

Abstract
Synaptic N-methyl-d-aspartate (NMDA) receptors detect coincident pre- and postsynaptic activity and play a critical role in triggering changes in synaptic strength at central synapses. Despite intensive study of synaptic plasticity, relatively little is known about the magnitude and duration of calcium accumulation caused by unitary events at individual synapses. We used fluorescence imaging to detect NMDA receptor-mediated miniature synaptic calcium transients (MSCTs) caused by spontaneous release of synaptic vesicles in dendrites of cultured hippocampal neurons. MSCTs originated focally in dendritic regions <1 μm in length and decayed with a time constant of 0.35 ± 0.03 s. Multiple occurrences of MSCTs recorded at single sites had fluctuating amplitudes, with a coefficient of variation of 0.34. From the reduction in the spatial spread of MSCTs with decreasing concentration of indicator dye, we estimated that the dominant endogenous calcium buffer in dendrites is relatively immobile (diffusion coefficient between 10 and 50 μm2/s). We conclude that calcium rise caused by spontaneous quantal synaptic NMDA receptor activation (i) is variable, (ii) lasts for a time period briefer than previous measurements indicate, and (iii) is confined by endogenous calcium buffers to local dendritic regions even when synapses are not on spines.
A consistent coincidence of pre- and postsynaptic activity can lead to an increase in synaptic strength; such increases are thought to underlie several forms of learning and memory (1, 2). The mechanistic basis for this coincidence detection is the activation of N-methyl-d-aspartate (NMDA) receptors, whose opening and passage of ions require both presynaptic activity and postsynaptic depolarization. However, the exact time window within which coincidence of pre- and postsynaptic activity needs to occur to induce long-term potentiation (LTP), a commonly studied form of synaptic plasticity, has not been clearly defined. An earlier study using photoactivated calcium chelators estimated that sustained calcium rise for more than 2 s was necessary for inducing LTP by tetanic stimulation (3). Two recent studies, however, have suggested that the permissive window is less than 100 ms (4, 5), which is in line with the expected peak duration of synaptic NMDA currents. Previous workers investigating calcium signals in spines have reported conflicting results on the duration of the increased postsynaptic calcium concentration after unitary synaptic activation. Several studies in different neuronal types have found calcium transients in spines that last for several seconds (6–11). In contrast, recent reports have indicated briefer signals in CA1 pyramidal neurons and cerebellar Purkinje neurons (12–15).
It was demonstrated recently that postsynaptic calcium transients resulting from spontaneous transmitter release at single release sites can be detected by using fluorescence imaging (8, 9). We have exploited this to determine the spatial and temporal profile of calcium rise because of activation of synaptic NMDA receptors at individual synapses. We demonstrate that these miniature synaptic calcium transients originate at focal points in the dendritic tree and spread laterally for distances of less than 2 μm on either side of the origin before decaying to 25% of the magnitude at the origin. Using FM1-43 labeling (16–18), we show directly that functional release sites are present at locations at which miniature synaptic calcium transient (MSCTs) occur. In contrast to rather prolonged calcium transients reported in most previous studies, we find that the average duration of MSCTs is less than a second. Additionally, by monitoring the extent of spread of indicator-bound calcium in dendrites, we inferred that the dominant endogenous calcium buffer in dendrites is relatively immobile and limits the spread of calcium to adjacent synaptic sites. Our study puts limits on the duration, spatial extent, and reliability of calcium signals in postsynaptic elements resulting from single synaptic releases.
Methods
Calcium Imaging.
Hippocampal neurons were cultured by using the methods described previously (19). Cultures were allowed to mature for 10–20 days before recordings were made. For imaging, coverslips containing neurons were transferred to a chamber mounted on a movable stage of a Zeiss WL microscope. The extracellular medium contained 137 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 10 mM Hepes (pH adjusted to 7.3–7.4 by adding NaOH), 10 mM glucose, 0.001 mM tetrodotoxin, 0.01 mM 6,7-dinitroquinoxaline-2,3-dione, and 0.01 mM picrotoxin. Patch pipettes containing 120 mM KGlu, 10 mM KCl, 5 mM Mg-ATP, 0.3 mM GTP, 10 mM NaCl, and 50, 200 or 400 μM fluo-3 were used to introduce dye into the cell and to obtain whole-cell recordings. Dye concentration (as estimated by fluorescence intensity) in all but the smallest processes appeared to equilibrate within 15 min after achieving whole-cell configuration. In some early experiments, coverslips containing neurons grown for 7–21 days were labeled by using the AM-ester form of fluo-3 (10 μM, for 20 min). Imaging was commenced after at least 30 min of wash, allowing for deesterification of the fluo-3 AM. No electrophysiological recordings were made in these experiments.
Images were acquired by using the Bio-Rad MRC-600 confocal laser-scanning microscope (488-nm line for excitation, 510-nm long-pass filter for fluorescence emission; ×40, 0.75-numerical aperture water immersion objective). After an area of interest was found, images were acquired in the line-scan mode, which allowed a temporal resolution of 4 ms. Images were analyzed with custom-written software by using matlab (Mathworks, Natick, MA). Regions of interest were defined and pixels were averaged for each line scan, yielding 1 point every 4 ms.
FM1-43 Labeling.
In some experiments, to visualize functional synapses, we used the styryl dye FM1-43. After imaging with fluo-3, a depolarizing solution (40 mM KCl) containing 10 μM FM1-43 was perfused locally over the neuronal processes within the field of view, using a puffer pipette for 60 s. Hyperkalemic solution causes release of synaptic vesicles and subsequent endocytosis. After washing the preparation for more than 5 min, synapses could be visualized as fluorescent spots. Another hyperkalemic challenge for more than 5 min was used to cause destaining of spots. The difference in fluorescence between initial and final images was used to calculate the releasable fluorescence for each bouton. Taking difference images also cancels the small fluorescence contamination from fluo-3 (which was 10 times lower than FM1-43 fluorescence).
Results
MSCTs in Hippocampal Dendrites.
Fig. 1a shows a dendritic field with a number of processes labeled with the indicator fluo-3. In this particular experiment, labeling was done by using the AM-ester form of the dye, and, therefore, all the cells including astrocytes were labeled, as can be seen in the background of Fig. 1. The fluorescence in the dendrites was sometimes nonhomogeneous, which is typical with the AM-ester loading protocol. When this field was observed in the frame-scanning mode of the confocal scanner, spontaneous transient increases in fluorescence were observed in some dendritic processes. A dendritic region that exhibited a transient fluorescence increase was chosen for further investigation in line-scanning mode. A line scan along the dendrite is shown in Fig. 1b, and the fluorescence intensity recorded at two sites, labeled 1 and 2, is shown in Fig. 1c. Transient rises in fluorescence were observed at different times in dendritic regions 1 and 2. The independence of the two transients at distinct locations indicates a local origin for each of them.
Figure 1.

Localized, spontaneous dendritic calcium transients. (a) Fluorescence image of a neuron. Fluo-3 was loaded into the cells by using the AM form of the dye, which accounts for the labeling of processes in the background (including astrocytes). The region enclosed by the rectangle is shown in b. (b) A scan line through the lower dendrite was chosen for fast imaging in the line-scan mode (4 ms per line). (c) Fluorescence changes in two regions indicated by 1 and 2 in b, plotted against time. Spontaneous increases in fluorescence intensity were observed independently at sites 1 and 2. Fluorescence intensity is in arbitrary digital units resulting from conversion of the photomultiplier output to numbers from 0 to 255.
MSCTs were infrequent, in accordance with the estimated rate of spontaneous excitatory postsynaptic currents (EPSCs) of one per min per synapse in these cells (20, 21). To increase the rate of spontaneous EPSCs, in some experiments the bathing solution was made hyperosmotic by adding 100 mM sucrose. This led to an increased frequency of occurrence of MSCTs, providing further evidence for a synaptic origin of MSCTs.
NMDA Receptor Activation Is Required for MSCTs.
Our principal evidence that MSCTs are caused by activation of NMDA receptors is that they were abolished by the addition of the antagonist d(−)-2-amino-5-phosphoropentanoic acid (D-APV) to the medium. Fig. 2a shows a typical example in which a field of dendrites was imaged in the frame-scan mode for 32 s (0.4 s between frames). Several sites exhibited MSCTs, sometimes more than once. When the same field was imaged for the same duration after perfusion with medium containing 50 μM D-APV, transients were not observed. Finally, upon reperfusion with normal saline, MSCTs were detected at the same sites. This block of MSCTs by D-APV was observed in four of four cells.
Figure 2.

MSCTs require NMDA receptor activation (a Inset) Fluo-3 image of a dendritic process. The average fluorescence intensity in the boxed areas 1 and 2 is shown during control period, during perfusion with medium containing 50 μM D-APV, and after washing out D-APV. Transient increases in fluorescence intensity observed in the control period was abolished during perfusion with D-APV and reappeared upon washing with control solution. Images were obtained in frame-scan mode (0.4 s between frames). (b) Fluo-3 image of a neuron with boxes drawn over areas where average fluorescence intensity was measured. Dye was loaded into the cell by using a patch pipette in the whole-cell recording mode. The perfusion medium contained 1.3 mM Mg2+ and 5 mM Ca2+. During the first 20 s of recording, the voltage at the soma was clamped at −70 mV. At the time indicated by the vertical arrow, the clamp voltage was switched to −30 mV. Note the appearance of MSCTs in the regions indicated. The increase in baseline fluorescence is due to depolarization-induced calcium influx.
A second line of evidence that suggests the requirement of NMDA receptor activation for MSCT is as follows. When a field of dendrites was imaged in the presence of 1.3 mM Mg2+ and 5 mM Ca2+, with the soma clamped at −70 mV, no transients were observed in dendrites close to the soma (observation was restricted to proximal dendrites, less than 30 μm from the soma, to minimize space clamp problems). At this voltage, the magnesium block of NMDA receptors should prevent calcium entry (19, 22). The same field was visualized after the clamp voltage was shifted to −30 mV. We used this voltage because according to equation 5 of Jahr and Stevens (23) and equation 1 of Jahr and Stevens (24), maximum calcium influx through NMDA receptor channels should occur at about −30 mV. At this holding potential, MSCTs were observed at multiple sites (Fig. 2b), further suggesting the involvement of NMDA receptors. This effect was observed in three of three cells.
In the previous experiments, we could not exclude the possibility that voltage-gated calcium channels contribute to calcium rises. To test whether voltage-gated calcium channels were required for the occurrence of MSCTs, we imaged dendrites in the presence of extracellular CdCl2 (50 μM). CdCl2 does not significantly affect the frequency or amplitude of miniature EPSCs (25). MSCTs continued to occur after addition of CdCl2, which indicated that the activation of voltage-gated calcium channels was not required for MSCTs. Under normal operating conditions, however, it is possible that voltage-gated calcium channels provide additional calcium influx if sufficient local depolarization occurs.
MSCTs Occur at Functional Release Sites.
In nine experiments, after visualizing calcium transients, we labeled the same dendritic field with FM1-43 to identify the location of functional synapses. Hyperkalemic solutions were used for staining and destaining synaptic terminals. Previous studies by us (18, 26) and others (17, 27) have demonstrated that the punctate fluorescent spots observed under these conditions correspond to synaptic release sites. In Fig. 3, a differential interference contrast image of a dendritic field is shown along with fluo-3 fluorescence image as well as FM1-43 fluorescence. The time course of fluorescence intensity in three boxed areas indicated that transients were observed in some areas of the dendrites (Fig. 3 Upper Right). An FM1-43 image of the same field revealed punctate fluorescence along the dendrite.
Figure 3.

FM1-43 staining at sites of origin of MSCTs. Differential interference contrast image of a neuron (Upper Left). The boxed region, magnified at bottom left (DIC), was scanned repeatedly in the frame-scan mode (265 ms per frame). Fluo-3 image of this region is shown (Lower Center), with the contrast inverted for clarity (Fluo-3). The time course of fluorescence in the three boxed regions is shown at Upper Right. After a number of images were obtained, the same dendritic region was labeled with FM1-43 to determine the locations of functional release sites (FM1-43). See Methods for details.
FM1-43 puncta were observed at dendritic sites that exhibited MSCTs in nine of nine experiments. In five cases, a single FM1-43 spot could be assigned to an MSCT. In the other four cases, two or more spots were within 3 μm of each other. Because the frame-scan mode was used for these experiments, the time resolution of MSCT imaging was not sufficient to allow a unique assignment of the MSCT to FM1-43 puncta. In all cases, the number of FM1-43 puncta within the imaged field exceeded the number of MSCT sites. There are at least three plausible reasons why this might be the case. (i) Most of the synapses have low spontaneous rates of release (21) and, therefore, might not have released any vesicle spontaneously during the period of MSCT imaging; however, these same synapses would be labeled with FM1-43 because they would have higher rates of release during the strong depolarizing stimulus used for FM1-43 labeling. (ii) The postsynaptic sites apposed to some of the FM1-43 labeled boutons might be lacking functional NMDA receptors. (iii) Finally, some sites might have produced MSCTs that were below the threshold of detection during fluo-3 imaging.
Time Course and Spatial Spread of MSCTs Along the Dendrite.
From 20 experiments, we obtained 38 MSCTs by using the line-scan imaging mode. Because this method provided high spatial and temporal resolution, we could measure the total duration of each MSCT at its origin (defined by a 1-μm length of dendrite). Fig. 4a shows an example of multiple MSCTs recorded at a single site. The detection criteria for MSCTs were an increase in fluorescence greater than 2 SDs above baseline noise, a rapid rise time, and sustained increase for more than 50 ms. MSCTs were assigned to the spatial site where the rise time was the fastest. Although many MSCTs exhibited exponential decay, others had a more complex time course, which may arise from delayed NMDA receptor channel openings (9). We estimated the time constant of decay for MSCTs that had a clear exponential decay phase. In addition, we estimated the total duration for all MSCTs as follows. Once an MSCT was detected, the onset was defined as the point when the calcium fluorescence deviated from baseline by more than 1 SD, and a similar criterion was applied for return to baseline. For 20 MSCTs with clear exponential decay, the average time constant was 0.35 ± 0.03 s, and the average duration of all 38 MSCTs was 0.84 ± 0.07 s. The distribution of MSCT duration is shown in Fig. 4b. For the 20 MSCTs that had a clear exponential decay, the time constant and total duration were tightly correlated (Fig. 4b Inset), validating the use of total duration as a meaningful measure. The less than a second time course is unlikely to be determined by dye kinetics because much faster calcium transients could be observed in these cells—for instance, action potential-evoked calcium rise, in the presence of synaptic blockers, lasted for less than 0.3 s (time constant, ≈0.1 s). In addition, there was only a small difference between duration measured at the MSCT origin with 50 and 200 μM fluo-3 (0.77 ± 0.11 and 0.92 ± 0.14 s, respectively). Note that, in contrast, the space-averaged calcium transients are longer and of lower amplitude with increasing indicator concentration.
Figure 4.

Time course of MSCTs. (a) MSCTs from a single site. The duration of five MSCTs originating at this site is indicated by the horizontal bars. Duration was measured as described in the text. Some fluctuations that are clearly above baseline noise (dashed lines) are not marked as MSCTs because their loci of origin were not at the measured site (this can also be seen by their slower rise times). (b) Distribution of MSCT duration. The total duration averaged 0.84 s. For 20 MSCTs with clear exponential decay, the total duration as measured by our method was tightly correlated with the time constant (Inset, correlation coefficient is 0.744, significant at P < 0.001).
We characterized the spatial and temporal dynamics of a MSCT in detail in a few favorable cases in which long regions of dendrites were included in a line scan, and MSCT originated close to the center of the imaged dendrite. Fig. 5 shows one such example. The line-scan image indicated that the transient rise in calcium originated focally in the middle of the imaged line and spread to more than 5 μm on either side of the origin. The location with the fastest rise time was defined to be the origin; the percentage change in fluorescence was also the largest at that location (Fig. 5b). At locations away from the origin, the transients were of lower amplitude and had slower rise times. These observations are consistent with the idea that the increase in calcium occurred focally and spread by diffusion to adjacent dendritic areas. Voltage spread because of passive or active electrotonic decay would occur within a fraction of milliseconds, so the observed spatial dispersion cannot be accounted for simply by electrotonic decay.
Figure 5.

Spread of calcium during MSCTs. (a) Fluo-3 image showing a field of dendrites (Upper). The line indicated in black along one of the dendrites was scanned in the line-scan mode. The occurrence of an MSCT during one such line scan is shown (Lower). Note the early increase fluorescence intensity in the middle of the scanned area, which then spreads to the neighboring area. (b) Spatial profiles of fluorescence intensity at various times after the onset of MSCT. The noisy traces are data, and the smooth lines are fits obtained by using Eq. 4, with the parameters specified in the text. Fluorescence is plotted as percent change in fluorescence from rest. (c) The data in b is replotted to show the time course of fluorescence at four points along the dendrite. The numbers within each graph refer to the spatial location with respect to the origin of MSCT. (d) Apparent diffusion coefficient as a function of buffer capacity of fluo-3 (obtained from its total concentration) plotted on logarithmic axes for 13 experiments. This dependence of Dapp on the buffer capacity of fluo-3 was used to estimate the endogenous buffer capacity and mobility. The solid line is Eq. 4 with De = 21 μm2/s and κe = 75.
We were able to describe the dynamics of calcium transients by using a simple model of diffusion in a uniform cylinder (see Appendix). This model assumes a single, dominant endogenous calcium buffer, in addition to the added buffer (the indicator dye). The apparent diffusion coefficient of dye-bound calcium estimated from this method was 97 ± 21 μm2/s (mean ± SD, five sites). To test whether the spread of calcium was significantly distorted by the mobility of the indicator dye, we repeated the diffusion measurements at a lower dye concentration. The apparent mobility of calcium measured by using fluorescence imaging will be dominated by the indicator dye if its buffer capacity is similar to or greater than the endogenous buffer capacity (28, 29). By decreasing the concentration of indicator, we decrease its buffer capacity and its influence on the mobility of calcium. From five experiments using 50 μM fluo-3, we estimated the average Dapp of 61 ± 19 μm2/s (mean ± SD), which was significantly smaller than the value at 200 μM fluo-3 (Fig. 5D). Three experiments using 400 μM fluo-3 yielded an average Dapp of 108 ± 14 μm2/s, which was not different from the value at 200 μM fluo-3. By extrapolating linearly, we determined that in the absence of exogenous buffers, the apparent diffusion coefficient of calcium (bound to endogenous buffers) will be less than 50 μm2/s. This is probably a conservative estimate, because more realistic considerations using a buffered diffusion model (see Appendix) provide an estimate for Dapp of 21 μm2/s (95% confidence interval of 10–36 μm2/s). This indicates that the dominant buffer in the dendrites of these neurons is not very mobile. With our estimates of endogenous buffer mobility, calcium entering through NMDA receptors at a single synaptic site will spread only to 2 μm on either side of the entry site before the concentration drops to less than 25% of the peak value.
Variability of MSCT Amplitudes.
Unitary EPSCs mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors at single hippocampal synapses show a large variability (30–34), and K. McAllister and C. Stevens (personal communication) find a similar large variability for NMDA receptor-mediated currents at single synapses. The variability of MSCTs will reflect, at least in part, the variability of NMDA responses. Although imaging in the frame-scan mode was desirable to obtain a large number of MSCTs, it did not afford enough temporal resolution to localize the origin of multiple of MSCTs to within 1–2 μm. In the absence of such localization, we could not differentiate between MSCTs originating less than 5 μm apart. Therefore, to ensure that we were looking at MSCTs from the same site, we performed fast line-scan imaging along the axis of the dendrite (see Fig. 5). This, however, precluded extended imaging because of photodynamic damage caused by repeated line scan at the same location. In 12 experiments, we were able to record 2 or more MSCTs at the same location. Because recordings were performed across different conditions in different neurons, we could not simply compare the amplitudes of fluorescent transients (which are affected by parameters such as local dye concentration) across cells. We calculated the coefficient of variation (CV) of MSCTs at each site and obtained the average CV across all sites. Assuming that the variability is homogeneous across sites, this average will provide a good estimate of the underlying true value.
The average CV from the 12 sites was 0.36. The observed variability is the sum of the measurement noise and the underlying biological variability (the variances are additive). Taking into account the measurement variability, which had a coefficient of variation of 0.13, the actual CV is 0.34, which we take as the best estimate of the CV of MSCTs. For comparison, the coefficient of variation of AMPA currents at single synaptic boutons of cultured hippocampal neurons has been estimated to be between 0.33 (ref. 34) and 0.5 (ref. 30). Remarkably, our measure of CV coincides with the estimate from another study measuring NMDA-mediated calcium transients in dendritic spines of CA1 pyramidal cells in slices (14).
Discussion
Spontaneous miniature EPSCs represent the postsynaptic response to single synaptic vesicles and constitute the fundamental unit of synaptic transmission. The evoked response at single active zones of central synapses, in many cases, is caused by the release of a quantum (31, 35). Therefore, a study of spontaneous synaptic activity can provide insights into the consequences of evoked release. We have imaged MSCTs in cultured hippocampal neurons and demonstrate that they are similar to those recently described in cultured neocortical neurons (8, 9). Using this technique, we addressed several issues regarding synaptic function. First, we find that the time course of the transients is more rapid than reported previously (8, 9) probably because of the low concentration of indicator dye that we used. Second, we provide quantitative estimates for the longitudinal spread of the calcium transient along the dendrite and for the mobility of endogenous buffers. Third, we show that the postsynaptic calcium signal at single synapses is variable. Finally, we provide direct evidence for the synaptic localization of these transients by using the styryl dye FM1-43 and demonstrate the feasibility of optically separating pre- and postsynaptic events at the same synapse.
Because we were able to image MSCTs at a temporal resolution of 4 ms, we could provide quantitative estimates for the range and speed of diffusion of calcium originating at a single synapse. We estimate that under native conditions, MSCTs will be confined locally, spreading only to 2 μm from the origin before calcium concentration is reduced to less than 25% of the maximal value at the origin. Because synapses are typically around 3 μm apart in our preparation (V.N.M. and C.F.S., unpublished observations), it appears that calcium rises because of activation of a single synapse can be restricted to individual sites even in the absence of spines. The spread will depend on the mobility and buffering capacity of endogenous calcium buffers. We have estimated that the endogenous buffers are fairly stationary, with a diffusion constant of 21 μm2/s. One previous report made a qualitative observation that dendritic calcium buffers are likely to be of low mobility based on the lack of washout of endogenous calcium buffering by whole-cell patch pipettes (36). In our experiments also, significant washout of fast, mobile buffers probably does not occur because the spatial spread of MSCTs is similar whether calcium indicator dye is loaded with patch pipette or when using the AM-ester form of the dye. A recent report has estimated the buffer mobility in large diameter axons of Aplysia californica to be low (De < 16 μm2/s; ref. 29).
In our study, because of the low dye concentrations used (necessary for good temporal resolution), in most cases we were unable to resolve spines. If, as seems likely, many of the MSCTs we imaged were from synapses made directly on dendritic shafts, our findings may be directly relevant to developing synapses, which occur mainly on dendritic shafts (37, 38), and to synapses on aspiny neurons. The dynamics of calcium at single synapses, in particular within spines, has been a subject of speculation (recently reviewed in ref. 39). Because calcium entry into postsynaptic elements after synaptic activation is a trigger for long-term changes in synaptic strength, the extent of spread of calcium could determine the extent to which neighboring synapses are independent. Several recent studies have indicated that calcium rises indeed can be confined to dendritic spines when synaptic activation is subthreshold for action potentials (12–15). The previous study of MSCTs in cultured cortical neurons concluded that the calcium rises were not confined to spines (8, 9). It seems likely that at least part of the apparently high mobility of calcium observed in that study might be attributed to added mobile buffer in the form of indicator dye that was 300 μM or greater. At this concentration, our estimates indicate that the apparent spread of calcium will be more than 5 μm on either side of the origin. The most recent studies of evoked calcium transients in spines of CA1 pyramidal neurons using multiphoton microscopy indicated that these are quite brief and are confined to individual spines (14, 15).
An important new finding of our study is the colocalization of the origin of MSCTs and FM1-43 puncta, which correspond to functional release sites (16–18). In all experiments, there were functional release sites close to the origin of MSCTs, supporting the idea that MSCTs are indeed synaptic in origin. Although in many cases we were able to identify a single release site at the origin of MSCT, in some cases several boutons were found in close proximity to the origin of MSCT. Preliminary data (not shown) indicated that the frequency of MSCTs at a given dendritic site is correlated with the intensity of FM1-43 fluorescence at that site. Because the FM1-43 intensity is a measure of the size of the recycling pool of presynaptic vesicles, our finding suggests that the frequency of spontaneous release may be determined in part by the size of the recycling pool of vesicles.
In summary, we have used an optical method for detecting the activation of a single synapse to infer the properties of unitary synaptic events. We find that the calcium rise induced by activation of a single synapse is localized, probably because of low mobility of calcium buffers. Calcium accumulation shows trial-to-trial variability. Finally, we show that functional pre- and postsynaptic properties can be studied at the same synapse.
Acknowledgments
Support for this work was from National Institutes of Health Grants NS12961 (C.F.S.) and MH46482 (T.J.S.). V.N.M. was a Helen Hay Whitney Foundation Fellow during the period of this work. C.F.S. and T.J.S. are Investigators of the Howard Hughes Medical Institute.
Abbreviations
- MSCT
miniature synaptic calcium transient
- NMDA
N-methyl-d-aspartate
- EPSC
excitatory postsynaptic current
- CV
coefficient of variation
- D-APV
d(−)-2-amino-5-phosphoropentanoic acid
Appendix
The following model was constructed to account for the observed spatiotemporal dynamics of calcium. Calcium entry was assumed to occur at a point source in a cylinder and follow the time course of the NMDA currents in these neurons (40). Assuming rapid equilibration in the radial direction (because of the small diameter of the dendrites), we need to consider only diffusion along the axis of the dendrite. Buffering by immobile and mobile buffers (including the indicator dye) was taken into account by introducing an effective diffusion coefficient for calcium (41). We also included a single kinetic parameter for irreversible removal of calcium and assumed that this process has simple first-order kinetics (42). The equation describing the concentration of calcium is
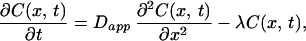 |
1 |
where C(x,t) is the concentration of calcium at position x and time t, Dapp is the effective diffusion constant for calcium, and λl is the rate of loss of calcium (pumps and sequestration mechanisms). The solution of this equation for a brief input of calcium such that at t = 0, C= C0 at x = 0, and everywhere else C = 0 is given by:
 |
2 |
If the time course of NMDA currents is
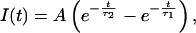 |
3 |
where A is a constant and τ1 = 10 ms and τ2 = 250 ms are time constants obtained from electrophysiological recordings in these neurons (40), then the time course of calcium concentration is given by convolving Eqs. 2 and 3.
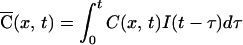 |
4 |
The solution (Eq. 4) has five free parameters: τ1 and τ2, the two time constants used to describe NMDA currents, Dapp, diffusion constant of calcium, λ, the rate of loss of calcium to buffers, and K, a normalizing factor (the products of all other constants including C0 and A). Of these, the time constants for NMDA current were fixed by using previous data (40), leaving three parameters. The data in Fig. 5 could be well fitted by the diffusion model, with the following parameters: Dapp = 90 μm2/s; λ = 8 s−1; K = 3.2. The average value of Dapp from five separate dendritic sites measured with 200 μM fluo-3 was 97 ± 21 μm2/s (mean ± SD). Similar measurements with 50 μM fluo-3 yielded a value of 61 ± 19 μm2/s (mean ± SD) for Dapp.
The apparent diffusion constant is a function of endogenous calcium buffers as well as added buffer in the form of fluo-3. We used the relation derived by Wagner and Keizer (41) to estimate the mobility and capacity of endogenous buffers:
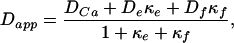 |
5 |
where DCa, De, and Df are the diffusion coefficients of free calcium, endogenous buffer, and fluo-3 respectively, and κe and κf are the calcium-binding ratios (28) of endogenous buffer and fluo-3.
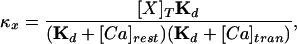 |
6 |
where [X]T is the total concentration of buffer X, Kd is the dissociation constant, [Ca]rest is the resting concentration of calcium, and [Ca]tran is the average concentration of calcium during an MSCT. The relation (5) holds if the following conditions are met: (i) there is a local equilibrium between the buffers and free calcium and (ii) the diffusion coefficients of the bound and unbound buffer are identical. The first condition essentially requires that the maximal calcium elevation is small compared with the dissociation constant of the mobile buffers. In Eq. 5, the values for some variables are known from previous experiments or from present data: 223 μm2/s for DCa (ref. 43); 130 μm2/s for Df (ref. 29). In our experiments, we estimated Dapp at three different concentrations of fluo-3, and the corresponding values of κf are 42, 167, and 333 (calculated from 50, 200, and 400 μM fluo-3 used in our experiments by using a dissociation constant of 0.4 μM, resting calcium concentration of 0.2 μM, and average calcium concentration during an MSCT of 0.4 μM). The least-squares fit of Eq. 5 to data in Fig. 5d using values indicated above allowed us to estimate De and κe, which are 21 μm2/s and 75, respectively. The 95% confidence interval for De was 10–36 μm2/s.
References
- 1.Stevens C F. Cell Neuron. 1993;72, Suppl. 10:55–63. doi: 10.1016/s0092-8674(05)80028-5. [DOI] [PubMed] [Google Scholar]
- 2.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Malenka R C, Lancaster B, Zucker R S. Neuron. 1992;9:121–128. doi: 10.1016/0896-6273(92)90227-5. [DOI] [PubMed] [Google Scholar]
- 4.Markram H, Lubke J, Frotscher M, Sakmann B. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 5.Bi G-q, Poo M-m. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller W, Connor J A. Nature (London) 1991;354:73–76. doi: 10.1038/354073a0. [DOI] [PubMed] [Google Scholar]
- 7.Guthrie P B, Segal M, Kater S B. Nature (London) 1991;354:76–80. doi: 10.1038/354076a0. [DOI] [PubMed] [Google Scholar]
- 8.Murphy T H, Baraban J M, Wier W G, Blatter L A. Science. 1994;263:529–532. doi: 10.1126/science.7904774. [DOI] [PubMed] [Google Scholar]
- 9.Murphy T H, Baraban J M, Wier W G. Neuron. 1995;15:159–168. doi: 10.1016/0896-6273(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 10.Segal M. J Physiol. 1995;486:285–296. doi: 10.1113/jphysiol.1995.sp020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eilers J, Augustine G J, Konnerth A. Nature (London) 1995;373:155–158. doi: 10.1038/373155a0. [DOI] [PubMed] [Google Scholar]
- 12.Yuste R, Denk W. Nature (London) 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 13.Denk W, Sugimori M, Llinas R. Proc Natl Acad Sci USA. 1995;92:8279–8282. doi: 10.1073/pnas.92.18.8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mainen Z F, Malinow R, Svoboda K. Nature (London) 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- 15.Koester H J, Sakmann B. Proc Natl Acad Sci USA. 1999;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Betz W J, Bewick G S. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- 17.Ryan T A, Reuter H, Wendland B, Schweizer F E, Tsien R W, Smith S J. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 18.Murthy V N, Sejnowski T J, Stevens C F. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 19.Nowak L, Bregestovski P, Ascher P, Herbert A, Prochiantz A. Nature (London) 1994;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 20.Geppert M, Goda Y, Hammer R E, Li C, Rosahl T W, Stevens C F, Südhof T C. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 21.Murthy V N, Stevens C F. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 22.Mayer M L, Westbrook G L, Guthrie P B. Nature (London) 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 23.Jahr C E, Stevens C F. J Neurosci. 1990;10:3178–3182. doi: 10.1523/JNEUROSCI.10-09-03178.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jahr C E, Stevens C F. Proc Natl Acad Sci USA. 1993;90:11573–11577. doi: 10.1073/pnas.90.24.11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scanziani M, Copogna M, Gahwiler B H, Thompson S M. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- 26.Murthy V N, Stevens C F. Nature (London) 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- 27.Ryan T A, Smith S J. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Z, Neher E. J Physiol. 1993;469:245–273. doi: 10.1113/jphysiol.1993.sp019813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabso M, Neher E, Spira M E. Neuron. 1997;18:473–481. doi: 10.1016/s0896-6273(00)81247-7. [DOI] [PubMed] [Google Scholar]
- 30.Bekkers J M, Richerson G B, Stevens C F. Proc Natl Acad Sci USA. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raastad M, Storm J F, Andersen P. Eur J Neurosci. 1992;4:113–117. doi: 10.1111/j.1460-9568.1992.tb00114.x. [DOI] [PubMed] [Google Scholar]
- 32.Bolshakov V Y, Siegelbaum S A. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 33.Liu G, Tsien R W. Nature (London) 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- 34.Forti L, Bossi M, Bergamaschi A, Villa A, Malgaroli A. Nature (London) 1997;388:874–878. doi: 10.1038/42251. [DOI] [PubMed] [Google Scholar]
- 35.Stevens C F, Wang Y. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- 36.Helmchen F, Imoto K, Sakmann B. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris K M, Stevens J K. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyer C, Schikorski T, Stevens C F. J Neurosci. 1998;18:5294–5300. doi: 10.1523/JNEUROSCI.18-14-05294.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denk W, Yuste R, Svoboda K, Tank D W. Curr Opin Neurobiol. 1996;6:372–378. doi: 10.1016/s0959-4388(96)80122-x. [DOI] [PubMed] [Google Scholar]
- 40.Bekkers J M, Stevens C F. Nature (London) 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 41.Wagner J, Keizer J. Biophys J. 1994;67:447–456. doi: 10.1016/S0006-3495(94)80500-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Markram H, Helm P J, Sakmann B. J Physiol. 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allbritton N L, Meyer T, Stryer T. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]