe PF. Translocation of cytochrome c from the mitochondria to the cytosol occurs during heat-induced programmed cell death in cucumber plants. FEBS Lett. 1999;463:151–154. doi: 10.1016/s0014-5793(99)01611-7. [DOI] [PubMed] [Google Scholar]
- Barrett AJ, Kembhavi AA, Brown MA, Kirschke H, Knight CG, Tamai M, Hanada K. l-trans-epoxysuccinyl-leucylamido(4-guanidino) butane and its analogues as inhibitors of cysteine proteinases including cathepsins B and L. Biochem J. 1982;201:189–198. doi: 10.1042/bj2010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobba A, Atlante A, Giannattasio S, Sgaramella G, Calissano P, Marra E. Early release and subsequent caspase-mediated degradation of cytochrome c in apoptotic cerebellar granule cells. FEBS Lett. 1999;457:126–130. doi: 10.1016/s0014-5793(99)01018-2. [DOI] [PubMed] [Google Scholar]
- Bolwell GP. Role of active oxygen species and NO in plant defense responses. Curr Opin Plant Biol. 1999;2:287–294. doi: 10.1016/S1369-5266(99)80051-X. [DOI] [PubMed] [Google Scholar]
- Braidot E, Petrussa E, Vianello A, Macri F. Hydrogen peroxide generation by higher plant mitochondria oxidizing complex I or complex II substrates. FEBS Lett. 1999;451:347–350. doi: 10.1016/s0014-5793(99)00616-x. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Kowaltowski AJ, Meinicke AR, Bechara EJH, Vercesi AE. Permeabilization of the inner mitochondrial-membrane by Ca2+ ions is stimulated by t-butyl hydroperoxide and mediated by reactive oxygen species generated by mitochondria. Free Radic Biol Med. 1995;18:479–486. doi: 10.1016/0891-5849(94)00166-h. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Kowaltowski AJ, Vercesi AE. The irreversibility of inner mitochondrial membrane permeabilization by Ca2+ plus pro-oxidants is determined by the extent of membrane protein thiol cross linking. J Bioenerg Biomembr. 1996;28:523–529. doi: 10.1007/BF02110442. [DOI] [PubMed] [Google Scholar]
- Chamnongpol S, Willekens H, Langebartels C, Vanmontagu M, Inzé D, Vancamp W. Transgenic tobacco with a reduced catalase activity develops necrotic lesions and induces pathogenesis-related expression under high light. Plant J. 1996;10:491–503. [Google Scholar]
- Day DA, Neuburger M, Douce R. Biochemical characterization of chlorophyll-free mitochondria from pea leaves. Aust J Plant Physiol. 1985;12:219–228. [Google Scholar]
- Day DA, Wiskich JT. Regulation of alternative oxidase activity in higher plants. J Bioenerg Biomembr. 1995;27:379–385. doi: 10.1007/BF02110000. [DOI] [PubMed] [Google Scholar]
- del Pozo O, Lam E. Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr Biol. 1998;8:1129–1132. doi: 10.1016/s0960-9822(98)70469-5. [DOI] [PubMed] [Google Scholar]
- Desikan R, Reynolds A, Hancock JT, Neill SJ. Harpin and hydrogen peroxide both initiate programmed cell death but have differential effects on defense gene expression in Arabidopsis suspension cultures. Biochem J. 1998;330:115–120. doi: 10.1042/bj3300115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortes F, Castilho RF, Catisti R, Carnieri EGS, Vercesi AE. Ca2+ induces a cyclosporin A-insensitive permeability transition pore in isolated potato tuber mitochondria mediated by reactive oxygen species. J Bioenerg Biomembr. 2001;33:43–51. doi: 10.1023/a:1005672623709. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Hautecler JJ, Slusegoffart CM, Evens A, Duyckaerts C, Sluse FE. Effect of aspartate and glutamate on the oxoglutarate carrier investigated in rat heart mitochondria and inverted submitochondrial vesicles. Biochim Biophys Acta. 1994;1185:153–159. doi: 10.1016/0005-2728(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Hernandez JA, Corpas FJ, Gomez M, Del Rio LA, Sevilla F. Salt-induced oxidative stress mediated by activated oxygen species in pea leaf mitochondria. Physiol Plant. 1993;89:103–110. [Google Scholar]
- Herz U, Schroder W, Liddell A, Leaver CJ, Brennicke A, Grohmann L. Purification of the NADH:ubiquinone oxidoreductase of the respiratory chain from the inner mitochondrial membrane of Solanum tuberosum. J Biol Chem. 1994;269:2263–2269. [PubMed] [Google Scholar]
- Jones A. Does the plant mitochondrion integrate cellular stress and regulate programmed cell death? Trends Plant Sci. 2000;5:225–230. doi: 10.1016/s1360-1385(00)01605-8. [DOI] [PubMed] [Google Scholar]
- Kesseler A, Diolez P, Brinkmann K, Brand MD. Characterization of the control of respiration in potato-tuber mitochondria using the top-down approach of metabolic control analysis. Eur J Biochem. 1992;210:775–784. doi: 10.1111/j.1432-1033.1992.tb17480.x. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Vercesi AE. Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med. 1999;26:463–471. doi: 10.1016/s0891-5849(98)00216-0. [DOI] [PubMed] [Google Scholar]
- Kroemer G. Mitochondrial implication in apoptosis: towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ. 1997;4:443–456. doi: 10.1038/sj.cdd.4400266. [DOI] [PubMed] [Google Scholar]
- Lam E, Pontier D, del Pozo O. Die and let live: programmed cell death in plants. Curr Opin Plant Biol. 1999;2:502–507. doi: 10.1016/s1369-5266(99)00026-6. [DOI] [PubMed] [Google Scholar]
- Lamb C, Dixon RA. The oxidative burst in plant disease resistance. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:251–275. doi: 10.1146/annurev.arplant.48.1.251. [DOI] [PubMed] [Google Scholar]
- Legendre L, Rueter S, Heinstein PF, Low PS. Characterization of the oligogalacturonide-induced oxidative burst in cultured soybean (Glycine max) cells. Plant Physiol. 1993;102:233–240. doi: 10.1104/pp.102.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemeshko VV. Mg2+ induces intermembrane electron transport by cytochrome c desorption is mitochondria with the ruptured outer membrane. FEBS Lett. 2000;72:5–8. doi: 10.1016/s0014-5793(00)01419-8. [DOI] [PubMed] [Google Scholar]
- Lemeshko VV. Failure of exogenous NADH and cytochrome c to support energy-dependent swelling of mitochondria. Arch Biochem Biophys. 2001;388:60–66. doi: 10.1006/abbi.2000.2214. [DOI] [PubMed] [Google Scholar]
- Levine A, Pennell R, Alvarez M, Palmer R, Lamb CJ. Calcium-mediated apoptosis in a plant hypersensitive disease resistance response. Curr Biol. 1996;6:427–437. doi: 10.1016/s0960-9822(02)00510-9. [DOI] [PubMed] [Google Scholar]
- Levine A, Tenhaken R, Dixon R, Lamb C. H2O2 from the oxidative burst orchestrates the plant hypersensitive disease resistance response. Cell. 1994;79:583–593. doi: 10.1016/0092-8674(94)90544-4. [DOI] [PubMed] [Google Scholar]
- May MJ, Leaver CJ. Oxidative stimulation of glutathione synthesis in Arabidopsis thaliana suspension cultures. Plant Physiol. 1993;103:621–627. doi: 10.1104/pp.103.2.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AL, Dry IB, Wiskich JT. Regulation of electron transport in plant mitochondria under state-4 conditions. Plant Physiol. 1991;95:34–40. doi: 10.1104/pp.95.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Wei MC, Buttle KF, Scorrano L, Panoutsakopoulou V, Mannella CA, Korsmeyer SJ. A reversible component of mitochondrial respiratory dysfunction apoptosis can be rescued by exogenous cytochrome c. EMBO J. 2001;20:661–671. doi: 10.1093/emboj/20.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorino JG, Tafani M, Rothman RJ, Marcineviciute A, Hoek JB, Farber JL. Functional consequences of the sustained or transient activation by Bax of the mitochondrial permeability transition pore. J Biol Chem. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- Piedras P, Hammond-Kosack KE, Harrison K, Jones JDG. Rapid Cf-9- and Avr9-dependent production of active oxygen species in tobacco suspension cultures. Mol Plant-Microbe Interact. 1998;11:1155–1166. [Google Scholar]
- Prasad TK, Anderson MD, Martin BA, Stewart CR. Evidence for chilling-induced oxidative stress in maize and a regulatory role for hydrogen peroxide. Plant Cell. 1994;6:65–74. doi: 10.1105/tpc.6.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AH, Taylor A, Ripley SJ, Griffiths A, Trewavas AJ, Knight MR. Oxidative signals in tobacco increase cytosolic calcium. Plant Cell. 1994;6:1301–1310. doi: 10.1105/tpc.6.9.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugin A, Frachisse JM, Tavernier E, Bligny R, Gout E, Douce R, Guern J. Early events induced by the elicitor cryptogein in tobacco cells. Plant Cell. 1997;9:2077–2091. doi: 10.1105/tpc.9.11.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea G, Laurenzi M, Tranquilli E, D'Ovidio R, Federico R, Angelini R. Developmentally and wound-regulated expression of the gene encoding a cell wall copper amine oxidase in chickpea seedlings. FEBS Lett. 1998;437:177–182. doi: 10.1016/s0014-5793(98)01219-8. [DOI] [PubMed] [Google Scholar]
- Royall JA, Ischiropoulos H. Evaluation of 2′,7′-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- Silva MAP, Carnieri EGS, Vercesi AE. Calcium transport by corn mitochondria: evaluation of the role of phosphate. Plant Physiol. 1992;98:452–457. doi: 10.1104/pp.98.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnoff N. Plant resistance to environmental stress. Curr Opin Biotechnol. 1998;9:214–219. doi: 10.1016/s0958-1669(98)80118-3. [DOI] [PubMed] [Google Scholar]
- Snell FD, Snell CT. Colorimetric methods of analysis. Inorganic. 1949;2:882–883. [Google Scholar]
- Solomon M, Belenghi B, Delledonne M, Levine A. The involvement of cysteine proteases and protease inhibitor genes in programmed cell death in plants. Plant Cell. 1999;11:431–444. doi: 10.1105/tpc.11.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. John JB. Determination of ATP in Chlorella with the luciferin-luciferase enzyme system. Anal Biochem. 1970;37:409–416. doi: 10.1016/0003-2697(70)90066-7. [DOI] [PubMed] [Google Scholar]
- Sun YL, Zhao Y, Hong X, Zhai ZH. Cytochrome c release and caspase activation during menadione-induced apoptosis in plants. FEBS Lett. 1999;462:317–321. doi: 10.1016/s0014-5793(99)01539-2. [DOI] [PubMed] [Google Scholar]
- Trewavas AJ, Malho R. Ca2+ signalling in plant cells: the big network! Curr Opin Plant Biol. 1998;1:428–433. doi: 10.1016/s1369-5266(98)80268-9. [DOI] [PubMed] [Google Scholar]
- Van Breusegem F, Slooten L, Stassart JM, Moens T, Botterman J, Van Montagu M, Inzé D. Overproduction of Arabidopsis thaliana FeSOD confers oxidative stress tolerance to transgenic maize. Plant Cell Physiol. 1999;40:515–523. doi: 10.1093/oxfordjournals.pcp.a029572. [DOI] [PubMed] [Google Scholar]
- Wojtaszek P. Oxidative burst: an early plant response to pathogen infection. Biochem J. 1997;322:681–692. doi: 10.1042/bj3220681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZX, Chen ZX. Salicylic acid induces rapid inhibition of mitochondrial electron transport and oxidative phosphorylation in tobacco cells. Plant Physiol. 1999;120:217–225. doi: 10.1104/pp.120.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Hanson MR. Programmed cell death during pollination-induced petal senescence in petunia. Plant Physiol. 2000;122:1323–1333. doi: 10.1104/pp.122.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]