

Abstract
Ataxia-telangiectasia is a hereditary multisystemic disease resulting from mutations of ataxia telangiectasia, mutated (ATM) and is characterized by neurodegeneration, cancer, immune defects, and hypersensitivity to ionizing radiation. The molecular details of ATM function in the nervous system are unclear, although the neurological lesion in ataxia-telangiectasia becomes apparent early in life, suggesting a developmental origin. The central nervous system (CNS) of Atm-null mice shows a pronounced defect in apoptosis induced by genotoxic stress, suggesting ATM functions to eliminate neurons with excessive genomic damage. Here, we report that the death effector Bax is required for a large proportion of Atm-dependent apoptosis in the developing CNS after ionizing radiation (IR). Although many of the same regions of the CNS in both Bax−/− and Atm−/− mice were radioresistant, mice nullizygous for both Bax and Atm showed additional reduction in IR-induced apoptosis in the CNS. Therefore, although the major IR-induced apoptotic pathway in the CNS requires Atm and Bax, a p53-dependent collateral pathway exists that has both Atm- and Bax-independent branches. Further, Atm- and Bax-dependent apoptosis in the CNS also required caspase-3 activation. These data implicate Bax and caspase-3 as death effectors in neurodegenerative pathways.
The human syndrome ataxia telangiectasia (A-T) results from mutations of the ATM gene and is characterized by progressive neurodegeneration that leads to severe ataxia (1). A-T is also typified by many other defects including immune deficiencies, cancer proneness, chromosomal instability, and ionizing radiation sensitivity (2). ATM is a large 370-kDa protein that contains a COOH-terminal domain similar to phosphatidylinositol 3-kinase and encodes a protein kinase activity specific for serine and threonine residues (2). This COOH region is conserved throughout a family of proteins that are involved in cellular responses to DNA damage and maintenance of genomic stability. Because a nervous system lesion is the most prevalent feature of A-T, ATM signaling in this tissue is particularly relevant for understanding ATM function in a biological context. The neurological defect(s) in A-T becomes apparent early in life, suggesting that it originates during development. Furthermore, Atm is highly expressed in the developing nervous system but only at low levels in the adult CNS (3). However, the mechanism of neuronal cell loss in A-T is unknown. To this end, we have investigated Atm signaling in the developing CNS of Atm-null mice. Apoptosis resulting from genotoxic damage of the nervous system requires Atm (4), suggesting Atm-dependent apoptosis may be important for the development and maintenance of the nervous system. However, there is a paucity of data concerning other death effectors in this signaling pathway. Therefore, we examined the death effector Bax and the caspases for their role in Atm-dependent apoptosis in the nervous system. Bax is a member of the Bcl-2 family of proteins and functions as a proapoptotic death effector (5–7). In the nervous system, Bax modulates some programmed cell deaths (8, 9) and can promote apoptosis in vitro in neuronal cultures after a variety of insults (10–12). Final integration of apoptotic signaling often involves activation of caspases, specific proteases that act downstream of the Bcl-2 family and facilitate the execution phase of apoptosis (13, 14). In this report, we show that Bax and caspase-3 are components of the Atm-signaling pathway and are required for Atm-dependent apoptosis in the developing nervous system.
Methods
Histology and Immunohistochemistry.
Mice were used on postnatal day 5 (P5; day of birth is P0). In all cases, experiments were done in triplicate by using wild-type (WT) littermates for each genotype. Mice were irradiated with 14 Gy from a cesium irradiator (delivered at a rate of 0.9 Gy/min) and allowed various times of recovery. Tissues were collected after fixation by transcardial perfusion with 4% paraformaldehyde, cryoprotected in 20% sucrose/PBS and cryosectioned (12-μm coronal sections). Sytox green (Molecular Probes) was dissolved in PBS to a final concentration of 1 μM, used on tissue sections permeabilized with 0.1% Triton X-100 in PBS, pH 7.4, and washed with PBS. In situ end labeling was performed as described (4). Quantitation of apoptosis was done by counting pyknotic figures in equivalent fields of the external granule layer (EGL) of the various mutant mice. Immunohistochemical detection of p53 was done by using the CM5 anti-p53 antibody (Vector Laboratories) at a dilution of 1/500 by using antigen retrieval as described previously (4). Antiactive caspase-3 was detected with affinity-purified CM1 antibody (0.66 mg/ml) (15) at a dilution of 1/1,500, also by using antigen retrieval. The Vectastain Elite ABC kit/VIP substrate avidin/biotin immunoperoxidase system (Vector Laboratories) was used for visualization of primary antibody binding. Sections were counterstained with 0.1% methyl green (Vector Laboratories) and mounted in permount.
Tissue Slice Cultures.
Organotypic slice cultures were performed by using a modification of Stoppini et al. (16). P5 cerebellum was extracted and placed in chilled artificial cerebrospinal fluid (0.12M NaCl/1.2 mM NaH2PO4/2.5 mM KCl/1 mM MgSO4/2 mM CaCl2/22 mM NaHCO3/20 mM glucose), cut into 300-μm slices, and placed on Millicell-CM membrane (Millipore). Tissue was bathed in Hanks' balanced salt solution (20 mM Hepes/0.45% glucose, pH 7.4) containing caspase inhibitors or DMSO vehicle alone irradiated and then incubated at 37°C for 5 hr. Caspase inhibitors (Calbiochem) in DMSO were used at a final concentration of 100 μM for benzyloxycarbonyl (z)-VAD-fluoromethyl ketone (FMK) and z-DEVD-FMK and 200 μM for all others. Tissue slices were then placed in 4% paraformaldehyde/PBS for 4 hr at 4°C, cryoprotected in 20% sucrose/PBS, and cryosectioned at 10 μm. Sections were stained with 1% neutral red (Aldrich) in 0.1 M acetic acid (pH 4.8) for 1 min followed by dehydration in ethanol.
Ribonuclease Protection Analysis (RPA) and Western Blot Analysis.
Tissues were harvested at various times after 14 Gy of irradiation. RNA was obtained by using Trizol reagent (Sigma). Twenty micrograms of total cerebellum RNA was used in a ribonuclease protection assay with a mouse Multiprobe mAPO-1 RPA set (PharMingen) by using an RPA II system (Ambion) according to the manufacturer's instructions. Protected RNA products were separated on a 5% acrylamide gel. Proteins were extracted from tissues by homogenization in SDS sample buffer, and equal quantities of protein were separated on 10% SDS/PAGE gels and transferred to nitrocellulose membranes. Membranes were stained with Ponceau S (Sigma) to confirm equal protein transfer. p53 was detected by using anti-p53 Ab-7 (Oncogene Science) at a dilution of 1/2,500 for 1 hr at room temperature. Antibody binding was detected by using enhanced chemiluminescence (Amersham). Caspase-3 was detected by using rabbit polyclonal anti-caspase-3 (PharMingen) at a dilution of 1/2,500 overnight at 4°C, and antibody binding was detected by using enhanced chemiluminescence (Amersham).
Caspase Assay.
Tissues were lysed in hypotonic lysis buffer (25 mM Hepes (pH 7.5)/5 mM MgCl2/2 mM DTT/5 mM EDTA/0.1% Triton X-100) containing protease inhibitors (Complete, Mini; Boehringer Mannheim). The supernatant fraction containing enzyme activity was obtained after centrifugation of tissue lysate at 16,000 × g for 20 min at 4°C. Caspase-1 and caspase-3 were measured by a fluorometric peptide cleavage assay by using the CaspACE Assay System (Promega). Assays were performed in 96-well plates by using a Fluoroskan II, Microwell Fluorescence Reader (Labsystems, Chicago). Fluorescence was measured by using an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Lysates were preactivated by incubating at 30°C for 1 hr and then were incubated with or without 50 μM caspase-1 inhibitor [Ac-YVAD-aldehyde (CHO)] or caspase-3 inhibitor (Ac-DEVD-CHO) for 30 min at 30°C before assay. Assays were performed in the presence of 50 μM of the corresponding substrate, according to the manufacturer's instructions. Caspase activity was expressed as pmol of 7-amino-4-methylcoumarin liberated per μg of protein at 30°C. Each measurement was performed in triplicate, and variation between assays was less than 10%.
Results
Because Bax is required for some forms of p53-dependent apoptosis (11, 17, 18), we used Bax knockout mice (19) to examine the in vivo role of Bax in Atm-dependent signaling in the nervous system. The developing CNS of Bax−/− mice was profoundly resistant to ionizing radiation-induced apoptosis. Although abundant cell death occurred in the P5 dentate gyrus of WT animals after IR (Fig. 1A, a and b), apoptosis was almost absent in the Bax−/− dentate gyrus (Fig. 1A, c and d). Because the decrease in IR-induced apoptosis of the Bax−/− dentate gyrus was similar to Atm−/− mice (4), we examined other regions of the CNS known to require Atm for IR-induced apoptosis. Although IR caused widespread apoptosis in the EGL in WT mice (Fig. 1B, a and e), the Bax−/− mice were resistant to apoptosis in this region of the cerebellum (Fig. 1B, d and h). Consistent with our previous studies, radioresistant neurons were also found in the Atm−/− EGL (Fig. 1B, b and f) and in the p53−/− EGL (Fig. 1B, c and g). Surprisingly, and in contrast to the cerebellum, normal levels of apoptosis occurred in the P5 Bax−/− retina at 18 hr after IR and were indistinguishable from WT retina (Fig. 1B, i and k). However, the central region of the Atm−/− P5 retina (and the p53−/− retina; not shown) was resistant to IR-induced death (Fig. 1B, j). Thus, Bax is not required for Atm-dependent IR-induced apoptosis in the retina. These data emphasize the importance of cellular context in neuronal apoptosis, because one region of the CNS (e.g., the cerebellum) requires Bax for apoptosis, whereas another (e.g., the retina) does not. Moreover, this difference also highlights the selectivity of p53 targets during apoptosis.
Figure 1.
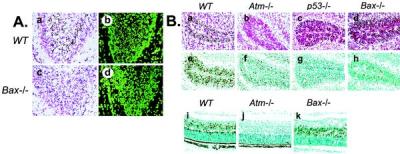
Bax is required for IR-induced apoptosis in the cerebellum and dentate gyrus. (A) Apoptosis is almost absent in the P5 Bax−/− dentate gyrus (c and d) compared with WT tissue (a and b) 18 hr after IR. Apoptosis was assessed by neutral red (a and c) and Sytox green (b and d). (B) Apoptosis occurs in the EGL of the P5 WT cerebellum (a and e) but is markedly reduced or absent in Atm−/− (b and f), p53−/− (c and g), and Bax−/− (d and h) tissue after IR. In contrast to in the cerebellum, apoptosis in the central region of the WT (i) and Bax −/− retina (k) are indistinguishable, whereas in the Atm−/− retina (j), it is absent. a–d are neutral red stained, and e–k are in situ end labeling-stained tissue 18 hr after IR. (×200.)
Stabilization and transactivation of p53 likely involves phosphorylation status (20). ATM is a regulator of p53, at least in part by modulating Ser-15 phosphorylation (21–23) and Ser-276 dephosphorylation (24). Stabilization of p53 after IR in the P5 cerebellum requires Atm (4). Therefore, dysfunctional Atm is defective in signaling to p53 upon genotoxic stress, and this presumably abrogates p53-dependent events leading to apoptosis. To order the Atm-dependent apoptotic pathway, we examined p53 stabilization in Bax−/− tissues. Normal levels of p53 stabilization occur after IR in the Bax−/− cerebellum (Fig. 2A) and retina (not shown), demonstrating that Bax is dispensable for p53 stabilization. Therefore, Atm-dependent apoptosis in the P5 CNS after IR is a result of p53-mediated events that require Bax. However, although p53 stabilization is found as early as 30 min after IR and is maintained through 5 hr, Bax levels do not change over this duration (Fig. 2B). There are also no differences in Bax levels in the Atm−/− or p53−/− mice compared with WT either before or after IR treatment (Fig. 2C.). Overall, these observations indicate Atm is an upstream signaling component of p53 responses, whereas Bax is a downstream component. Therefore, it is likely these components function sequentially and are in a linear pathway.
Figure 2.

p53 stabilization occurs in Bax−/− cerebellum, whereas Bax protein levels are unaffected by IR. (A) Examination of p53 stabilization by using immunohistochemistry shows equivalent p53 stabilization in the EGL of WT and Bax−/− cerebellum after IR but reduced stabilization in Atm−/− and an absence in p53−/− tissue. (×200.) (B) Bax levels do not alter up to 5 hr after IR. After identification of p53, the immunoblot was reprobed with an antibody to Bax; 1–4 are P5 cerebellum samples 0, 0.5, 1, and 5 hr after IR, respectively. (C) p53 is induced in WT and Bax−/− eye after IR (lanes 5 and 8, respectively), whereas induction is reduced in Atm−/− and absent from p53−/− tissue (lanes 6 and 7, respectively). The induction of p53 in the Atm−/− retina occurs in multipotential cells of the periphery, where IR-induced apoptosis is independent of Atm (4). Bax levels are similar in WT, Atm−/−, and in p53−/− cerebellum and eye (1–3, respectively) and are not obviously affected by IR (WT; Atm and p53 are 5, 6, and 7, respectively). Bax−/− tissues (4 and 8) have no detectable Bax. Lanes 1–4 and 5–8 are extracts from unirradiated or irradiated animals, respectively. Control is a region of the Ponceau S-stained membrane used for the anti-Bax immunoblot of the cerebellum to illustrate protein loading and transfer.
To confirm that Bax and Atm reside in the same pathway, we interbred heterozygotes to generate animals that were nullizygous for both Atm and Bax. Surprisingly, these mice showed a further reduction in IR-induced apoptosis, implying that both Atm-independent and Bax-independent pathways also exist in these tissues (Fig. 3A). Similar reductions in apoptosis in the Atm/Bax double-null compared with either the Atm- or Bax-null after IR were also observed in the dentate gyrus and olfactory bulb of double-null animals (not shown). However, these independent pathways are quantitatively a minor component of IR-induced apoptosis, because in either Atm- or Bax-null animals, only ≈5% of the EGL cells die and in the double-null animals, ≈0.2% (Fig. 3B). Possibly, sufficient DNA damage can signal to p53 independently of Atm, and distinct Bax-independent effector pathways may exist. Because p53 is critical for all IR-induced death in the developing P5 CNS, these Atm- and Bax-independent pathways must still operate through p53.
Figure 3.
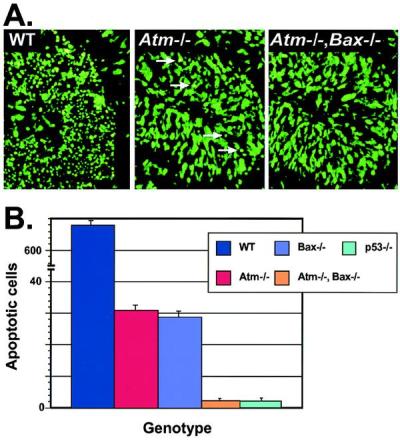
Apoptosis is further reduced in Atm/Bax double-null CNS tissue after IR. (A) Sytox green staining of the P5 cerebellum from WT, Atm-null, and Atm/Bax double-null animals shows a pronounced reduction in the number of apoptotic EGL cells 18 hr after IR in the Atm-null compared with WT. A further reduction was seen in Atm/Bax double-null EGL. Arrows in the Atm−/− section indicate apoptotic cells. (×400.) (B) Quantitative comparison of irradiated cerebellum from various genotypes scored for apoptotic cells. Although the WT cerebellum had abundant cell deaths, the Atm-null and Bax-null cerebellum showed only low numbers of dead cells, whereas the Atm/Bax double-null cerebellum was indistinguishable from the p53-null cerebellum, with an almost complete absence of apoptosis.
Caspases are known downstream effectors of apoptosis and are responsible for the commitment to apoptosis (13, 14). Therefore, we examined whether Atm signaling involved caspase activation. Initially, we determined the kinetics of caspase activation during IR-induced apoptosis in the WT CNS. Although no detectable apoptosis was present at 2 hr after IR, at 3 hr apoptotic cells were observed in the margin of the outer proliferating region of the cerebellar EGL, and by 4 hr substantial deaths were seen throughout the entire region (Fig. 4A). To establish which caspases may be causal in Atm-dependent apoptosis, we used RPA to determine the caspase profile in the cerebellum. Of those present, caspase-3 was at least an order of magnitude more abundant than any others. Only low expression levels were found for caspases-2, -6, and -8, whereas caspases-1, -7, -10, -11, and -12 were not detected (data not shown, but see Fig. 5). Caspases-4, -5, -13, and -14 are reportedly not present in the brain (25–27), although caspase-9 is present and is clearly required for nervous system function (28, 29). To determine the involvement of caspase-3 in IR-induced apoptosis in the CNS, we used an antibody (CM1) specific for the cleaved 17-kDa form of active caspase-3 (15). At 2 hr after IR, CM1 staining of irradiated cerebellum showed a pronounced ridge of cleaved caspase-3 in the outer proliferative zone of the EGL and by 3 hr, a wave of caspase-3 activation spread throughout the EGL (Fig. 4A). Importantly, this wave of activation precedes the occurrence of apoptosis in the P5 cerebellum. A fluorogenic peptide assay also showed a marked increase in caspase-3, but not caspase-1, activity between 2 and 4 hr after IR in the cerebellum (Fig. 4B). Thus, caspase-3 cleavage and activation precedes and coincides with the hallmark morphological changes of apoptosis. Finally, we used specific peptide inhibitors to block caspase activity elicited by IR treatment of the cerebellum. Using slice culture methodology to preserve the biological context of the granule cells in the cerebellum, we recapitulated IR-induced apoptosis with identical kinetics to that found in vivo. IR-induced apoptosis was prevented by caspase inhibitors with pan-caspase (z-VAD-FMK) and caspase-3 specificity (z-DEVD-FMK) but not caspase-1 specificity (z-YAVD-CHO) (Fig. 4C). Although the caspase-3 inhibitor z-DEVD-FMK can also inhibit caspases-7 and -10, the inability to detect expression of these by using RPA makes it unlikely that they contribute to Atm-dependent apoptosis in the P5 CNS. Consistent with a requirement for caspase-9 to activate caspase-3 (30, 31), caspase-9 inhibitor (z-LEHD-FMK) also blocked IR-induced death. In contrast, caspases-2, -6, or -8 inhibitors did not block apoptosis in cerebellar slices after IR (data not shown). Taken together, these data indicate caspase-3 activation is necessary for apoptosis of cerebellar granule cells.
Figure 4.
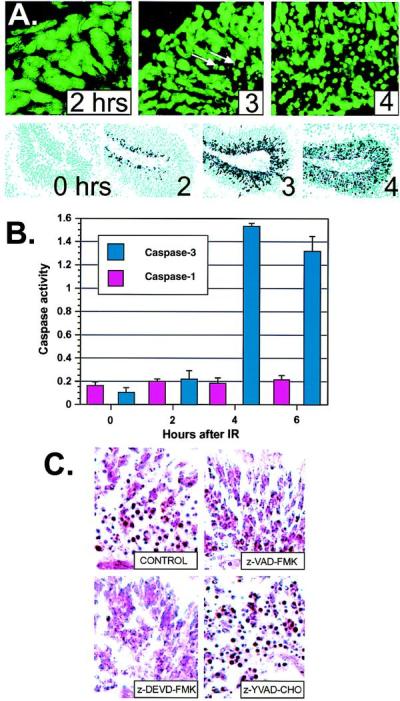
Caspase-3 activation coincides with apoptosis in the CNS of WT mice. (A) Apoptotic cells appeared between 2 and 3 hr in the cerebellar EGL after IR and by 4 hr, apoptotic figures were widely distributed throughout the EGL. Sections are stained with Sytox green to visualize pyknotic cells; arrows indicate apoptotic cells. (×400.) Immunohistochemistry by using antibodies specific for the cleaved subunit of caspase-3 (15) show caspase-3 activation precedes the apoptotic morphological changes. Caspase-3 cleavage appears initially at 2 hr in the outer EGL, and abundant staining throughout the EGL occurs at 3 and 4 hr. (×200.) (B) Caspase-3 enzyme activity coincides with caspase-3 cleavage and apoptosis seen in A. An increase in caspase-3 activity (pmol of 7-amino-4-methylcoumarin liberated per μg of protein) occurred between 2 and 4 hr after IR in the cerebellum. Caspase-1 activity was insensitive to radiation. (C) Cerebellar slice cultures were irradiated, incubated with various caspase inhibitors, cryosectioned, and stained with neutral red to identify apoptotic cells. Incubation with general caspase inhibitors (z-VAD-FMK) or caspase-3 inhibitor (z-DVED-FMK), but not caspase-1 inhibitor (z-YVAD-CHO) or DMSO vehicle alone (CONTROL), prevented IR-induced apoptosis in the cerebellar EGL. No apoptosis was observed in unirradiated cultures. Sections are from P5 cerebellum. (×400.)
Figure 5.
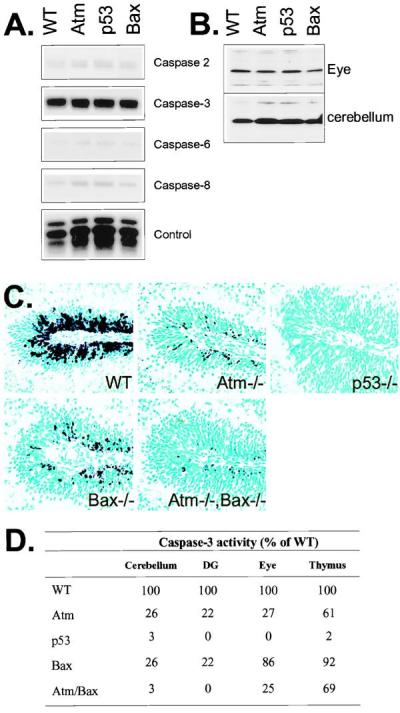
Caspase expression and proteolysis after IR. (A) RPA analysis of the P5 WT, Atm, p53, and Bax-null cerebellum shows a high level of expression for caspase-3 and only low levels for caspases-2, -6, and -8. (B) Protein immunoblot analysis with anticaspase-3 shows equal levels of caspase-3 proenzyme are present in WT, Atm−/−, p53−/−, and Bax−/− tissues. (C) Caspase-3 activation was reduced in the Atm- and Bax-null cerebellar EGL and further reduced in the Atm/Bax double-null animals after irradiation. No caspase-3 activation was observed in the irradiated p53-null cerebellum. (×200.) (D) The relative levels of caspase-3 activity in various mutant tissues 6 hr after IR as a percentage of WT activity. In all cases, samples were normalized for caspase-3 activity present in unirradiated tissues. DG, dentate gyrus.
Because Atm-dependent apoptosis after IR in the cerebellum and dentate gyrus requires p53 and Bax, we assessed caspase-3 activation after irradiation in Atm-, p53-, and Bax-null mice. Initially, we surveyed caspase gene expression in each of the genotypes to determine that there were no conspicuous profile differences. Caspase RNA expression profiles were qualitatively and quantitatively identical in each genotype (Fig. 5A) as were caspase-3 zymogen levels (Fig. 5B). In WT animals, caspase-3 activation involves both the outer proliferative and migratory populations of the EGL, whereas in the Atm- and Bax-null tissues, activation occurs only in occasional cells in the proliferative region (Fig. 5C). The Atm/Bax double-null tissue shows a greater reduction in caspase-3 cleavage than either mutant alone, consistent with increased attenuation of apoptosis in the double-null nervous system. Although some limited cell death is associated with the proliferative EGL in the Atm-null and Bax-null animals, the absence of caspase-3 activation in p53-null animals highlights the requirement for p53 in all IR-induced apoptosis and caspase-3 activation in the cerebellum. Therefore, Atm, p53, and Bax all participate in the activation of caspase-3 after IR.
Caspase-3 activity also closely paralleled the extent of caspase-3 cleavage and the relative levels of apoptosis seen in the CNS of the mutant mice after IR (Fig. 5D). In contrast, the thymus of all genotypes except the p53−/− animals showed high caspase-3 activity after IR. These data support previous reports describing a requirement for p53, but not Atm or Bax, for IR-induced death of thymocytes (4, 19, 32, 33). However, the Atm and Atm/Bax double-null thymus have a reduction in caspase-3 activity by ≈30% compared with the WT and Bax thymus, which may reflect radioresistant Atm-null CD4+/CD8+ thymocytes described previously (33). Caspase-3 activation in the eye also reflects the apoptotic profile after IR. Bax is not required for IR-induced apoptosis in the retina (Fig. 1B), and concordantly caspase-3 levels were similar to WT retina. Consistent with the occurrence of Atm-dependent death being restricted to the central region of the retina, but not the periphery (4), caspase-3 activity is substantially reduced in Atm−/− retina. The Atm/Bax double-null eye shows levels of caspase 3 similar to the Atm-null, whereas, and as with all other regions of the P5 CNS, no IR-induced caspase activity was found in the p53-null eye.
Discussion
Dysfunctional ATM results in neurodegeneration. However, the molecular events leading to ATM-dependent cell death in the nervous system are unclear. Because ionizing radiation activates ATM, this agent is a useful tool for delineating ATM signaling. With this approach, Atm was found to be required for genotoxic damage-induced apoptosis in the nervous system, suggesting that Atm is required at a developmental checkpoint to eliminate neurons that have acquired genetic damage (4). In this report, we show that in addition to Atm and p53, Bax and caspase-3 are also required for apoptosis in the developing CNS after ionizing radiation. Because Bax acts downstream of p53, and p53 is a pivotal apoptotic signal integrator in the CNS (4, 34, 35), involvement of both p53 and Bax may be widespread in neurodegeneration. A surprising finding from this work was that, in contrast to other regions of the CNS, apoptosis after IR in the developing retina did not require Bax as a death effector. This was despite Bax being present in this tissue at comparable levels to other CNS regions. Consistent with this, degeneration of retinal photoreceptors occurring in the rd mutant mouse is not rescued in a Bax-null background (36). Because IR-induced apoptosis in the retina does involve similar signaling intermediates to other regions of the CNS, including Atm, p53, and caspase-3, the dispensability of Bax is an unexpected selective fine-tuning of apoptosis. Moreover, because p53 is required for Atm-dependent apoptosis in the retina as well as other CNS regions, these data further underscore the specificity of p53 targets during apoptosis. Although Bax has been implicated as a direct target of p53 (37), recent data suggest an indirect mechanism of activation (38). Given the linearity of Atm and Bax signaling described here, the pronounced reduction of apoptosis in the Atm/Bax double-null CNS after IR compared with single nullizygous mice reveals additional complexity in the utilization of Atm- and Bax-dependent apoptotic pathways. Moreover, this occurs within apparently uniform populations of cells, such as the EGL of the developing cerebellum, a structure that gives rise almost exclusively to granule neurons (39). Thus, Atm- and Bax-independent pathways in the EGL suggest either an unappreciated cellular heterogeneity, that sufficient DNA damage can signal to p53 independently of Atm [e.g., through Atr (40)] or, like the retina, distinct Bax-independent effector pathways exist.
A central issue in understanding ATM function in the nervous system is what etiological agent activates ATM. One obvious possibility is DNA damage. Compelling evidence that DNA damage does occur in the nervous system during development has recently been found in mice with gene ablations for certain components of the V(D)J recombination machinery. Mice nullizygous for DNA ligase IV or XRCC4 (a DNA ligase IV-associated protein) have striking developmental defects of the nervous system resulting in embryonic lethality (41–43). Because DNA ligase IV is known to function in end joining of DNA strand breaks, these results provide direct support for an interrelationship between DNA processing and nervous system development. This raises the possibility that Atm may also be involved in the damage response that requires ligase IV or XRCC4. It will be important to establish whether Atm does indeed cooperate with DNA processing machinery like DNA ligase IV or XRCC4 during nervous system development. Although the ligase IV/XRCC4-null mice implicate DNA damage as a feature of nervous system development, the source of the damage is unclear. Possibly these DNA lesions happen nonspecifically as a consequence of cellular growth, or an as-yet-unidentified process akin to V(D)J recombination occurs during neurogenesis. Other physiological inducers may also contribute to DNA damage and may be important for ATM function. For example, glutamate-mediated excitotoxicity of cultured neurons can lead to DNA strand breaks (44), and if this occurred in vivo, the DNA repair and response machinery may be required to react to this damage. Alternatively, DNA damage may not be central to ATM function in the nervous system. The recent report of a specific association of ATM with the cytoplasmic vesicular protein β-adaptin (45) points to a role in protein trafficking.
Our data also demonstrate caspase-3 activation is required for Atm-dependent apoptosis in the CNS. Caspase activation may be a common end point for many situations involving neurological dysfunction (46). The involvement of p53, Bax, and caspase-3 in Atm signaling in the CNS suggests a potential for these proteins as apoptotic effectors in other neuropathological conditions. Further elucidation of the molecular details of signaling interactions in neuronal apoptosis will be required to provide a framework for understanding and treating these neuropathologies.
Acknowledgments
We thank Drs. Suzanne Baker and John Cleveland for advice and comments on the manuscript. These studies were supported by National Institutes of Health (NS-37956), the A-T Children's Project, and the American Lebanese and Syrian Associated Charities of St. Jude Children's Research Hospital.
Abbreviations
- A-T
ataxia telangiectasia
- CNS
central nervous system
- ATM
ataxia telangiectasia, mutated
- Pn
postnatal day n
- WT
wild type
- RPA
ribonuclease protection analysis
- EGL
external granule layer
- z
benzyloxycarbonyl
- FMK
fluoromethyl ketone
- CHO
aldehyde
- R
ionizing radiation
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Sedgwick R P, Boder E. In: Handbook of Clinical Neurology. Vinken P, Bruyn G, Klawans H, editors. Vol. 60. New York: Elsevier; 1991. pp. 347–423. [Google Scholar]
- 2.Lavin M F, Shiloh Y. Annu Rev Immunol. 1997;15:177–202. doi: 10.1146/annurev.immunol.15.1.177. [DOI] [PubMed] [Google Scholar]
- 3.Soares H D, Morgan J I, McKinnon P J. Neuroscience. 1998;86:1045–1054. doi: 10.1016/s0306-4522(98)00117-1. [DOI] [PubMed] [Google Scholar]
- 4.Herzog K H, Chong M J, Kapsetaki M, Morgan J I, McKinnon P J. Science. 1998;280:1089–1091. doi: 10.1126/science.280.5366.1089. [DOI] [PubMed] [Google Scholar]
- 5.Adams J M, Cory S. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 6.Chao D T, Korsmeyer S J. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 7.Oltvai Z N, Milliman C L, Korsmeyer S J. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 8.Deckwerth T L, Elliott J L, Knudson C M, Johnson E M, Jr, Snider W D, Korsmeyer S J. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 9.White F A, Keller-Peck C R, Knudson C M, Korsmeyer S J, Snider W D. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitada S, Krajewski S, Miyashita T, Krajewska M, Reed J C. Oncogene. 1996;12:187–192. [PubMed] [Google Scholar]
- 11.Xiang H, Kinoshita Y, Knudson C M, Korsmeyer S J, Schwartzkroin P A, Morrison R S. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson M D, Xiang H, London S, Kinoshita Y, Knudson M, Mayberg M, Korsmeyer S J, Morrison R S. J Neurosci Res. 1998;54:721–733. doi: 10.1002/(SICI)1097-4547(19981215)54:6<721::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 13.Cryns V, Yuan J. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 14.Thornberry N A, Lazebnik Y. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 15.Srinivasan A, Roth K A, Sayers R O, Shindler K S, Wong A M, Fritz L C, Tomaselli K J. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- 16.Stoppini L, Buchs P A, Muller D. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 17.Yin C, Knudson C M, Korsmeyer S J, Van Dyke T. Nature (London) 1997;385:637–640. doi: 10.1038/385637a0. [DOI] [PubMed] [Google Scholar]
- 18.McCurrach M E, Connor T M, Knudson C M, Korsmeyer S J, Lowe S W. Proc Natl Acad Sci USA. 1997;94:2345–2349. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knudson C M, Tung K S, Tourtellotte W G, Brown G A, Korsmeyer S J. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 20.Giaccia A J, Kastan M B. Genes Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 21.Canman C E, Lim D S, Cimprich K A, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan M B, Siliciano J D. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 22.Banin S, Moyal L, Shieh S, Taya Y, Anderson C W, Chessa L, Smorodinsky N I, Prives C, Reiss Y, Shiloh Y, et al. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 23.Khanna K K, Keating K E, Kozlov S, Scott S, Gatei M, Hobson K, Taya Y, Gabrielli B, Chan D, Lees-Miller S P, et al. Nat Genet. 1998;20:398–400. doi: 10.1038/3882. [DOI] [PubMed] [Google Scholar]
- 24.Waterman M J, Stavridi E S, Waterman J L, Halazonetis T D. Nat Genet. 1998;19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- 25.Hu S, Snipas S J, Vincenz C, Salvesen G, Dixit V M. J Biol Chem. 1998;273:29648–29653. doi: 10.1074/jbc.273.45.29648. [DOI] [PubMed] [Google Scholar]
- 26.Munday N A, Vaillancourt J P, Ali A, Casano F J, Miller D K, Molineaux S M, Yamin T T, Yu V L, Nicholson D W. J Biol Chem. 1995;270:15870–15876. doi: 10.1074/jbc.270.26.15870. [DOI] [PubMed] [Google Scholar]
- 27.Humke E W, Ni J, Dixit V M. J Biol Chem. 1998;273:15702–15707. doi: 10.1074/jbc.273.25.15702. [DOI] [PubMed] [Google Scholar]
- 28.Kuida K, Haydar T F, Kuan C Y, Gu Y, Taya C, Karasuyama H, Su M S, Rakic P, Flavell R A. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 29.Hakem R, Hakem A, Duncan G S, Henderson J T, Woo M, Soengas M S, Elia A, de la Pompa J L, Kagi D, Khoo W, et al. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 30.Slee E A, Harte M T, Kluck R M, Wolf B B, Casiano C A, Newmeyer D D, Wang H G, Reed J C, Nicholson D W, Alnemri E S, et al. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srinivasula S M, Ahmad M, Fernandes-Alnemri T, Alnemri E S. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 32.Barlow C, Brown K D, Deng C X, Tagle D A, Wynshaw-Boris A. Nat Genet. 1997;17:453–456. doi: 10.1038/ng1297-453. [DOI] [PubMed] [Google Scholar]
- 33.Westphal C H, Rowan S, Schmaltz C, Elson A, Fisher D E, Leder P. Nat Genet. 1997;16:397–401. doi: 10.1038/ng0897-397. [DOI] [PubMed] [Google Scholar]
- 34.Sakhi S, Bruce A, Sun N, Tocco G, Baudry M, Schreiber S S. Proc Natl Acad Sci USA. 1994;91:7525–7529. doi: 10.1073/pnas.91.16.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morrison R S, Wenzel H J, Kinoshita Y, Robbins C A, Donehower L A, Schwartzkroin P A. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosinger Ogilvie J, Deckwerth T L, Knudson C M, Korsmeyer S J. Invest Ophthalmol Visual Sci. 1998;39:1713–1720. [PubMed] [Google Scholar]
- 37.Miyashita T, Reed J C. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt T, Korner K, Karsunky H, Korsmeyer S, Muller R, Moroy T. Cell Death Differ. 1999;6:873–882. doi: 10.1038/sj.cdd.4400562. [DOI] [PubMed] [Google Scholar]
- 39.Goldowitz D, Hamre K. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 40.Tibbetts R S, Brumbaugh K M, Williams J M, Sarkaria J N, Cliby W A, Shieh S Y, Taya Y, Prives C, Abraham R T. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnes D E, Stamp G, Rosewell I, Denzel A, Lindahl T. Curr Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 42.Gao Y, Sun Y, Frank K M, Dikkes P, Fujiwara Y, Seidl K J, Sekiguchi J M, Rathbun G A, Swat W, Wang J, et al. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- 43.Frank K M, Sekiguchi J M, Seidl K J, Swat W, Rathbun G A, Cheng H L, Davidson L, Kangaloo L, Alt F W. Nature (London) 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 44.Didier M, Bursztajn S, Adamec E, Passani L, Nixon R A, Coyle J T, Wei J Y, Berman S A. J Neurosci. 1996;16:2238–2250. doi: 10.1523/JNEUROSCI.16-07-02238.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim D S, Kirsch D G, Canman C E, Ahn J H, Ziv Y, Newman L S, Darnell R B, Shiloh Y, Kastan M B. Proc Natl Acad Sci USA. 1998;95:10146–10151. doi: 10.1073/pnas.95.17.10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friedlander R M, Yuan J. Cell Death Differ. 1998;5:823–831. doi: 10.1038/sj.cdd.4400433. [DOI] [PubMed] [Google Scholar]