

Abstract
Chemosensory proteins (CSPs) have been proposed to transport hydrophobic chemicals from air to olfactory or taste receptors. They have been isolated from several sensory organs of a wide range of insect species. The x-ray structure of CSPMbraA6, a 112-aa antennal protein from the moth Mamestra brassicae (Mbra), was shown to exhibit a novel type of α-helical fold. We have performed a structural and binding study of CSPMbraA6 to get some insights into its possible molecular function. Tryptophan fluorescence quenching demonstrates the ability of CSPMbraA6 to bind several types of semio-chemicals or surrogate ligands with μM Kd. Its crystal structure in complex with one of these compounds, 12-bromo-dodecanol, reveals extensive conformational changes on binding, resulting in the formation of a large cavity filled by three ligand molecules. Furthermore, binding cooperativity was demonstrated for some ligands, suggesting a stepwise binding. The peculiar rearrangement of CSPMbraA6 conformation and the cooperativity phenomenon might trigger the recognition of chemicals by receptors and induce subsequent signal transduction.
Chemosensory proteins (CSPs) have been identified in antennae from Drosophila melanogaster and in antennae, proboscis, tarsi, and labrum from several insects (1–7). Several CSPs from antenna or proboscis have also been isolated in the moth Mamestra brassicae (3). Due to their localization, these proteins have been proposed to be involved in chemical signal transduction or in chemo-perception, either olfaction or taste (2–6). They might transport chemicals from air to receptors through the aqueous medium surrounding them, but their exact physiological roles have still to be assigned. CSPs are shorter (110–115 aa) than the other classes of odorant transport proteins, pheromone-binding proteins (PBPs) or general odorant binding proteins (8) and share no sequence homology with them. They contain only four conserved cysteines forming two disulfide bridges (6) instead of three in the other classes. The structure of the PBP from Bombyx mori has been solved, revealing a 6 α-helical protein that undergoes large conformational changes on pH shift (9, 10). Very recently, a PBP function has been deciphered by Krieger and Ross (11), who identified in fire ants two PBP alleles governing alternative social behaviors. Their findings indicate that, at a molecular level, different receptors might be activated by a specific PBP allele/social-pheromone complex.
Less is known, however, about CSPs as compared with the PBPs. We have, therefore, initiated a structural and biophysical study of CSPs to explore their role in ligand transport and signal transduction. We have reported that the unbound CSPMbraA6 is remarkably different from PBP, either unbound or bound, and displays an original fold (12). Helices A and B as well as helices D and E form two V-shaped structures 12 Å apart, whereas helix C is perpendicular and in between them (Fig. 1). The C-terminal helix F is packed against the external face of the D-E helices, and does not take part in the core assembly. A narrow channel starting from the surface region extends 14 Å within the core of the protein. This channel seemed suitable for binding alkyl compounds, a hypothesis that has been tested by using tryptophan fluorescence quenching with bromo-alkyl alcohols or acids (12). Here, we report the x-ray structure of CSPMbraA6 in complex with a surrogate ligand, 12-bromo-dodecanol (BrC12OH). As suspected from a previous NMR study (13), drastic changes have occurred on complexation, and three ligands are found in the internal cavity. We also report binding assays of native or mutated CSPMbraA6 with several linear or bulky chemicals, some displaying a cooperative behavior, and we propose that a stepwise mode of binding of chemicals to CSPMbraA6 may occur.
Figure 1.

X-ray structure of the complexed form of CSPMbraA6. (A) Ribbon view of the Cα tracing and disulfide bridges (green) arrangement. Helices (red) are numbered a to f. The turns and coiled elements are in yellow. The ligands are not displayed here. (B) Superimposition of the apo (blue) and complexed (red) CSPMbraA6 structures. Views were prepared with MOLSCRIPT (32).
Methods
Protein Purification and Crystallization.
CSPMbraA6 expression in the Escherichia coli periplasm has already been published (14). In brief, Bl21(DE3) bacteria transformed with pET22b(+)/CSPMbraA6 plasmid were grown at 37°C without induction. After centrifugation of bacteria, the periplasmic CSPMbraA6 was released by osmotic shock and purified by anionic exchange on a resourceQ column followed by gel filtration on a preparative Superdex 200.
Besides the two crystal forms obtained with the apo-protein [forms 1 and 2 (12)], two crystal forms of CSPMbraA6 in complex with BrC12OH were obtained in 34% MPEG 2000, 0.1 M sodium-cacodylate (pH 6.5). Form 3 is monoclinic (P21, a = 33.6 Å, b = 49.7 Å, c = 50.3 Å, β = 93.7°) and contains two molecules per asymmetric unit. Form 4 is monoclinic centered (C2, a = 61.5 Å, b = 54.6 Å, c = 33.2 Å, β = 116.6°) and contains one molecule per asymmetric unit.
Site-Directed Mutagenesis of CSPMbraA6.
The QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used to introduce the different mutations. Briefly, the pET22b(+)/CSPMbraA6 was entirely amplified by the PfuTurbo DNA polymerase by using two complementary mutated primers in which a Trp codon (TGG) was replaced by a Cys codon (TGC) at position 94 (W94C) or at position 81 (W81C) and a Tyr codon (TAC) by a Val codon (GTC) or a Phe codon (TTC) at position 26 (Y26V or Y26F, respectively). Parental plasmid was digested by DpnI, and the vector containing the mutation was transformed in XL1-Blue cells. Transformants were selected onto LB carbenicillin (50 μg/ml) agar plates. Mutations were verified by automated DNA sequencing (ESGS, Evry, France), and the resulting constructs were referred to as pET22b(+)/W94C, pET22b(+)/W81C, pET22b(+)/Y26V, and pET22b(+)/Y26F. The mutant proteins were purified in the same way as the native protein. The different mutant proteins were characterized by mass spectrometry. Mass analyses were obtained by the matrix-assisted laser desorption ionization-time of flight method with Voyager-DE RP (PerSeptive Biosystems, Framingham, MA). Samples (0.7 μl containing 15 pmols) were mixed with an equal volume of sinapinic acid matrix solution and spotted on the target, then dried at room temperature for 10 min. The mass standard was apo-myoglobin (molecular mass of 16,951.6 Da). The mutant proteins proper folding was verified by circular dichroism. Spectra were recorded on a Jasco (Easton, MD) J-810 spectrophotometer in 10 mM Na/Na2 PO4 (pH 7.5) at 20°C by using a cell path of 0.1 cm and a concentration of protein of 0.1 mg/ml.
X-Ray Structure Determination and Refinement.
For data collection, crystals of CSPMbraA6 in complex with BrC12OH were flash frozen at 100 K in their mother liquor without cryoprotectant. The x-ray fluorescence from each crystal was measured as a function of the incident x-ray energy in the vicinity of the Br-K edge. A single wavelength anomalous diffraction dataset was collected on ID-29 at the European Synchrotron Radiation Facility (Grenoble) at the Br-K edge (λ = 0.91965 Å, maximum f"; Table 1). Data were collected from a single crystal in an arbitrary setting by using a ADSC (Poway, CA) Quantum 4 detector after optimizing the oscillation range by using strategy (15). All data were processed and reduced by using denzo (16) and the ccp4 program suite (17). The bromine substructure was solved at 4.5-Å resolution by using difference Patterson methods and expanded by using difference Fourier methods. Heavy atom refinement and phase calculation with mlphare (17) and dm (18) to 3.0-Å resolution gave poor but partly encouraging maps. Local scaling and re-refinement of scattering factors given a heavy atom model as implemented in solve (19) (analyze mode) together with maximum-likelihood density modification and fragment fitting implemented in resolve (20) gave a starting model for warp (21) that could eventually build the complete model by using data to 1.5-Å resolution. Form 4 was solved at 2.0-Å resolution by molecular replacement with amore (22) by using the refined form 3 model. Refinement was performed with cns (23) followed by refmac5 (24). The final Rfree are 0.202 and 0.245 for forms 3 and 4, respectively (Table 1). Protein geometry was assessed by using procheck (25) showing 93.0% and 91.2% residues in the most favorable region for forms 3 and 4, respectively. The coordinates and structure factors have been deposited in the Protein Data Bank at the Research Collaboratory for Structural Bioinformatics as entries 1N8U and 1N8V.
Table 1.
Data collection and refinement statistics of CSPMbraA6 crystal forms 3 and 4
| Form 3 | Form 4 | |
|---|---|---|
| Data collection* | ||
| Beamline (technique) | ID14-EH4 (SAD)† | ID14-EH2 (MR)‡ |
| Space group/Nb mol per AU | P21/2 | C2/1 |
| Cell, Å, ° | 33.5, 54.2, 56.4 β = 93.9 | 61.5, 54.6, 33.2 β = 116.6 |
| λ, Å* | f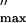 : 0.9197 : 0.9197 |
0.933 |
| Resolution, Å | 55.0–1.39 | 28.0–1.8 |
| Unique refs./multiplicity | 38,354/2.9 | 7,693/1.6 |
| Rsym, %* | 5.4 (24.7) | 3.1 (8.7) |
| Ranom, %* | 5.1 (22.1) | NA |
| I/σ(I)* | 5.7 (2.6) | 11.8 (6.9) |
| Completeness, % | 95.7 | 81.3 |
| Refinement statistics | ||
| Resolution, Å | 55.0–1.39 | 28.0–1.80 |
| Reflections/atoms | 36,789/2,156 | 6,730/973 |
| Rwork/Rfree | 0.18/0.20 | 0.18/0.24 |
| Mean B, Å2 | 14.2 | 18.3 |
| rms deviation bonds, Å/angles, ° | 0.011/1.37 | 0.014/1.48 |
| Ramachandran regions 1/2, % | 93.0/7.0 | 91.2/8.8 |
Values for the last resolution shell are in parentheses. Form 3, 1.44–1.39 Å; form 4, 1.9–1.8 Å.
SAD, single wavelength anomalous diffraction.
MR, molecular replacement.
Fluorescence Spectroscopy.
Brominated compounds and pheromones were obtained from Chemtech BV (Amsterdam) and from Sigma. One hundred percent methanolic solutions were freshly prepared. Fluorescence quenching was measured by using a Cary Eclipse (Varian), and measurements were made in a right angle configuration, at 20°C, by using 1-nm excitation and 10-nm emission bandwidths. The excitation wavelength was 280 nm, and the emission spectra were measured between 290 and 540 nm. In all experiments, the final methanol concentration in the cuvette was kept below 1%. Samples contained 1 μM protein in 10 mM Tris buffer, 25 mM NaCl (pH 8.0); ligands were used at concentrations between 0.02 and 22.5 μM. To estimate the affinity of brominated compounds to CSPMbraA6, the fluorescence intensities at 343 nm at increasing concentrations of quencher were plotted vs. the quencher concentration (Table 2, method a).
Table 2.
Dissociation constants of CSPMbraA6 with different compounds
| Method | Kdapp0.5, μM | Hill coeff. | Kd1, μM | Kd2app, μM | |
|---|---|---|---|---|---|
| Aliphatic compounds | |||||
| Native protein | |||||
| a | Br-C12-OH | 0.93 | 2.2 | 5.0 | 0.36 |
| a | Br-C15-acid | 1.6 | 2.6 | 14 | 0.3 |
| a | Br-C18-acid | 0.4 | 1.4 | 1.8 | 0.2 |
| b | C16-ol | 0.85 | |||
| a | C16-acid | 0.35 | 2.2 | 1.6 | 0.25 |
| b | C16-acetate-Me | 0.70 | |||
| b | Z11-C16-ol | 0.68 | |||
| a | Z11-C16-ald | 1.2 | 1.4 | ||
| b | Z11-C16-acetate-Me | 0.51 | |||
| b | Z7,Z11-C16-acetate-Me | 0.52 | |||
| b | E6,Z11-C16-acetate-Me | 0.19 | |||
| a | Z9-C16-ald | 1.1 | 1.1 | ||
| a | C11-ald | 0.6 | 1.4 | ||
| Bulky compounds | |||||
| a | Cineol | 3.2 | 2 | ||
| a | AMA | NB | |||
| a | ANS | NB | |||
| a | 2-Bromo-naphtalene | NB | |||
| a | Benzyl-benzoate | NB | |||
| Trp94Cys | |||||
| a | Br-C12-OH | 2.3 | 1.1 | ||
| a | Br-C15-acid | 1.5 | 1.4 | ||
| a | Br-C18-acid | 2.5 | 1.3 | ||
| Trp81Cys | |||||
| a | Br-C12-OH | 3.3 | 1.9 | ||
| a | Br-C18-acid | 1.25 | 2.2 | ||
| Tyr26Val | |||||
| a | Br-C12-OH | 3.5 | 2.3 | 33 | 0.4 |
| Tyr26Phe | |||||
| a | Br-C12-OH | 4.2 | 2.3 | 23 | 2 |
Methods a and b are described in Methods. NB, no binding.
The data of fluorescence quenching by the ligands have been analyzed in terms of Hill plot, log(Y/(1 − Y)) = f(log[X]), where Y is the normalized saturation value and [X] is the total ligand concentration, the slope of the function being the Hill coefficient. The fluorescence experimental curves have also been fitted by Adair equation (26) with a number of sites n = 1 to 3, K being the association constants: for a single site Y = K⋅[X]/(1 + K⋅[X]), for two sites equivalent and independent Y = (K1⋅[X] + 2K1⋅K2⋅[X]2)/(1 + K⋅[X] + (K1⋅K2⋅[X]2), for two sites equivalent and interdependent Y = (2K1⋅[X] + K1⋅K2⋅[X]2)/(1 + K⋅[X] + K1⋅K2⋅[X]2), and for three sites equivalent and interdependent Y = (3K1⋅[X] + 2K1⋅K2⋅[X]2 + K1⋅K2⋅K3⋅[X]3)/(1 + K⋅[X] + K1⋅K2⋅[X]2 + K1⋅K2⋅K3⋅[X]3). In all cases, except for Z9-C16-al with the wild-type protein and BrC12OH with the mutant W94C, the curve fit well with the model of two interdependent sites and in some cases also with three interdependent sites. All calculations were carried out with prism 3.02 (GraphPad, San Diego).
The Kapp0.5 values were estimated manually from the concentration of ligand producing half the maximal effect. The chasing of BrC12OH (5 μM) was performed under the same conditions as described above by adding increasing amount of competitor (Table 2, method b). The Kdapp values were calculated taking into account the Kapp0.5 values for BrC12OH with the following formula: Kdapp = [Kapp0.5]/(1 + [BrC12-OH]/Kapp0.5BrC12-OH), in which [BrC12-OH] = free concentration of the BrC12-OH and Kapp0.5BrC12-OH = dissociation constant for CSP2/BrC12-OH. The dissociation constants, calculated as the inverse of Kx values obtained by fitting, are reported in Table 2 together with the Kapp0.5 and the Hill coefficients.
Results
Overall Fold and Comparison with the apo-Protein.
Based on the fluorescence and NMR experiments, CSPMbraA6 was crystallized in the presence of BrC12OH, yielding two new crystal forms as compared with the apo-protein crystal forms 1 and 2 (12). The structure of the new crystal form 3 could not be solved by molecular replacement with the apo-protein coordinates, but could be solved by using single wavelength anomalous diffraction at the bromine edge. The BrC12OH CSPMbraA6 complex structure has undergone large conformational changes on complexation. The N terminus (residues 1–11) becomes visible in the electron density map, adopting an ordered extended conformation (Fig. 1A). Helix A is reduced to 6 residues (residues 13–18) and is shifted outwards from the protein core. The positions of helices B, D, and E have undergone moderate orientation changes, and helix F is slightly pushed away from the protein core. The last 10 residues, including the end of helix F, and the extended C terminus have become disordered. The most striking changes occur at helix C (residues 38–53), which is pushed outwards by ≈5 Å and is split into two shorter helices at residue 47 (Fig. 1B). The CSPMbraA6 structure in crystal form 4 is very close to that of form 3, both for the ligands and the protein: the rms deviation between the Cα atoms is 0.35 Å.
The Ligand-Binding Site.
Unexpectedly, three elongated electron densities are observed inside the protein, compatible with the presence of three bound BrC12OH ligand molecules (Fig. 2A). They are packed one against the other (Fig. 2 A and B), and their orientation has been easily assigned at the resolution of the present study considering the anomalous difference maps and the electron content difference between their hydroxyl and bromine extremities. Due to the large conformational changes, a wide and solvent accessible cavity is formed (Fig. 2C). Furthermore, one of the ligand molecules occupies in the complex the position of helix C in the apo-protein.
Figure 2.

X-ray structure of CSPMbraA6 in complex with BrC12OH [carbons, yellow; bromines (Br), orange; oxygens, red]. (A) The 2Fo-Fc electron density map of the three ligand molecules. (B) The stereo view of the structure of the complexed form of CSPMbraA6 (red) with the three BrC12OH molecules inside. (C) Water-accessible surface of the CSPMbraA6 internal cavity. The three openings to the exterior are indicated by red arrows. Views prepared with TURBO-FRODO (ref. 33; A), MOLSCRIPT (ref. 32; B), and GRASP (ref. 34; C).
This internal cavity (Fig. 2C) has a volume of 1,440 Å3, a value about three times that observed for other transport proteins, such as lipocalins (27). Of the 880 Å2 of its water-accessible surface area, 580 Å2 are covered by the ligand molecules; 85% of the 30 residues forming the cavity wall are aromatic or apolar residues. The most drastic surface area changes occur at Leu-43, Leu-47, Ile-51, Gln-62, Ala-66, Val-69, Ile-70, and Leu-84, which become completely buried, their accessible surface decreasing from 412 Å2 to 0. Leu-13, Tyr-26, Tyr-98, and a few other residues lose 180 Å2 of accessible surface value but are still partially accessible to water. Tryptophans 81 and 94 are close to the ligands, each being at 3.8 Å from a distinct BrC12OH molecule.
Three openings make it possible for the cavity to communicate with the bulk solvent (Fig. 2C): the two larger ones are surrounded by residues Tyr-98, Trp-94, and Gly-51 and by residues Leu-13 and Asn-61, respectively. The third one is smaller and located between residues Tyr-4 and His-46. Through them, ligands with longer alkyl chains, such as stearic acid (C18), could point out of the cavity, reach the bulk solvent, and thus establish interactions with positively charged residues at the protein surface.
Ligands Binding to Native and Mutated CSPMbraA6 Assayed by Using Tryptophan Fluorescence.
All of the binding experiments have a Hill coefficient >1 and in the range 1.1–2.6 (Table 2). This result indicates that binding takes place at more than one site and with an apparent positive cooperativity. The model of three interdependent sites does not provide a valuable fit; in particular, the Kd values are often not reliable or negative. We have therefore performed the analysis with two dissociation constants, assuming the coalescence of two equilibria (the addition of the 2d and 3d ligand) as indicated by the scheme in Fig. 3. The dissociation constants K2 are therefore considered as apparent (K2app). In some cases, the above model could not be distinguished from the one site model, and only the Kapp0.5 constants were taken into account (Table 2).
Figure 3.

(A) Schematic representation of the putative binding pathway of BrC12OH to CSPMbraA6. Form A shows the unbound protein (red circles are water molecules; the Tyr-26 side-chain is inside). Form B shows the hypothetical first intermediate with a 1:1 stoichiometry as modeled in ref. 1. Form C shows the x-ray complex with a 3:1 stoichiometry. (B) Intrinsic fluorescence quenching of CSPMbraA6 in the presence of BrC12OH. Experimental points correspond to three independent experiments; fittings with the Adair equation corresponding to a single site Y = K⋅[X]/(1 + K⋅[X]) (black line) and two sites equivalent and interdependent Y = (2K1⋅[X] + K1⋅K2⋅[X]2)/(1 + K⋅[X] + K1⋅K2⋅[X]2) (red line). (Inset) Hill plot of the same experiments.
Taking now in particular the case of BrC12OH and the wild-type protein, for which we have the x-ray structure, the Hill coefficient is 2.2, and the first two dissociation constants given by the model of two interdependent sites are 5 μM and 0.4 μM (K1 and K2app, respectively; Fig. 3B). The first molecule encounters more difficulty in binding than the subsequent ones. This result has also been found to be true for the other compounds that could be analyzed with the same model, whether it would be with the native protein or with the Tyr-26 mutants (Table 2).
We have determined the Kapp0.5 for a series of alkyl or bulky compounds. For the alkyl compounds, the Kapp0.5 values range between 0.19 μM and 1.6 μM. Alkyl compounds bind to CSPMbraA6 independently from their unsaturation, length, or chemical function. The acetate compounds seem to exhibit a better affinity, with an average Kapp0.5 of 0.5 ± 0.3 μM, compared with the average Kapp0.5 values for alcohols (0.8 ± 0.1 μM), acids (0.8 ± 0.4 μM), and aldehydes (1.0 ± 0.4 μM). The saturated compounds exhibit an average Kapp0.5 value of 0.75 μM, identical to that of unsaturated compounds. Among the five bulky compounds, only one (cineol) binds with a weak affinity constant, whereas the others do not bind at all.
To assess the involvement of the tryptophans in binding, we have mutated each of them into cysteine and measured the binding with the same brominated alkyl compounds. In both cases, tryptophan fluorescence varies on complexation: it increases for the Trp-81 → Cys mutant protein (Trp-94 involvement) and decreases for the Trp-94 → Cys mutant protein (Trp-81 involvement) (Table 2). It has previously been shown with PBPs that intrinsic fluorescence of tryptophans can either decrease or increase, depending on the way the hydrophobicity of their microenvironment is modified both by the ligand and by conformational changes (28, 29).
We have also investigated the possible role of Tyr-26 in ligand binding because this residue has shown a large mobility in different native forms of CSPMbraA6 and between the native and the complexed forms. Two mutant proteins were designed, where Tyr-26 was replaced by a valine or by a phenylalanine residue. The affinity of these mutant proteins for BrC12OH was tested. They both exhibit fluorescence quenching with an intensity comparable to that of the native for the Val mutant protein and limited to 30% of the native for the Phe mutant protein. The overall affinity of BrC12OH for both mutant proteins is lower than that for the native protein, and the K1 constant is four to six times larger for the mutants than for the native (Table 2).
Discussion
Putative Binding Mechanism.
The conformational changes observed on complexation are likely not induced by the crystal packing because they are identical in the two different crystal forms. Furthermore, we have recorded 15N heteronuclear sequential quantum correlation spectra of CSPMbraA6 alone or in complex with BrC12OH (13). The superimposition of the spectra of the apo and complexed protein spectra has revealed >50% changes in the peak displacements, indicating that the extensive conformational changes observed in the crystal also occur in solution (13). Finally, both tryptophans have their fluorescence modified on ligand binding. This observation is in agreement with the CSPMbraA6 x-ray complex structure, in which each of the tryptophans is close to the ligands, which is not the case for the 1:1 model (12).
An elongated channel, filled with water molecules but blocked half-way by the side-chain of Tyr-26, has been observed in the native structure (12). Rotating Tyr-26 side-chain made it possible to model a tight binding of long aliphatic molecules. The crystal structure of the complex reveals, however, that the docking stoichiometry is 3:1 instead of 1:1, and that the binding cavity becomes much larger on binding. Fluorescence quenching spectroscopy confirms that more than one ligand molecule binds to CSPMbraA6, and indicates that the binding is cooperative.
Two hypotheses may describe the complexation pathway between the unbound and complexed protein. First, the two conformations exist in preequilibrium, and the three ligands may enter quasi-simultaneously in the open form of CSPMbraA6. Second, the entry of the ligands occurs stepwise, the modeled 1:1 complex (12) being thus a transient intermediate on the way of complete complexation (Fig. 3A, form B of scheme). The entry of the first ligand might then destabilize the CSPMbraA6 structure and favor access of the two successive ligands (Fig. 3A, form C of scheme). Because Tyr-26 seems to block the tunnel opening, its rotation might very well be the trigger for the conformational change switched by the binding of the first ligand.
Our results of fluorescence quenching confirm the second hypothesis of ligand binding in successive steps. Tyr-26 seems to play a role in the binding of the first substrate, because the K1 value is largely increased with both the Tyr-26 mutant proteins; however, the fact that the first ligand binding is more difficult for the mutant Tyr-26 → Val seems somehow in contrast with the above proposal.
Implications of the Three Ligands Binding.
Using a fluorescent probe, Ban et al. (30) have demonstrated that a CSP from Schistocerca gregaria failed to bind long linear molecules, such as the pheromones of the same species, but also carboxylic acids and linear alcohols of 12, 14, and 18 carbon atoms. However, some compounds with 8–9 carbon atoms in the main chain and two compounds with the same shape of the probe and bearing an aldehyde function could displace the exogenous chromophore in competition studies. This finding is in contrast with the binding specificity of CSPMbraA6 and shows that CSPs from different species exhibit very different binding properties. Depending on their localization, CSPs could also bind a large variety of chemical compounds (differing in length, function, and shape) involved in the chemosensory process. In contrast with lipocalins, CSPMbraA6 has an all α-helical structure, intrinsically more flexible than a β-barrel, and exhibits a ligand accommodation mechanism based in a large part on protein backbone flexibility and not only on internal side-chains fluidity.
The observation of three ligand molecules present in the same cavity and of a cooperative binding are, to our knowledge, unique features among lipid transport proteins. They may indicate either that the physiological ligand of CSPMbraA6 is much larger than BrC12OH or that the binding of several molecules might be a molecular trick to induce considerable conformational changes readily recognized by a specific receptor. These conformational changes suggest that CSPs might be considered as a first acceptor for chemical compounds (food or odors), and that the special conformation reached on ligand binding might be used to trigger receptor (31) recognition and activation.
Acknowledgments
We thank Philippe Cantau and Eric Blanc for technical assistance. The European Synchrotron Radiation Facility is greatly acknowledged for beam time allocation. This study was supported in part by the Provence-Alpes-Côte d'Azur region (9811/2177) and by European Union BIOTECH Structural Biology Project Grant BIO4-98-0420.
Abbreviations
- CSP
chemosensory protein
- PBP
pheromone-binding protein
- BrC12OH
12-bromo-dodecanol
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1N8U and 1N8V).
References
- 1.Picimbon J F, Dietrich K, Angeli S, Scaloni A, Krieger J, Breer H, Pelosi P. Arch Insect Biochem Physiol. 2000;44:120–129. doi: 10.1002/1520-6327(200007)44:3<120::AID-ARCH3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 2.Adams M D, Celniker S E, Holt R A, Evans C A, Gocayne J D, Amanatides P G, Scherer S E, Li P W, Hoskins R A, Galle R F, et al. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- 3.Nagnan-Le Meillour P, Cain A H, Jacquin-Joly E, François M C, Ramachandran S, Maida R, Steinbrecht R A. Chem Senses. 2000;25:541–553. doi: 10.1093/chemse/25.5.541. [DOI] [PubMed] [Google Scholar]
- 4.Jacquin-Joly E, Vogt R G, Francois M C, Nagnan-Le Meillour P. Chem Senses. 2001;26:833–844. doi: 10.1093/chemse/26.7.833. [DOI] [PubMed] [Google Scholar]
- 5.Pikielny C W, Hasan G, Rouyer F, Rosbash M. Neuron. 1994;12:35–49. doi: 10.1016/0896-6273(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 6.Angeli S, Ceron F, Scaloni A, Monti M, Monteforti G, Minnocci A, Petacchi R, Pelosi P. Eur J Biochem. 1999;262:745–754. doi: 10.1046/j.1432-1327.1999.00438.x. [DOI] [PubMed] [Google Scholar]
- 7.Marchese S, Angeli S, Andolfo A, Scaloni A, Brandazza A, Mazza M, Picimbon J, Leal W S, Pelosi P. Insect Biochem Mol Biol. 2000;30:1091–1098. doi: 10.1016/s0965-1748(00)00084-9. [DOI] [PubMed] [Google Scholar]
- 8.Krieger J, von Nickisch-Rosenegk E, Mameli M, Pelosi P, Breer H. Insect Biochem Mol Biol. 1996;26:297–307. doi: 10.1016/0965-1748(95)00096-8. [DOI] [PubMed] [Google Scholar]
- 9.Sandler B H, Nikonova L, Leal W S, Clardy J. Chem Biol. 2000;7:143–151. doi: 10.1016/s1074-5521(00)00078-8. [DOI] [PubMed] [Google Scholar]
- 10.Horst R, Damberger F, Luginbuhl P, Guntert P, Peng G, Nikonova L, Leal W S, Wuthrich K. Proc Natl Acad Sci USA. 2001;98:14374–14379. doi: 10.1073/pnas.251532998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krieger M J, Ross K G. Science. 2002;295:328–332. doi: 10.1126/science.1065247. [DOI] [PubMed] [Google Scholar]
- 12.Lartigue A, Campanacci V, Roussel A, Larsson A M, Jones T A, Tegoni M, Cambillau C. J Biol Chem. 2002;277:32094–32098. doi: 10.1074/jbc.M204371200. [DOI] [PubMed] [Google Scholar]
- 13.Mosbah A, Campanacci V, Lartigue A, Tegoni M, Cambillau C, Darbon H. Biochem J. 2003;369:39–44. doi: 10.1042/BJ20021217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campanacci V, Mosbah A, Bornet O, Wechselberger R, Jacquin-Joly E, Cambillau C, Darbon H, Tegoni M. Eur J Biochem. 2001;268:4731–4739. doi: 10.1046/j.1432-1327.2001.02398.x. [DOI] [PubMed] [Google Scholar]
- 15.Ravelli R B G, Sweet R M, Skinner J M, Duisenberg A J M, Kroon J. J Appl Crystallogr. 1997;30:551–555. [Google Scholar]
- 16.Otwinowski Z. In: Data Collection and Processing. Sawyer L, Isaacs N W, Bailey S, editors. Daresbury, U.K.: Daresbury Laboratory; 1993. pp. 56–63. [Google Scholar]
- 17.Collaborative Computational Project, Number 4. Acta Crystallogr D. 1994;50:760–766. [Google Scholar]
- 18.Cowtan K, Main P. Acta Crystallogr D Biol Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 19.Terwilliger T C, Berendzen J. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terwilliger T C. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perrakis A, Morris R, Lamzin V S. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 22.Navaza J. Acta Crystallogr A. 1994;50:157–163. [Google Scholar]
- 23.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 24.Murshudov G N, Vagin A A, Dodson E J. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 26.Segel I H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. New York: Wiley; 1993. [Google Scholar]
- 27.Spinelli S, Vincent F, Pelosi P, Tegoni M, Cambillau C. Eur J Biochem. 2002;269:2449–2456. doi: 10.1046/j.1432-1033.2002.02901.x. [DOI] [PubMed] [Google Scholar]
- 28.Wojtasek H, Leal W S. J Biol Chem. 1999;274:30950–30956. doi: 10.1074/jbc.274.43.30950. [DOI] [PubMed] [Google Scholar]
- 29.Bette S, Breer H, Krieger J. Insect Biochem Mol Biol. 2002;32:241–246. doi: 10.1016/s0965-1748(01)00171-0. [DOI] [PubMed] [Google Scholar]
- 30.Ban L, Zhang L, Yan Y, Pelosi P. Biochem Biophys Res Commun. 2002;293:50–54. doi: 10.1016/S0006-291X(02)00185-7. [DOI] [PubMed] [Google Scholar]
- 31.Krieger J, Raming K, Dewer Y M, Bette S, Conzelmann S, Breer H. Eur J Neurosci. 2002;16:619–628. doi: 10.1046/j.1460-9568.2002.02109.x. [DOI] [PubMed] [Google Scholar]
- 32.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 33.Roussel A, Cambillau C. Silicon Graphis Geometry Partners Directory. Vol. 81. Mountain View, CA: Silicon Graphics; 1991. [Google Scholar]
- 34.Nicholls A, Sharp K A, Honig B. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]