

Abstract
Strenuous exercise leads to an increase in metabolic rate, increased production of reactive oxygen species, and compromised antioxidant defense systems. To study the effects of oxidative stress during strenuous exercise, a homogeneous group of 31 male subjects participated in a 6-month, 5 days/week training schedule involving two extreme marches of 50 km and 80 km at sea level, separated by 2 weeks of regular training. Each participant carried 35 kg of extra weight. Blood samples were drawn imediately before and after each march. Twenty-nine subjects completed the 50-km march, and only 16 completed the 80-km march. Plasma levels of reduced ascorbic acid, total ascorbate, and dehydroascorbate did not undergo significant changes during either march. However, the 50- and 80-km marches led to 25% and 37% increases, respectively, in plasma levels of uric acid; due presumably to increases in the metabolic rate and consequent pyrimidine nucleotide metabolism. Both marches led to ≈10-fold increase leakage of creatine phosphokinase into the plasma. Likewise, plasma levels of aspartate transaminase, a characteristic marker of liver injury, increased ≈4-fold. Plasma levels of bilirubin, creatine, urea, and glucose also increased. Plasma protein carbonyl content, a marker of protein oxidative damage, decreased significantly during each march. These results are discussed with respect to the consideration that elevation of the respiration rate during exercise leads to production of more reactive oxygen species than the antioxidant systems can scavenge. Plausible explanations for leakage of molecules into the plasma are discussed.
Body oxygen uptake is greatly increased during intense exercise (1, 2). Muscles oxygen utilization during strenuous exercise can increase as much as 100–200 times than at rest (3, 4). Consequently, increased electron flux through the rapidly respiring mitochondria in the active muscle may lead to an enhancement of electron leakage and consequent reactive oxygen species (ROS) production.
ROS may be generated during and after physical exercise in working muscles, and in the tissues that undergo ischemia-reperfusion (4–8). It is estimated that 2–5% of the total electron flux leaks to form primary short-lived ROS such as superoxide radical anion (⋅O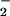 ), hydrogen peroxide (H2O2), and hydroxyl radical (⋅OH) (9). Sources of ROS during exercise include enhanced purine oxidation, damage to iron-containing proteins, disruption of Ca2+ homeostasis (10), and flow-induced endothelium ROS production (11). Neutrophil activation infiltrating the muscle after injury by exercise may also produce ROS (12). ROS may cause oxidative tissue damage (13), e.g., DNA injury (14), lipid peroxidation (15) and protein oxidation (16–18). These processes are associated with the appearance of and increase in the severity of many diseases (19, 20).
), hydrogen peroxide (H2O2), and hydroxyl radical (⋅OH) (9). Sources of ROS during exercise include enhanced purine oxidation, damage to iron-containing proteins, disruption of Ca2+ homeostasis (10), and flow-induced endothelium ROS production (11). Neutrophil activation infiltrating the muscle after injury by exercise may also produce ROS (12). ROS may cause oxidative tissue damage (13), e.g., DNA injury (14), lipid peroxidation (15) and protein oxidation (16–18). These processes are associated with the appearance of and increase in the severity of many diseases (19, 20).
The antioxidant defense system against ROS consists of enzymes and of low molecular weight antioxidants (LMWA), such as ascorbic acid (AA). LMWA easily penetrate into cells and accumulate in specific locations near targets of ROS attack. They are disipated during their reaction with various ROS and can be replenished through the diet (21). Previous studies claim that in human, as in other species subjected to unusual physical activity, an imbalance may occur between free radical production and antioxidant levels leading to oxidative stress (22–25). This stress may produce metabolic alterations and affect physical performance (26, 27). Some studies, however, reject this assertion (28, 29).
If the production of free radicals is excessive as observed during strenuous aerobic exercise (8, 30) or if the antioxidant defense mechanisms are impaired, the balance between prooxidants and antioxidants is lost. Thus, there is an apparent paradox between the benefits of normal (moderate) and strenuous aerobic exercise.
The present study was undertaken to determine whether the ability of highly trained soldiers to withstand oxidative stress, provoked by strenuous exercise, is related to the plasma antioxidant status, and to determine also the effect of such stress on the extent of muscle injury, as monitored by leakage of large muscle proteins into the plasma.
Materials and Methods
Subjects and Study Description.
The study group included 31 young healthy males (19 ± 1 years old; 181 ± 4 cm height; and 80 ± 6 kg weight), characterized by a uniform lifestyle, balanced nutrition, and physical fitness (Vo2max = 65.7 ± 0.6 ml/kg/min), yielding a high degree of reproducibility within the group. The subjects had been closely monitored for 2 weeks, during two marches of 50 and 80 km, which was part of a 6-month, 5 days/week training regime.
Each participant carried a back load of 35 kg, walking at high pace on unpaved roads at sea level (10–85 m above sea level), often on deep sand. To our best knowledge, the efforts carried out by the group are more demanding than other published studies. This study was approved by the Institutional Review Board, Helsinki Committee.
Diet.
Subjects consumed a daily balanced diet that provided ≈3,500 kcal. Otherwise, the subjects did not consume any extra antioxidants or other food additives. During the marches, water consumption was unlimited and the subjects were encouraged to drink water to ensure that no dehydration developed.
Blood Collection.
Blood samples were drawn in heparinized vacutainers. Blood samples were collected at the start and immediately at the end of 50- and 80-km marches. Plasma was separated immediately and kept at −80°C until analyzed.
HPLC Analyses.
Ascorbate and urate were quantified by a modification of the HPLC-electrochemical detection method as described (31, 32).
Standards of uric acid (UA) and AA were prepared in a solution of metaphosphoric acid (MPA) (5% wt/vol; Aldrich Chem, Metuchen, NJ) containing 0.54 mM Na2EDTA (BDH). All MPA solutions were freshly prepared each day. UA standards (4.6 μg/ml) were prepared from UA sodium salt and either used immediately or stored at −70°C for several weeks. Similarly, AA standards were prepared from the sodium salt in the same solution as for UA. Stock solutions were diluted with the mobile phase before injection. Sixty microliters of plasma were added to an equal volume of 10% (wt/vol) MPA (containing Na2EDTA), mixed on a vortex mixer and centrifuged for 3 min to precipitate plasma proteins. The supernatant was stored at −70°C until analyzed. Total AA (AA plus dehydroascorbic acid) was determined by diluting the MPA-stabilized samples 50-fold with a mobile phase containing 3.9 mg of cysteine (Nutritional Biochemicals, Cleveland) in a 25-ml volumetric flask. The diluted sample was incubated for 5 min at 4°C.
Analyses were carried out on a Spectra-Physics SP8810 liquid chromatograph connected to a Superpher 100RP-18, 5-μm column 250 × 4 mm (E. Merck, Darmstadt, Germany). The column was connected to a LC-4A amperometric detector (BAS, West Lafayette, IN) with a glassy-carbon working electrode and an Ag/AgCl reference electrode. The applied potential was set at +0.5V. The mobile phase consisted of 40 mM sodium acetate buffer (pH 4.75), 0.54 mM Na2EDTA and 1.5 mM tetrabutylammonium hydroxide (Aldrich Chemicals) as an ion-pairing agent. The separations were performed at a flow rate of 1.1 ml/min with a back pressure of 100 torr (1 torr = 133 Pa). The chromatograms were recorded on a PC-based data acquisition and processing system (Chrom-A-Set, Barspec, Israel). Freshly prepared human plasma diluted with mobile phase containing 1.0 mM cysteine produced three prominent peaks that correspond to cysteine (retension time = 163 s), AA (238 s), and UA (319 s), respectively.
The sensitivity for AA and UA was 20 pg in a loop of 20 μl.
Oxidative Stress by Ascorbic Acid (OSAA).
The ratio between the dehydroascorbate (DHAA) and the total ascorbate (TAA) concentrations (TAA = AA + DHAA) was used as a marker of OSAA (33): OSAA = [DHAA]/[TAA]%.
Deternination of Protein Carbonyl Content (PCC).
The carbonyl content of total plasma protein was determined spectrophotometically with 2,4-dinitrophenylhydrazine as described (34). Absorbance was measured in a 96-well plate reader (Spectramax 384 Plus, Molecular Devices); protein was determined with the bicinchoninic acid method (Pierce, Rockford, IL).
Creatine Phosphokinase (CPK), Aspartate Transaminase (AST), Bilirubin, Creatinine, Urea, and Glucose.
CPK and AST were measured by standard procedures used in clinical biochemistry laboratory.
Osmolarity.
Osmolarity was monitored by a 5500 Vapor Pressure Osmometer (Wescor, Logan, UT).
Statistical Analyses.
Statistical analyses within before and after each march were done by using a two-tailed paired t test, and between the basal values of the marches by using a two-tailed unpaired t test. Statistical significance was set at P < 0.05. Values are given as means ± SE.
Results
A group of 31 highly homogenous group of young subjects, as described in Table 1, was monitored for a 2-week period. This period was a part of their 6-month, strenuous, well planned, 5 days/week training schedule. All subjects shared the same board and received the same balanced nutrition, and enjoyed an identical lifestyle. Within these 2 weeks the group had two marches of 50 and 80 km, at altitude of 10–85 m above sea level, and pack weight of 35 kg. Blood samples were collected from each participant just before the start and immediately after the completion of each march.
Table 1.
Subjects description and plasma osmolarity
| 50 km
|
80 km
|
|||
|---|---|---|---|---|
| Before | After | Before | After | |
| n | 31 | 29 | 31 | 16 |
| Age, years | 19 ± 1 | |||
| Weight, kg | 81 ± 1.2 | 80 ± 1.2 | 83 ± 1.3 | 82 ± 1.3 |
| Height, cm | 181 ± 1 | |||
| Plasma osmolarity, mmol/kg | 293 ± 1.5 | 285 ± 2 | 287 ± 1.6 | 282 ± 1.9 |
The group was on a regular training process. They were monitored before and after each of two extensive marches of 50 and 80 km for body weight and plasma osmolarity. Data are given as values ± SE.
The duration of the marches was ≈10 and 20 h for the first (50-km) and second (80-km) march, respectively. Twenty-nine subjects completed the first march, and only 16 (≈52%) completed the second march. No significant changes in body weight or in plasma osmolarity could be detected between the start and end of each march.
Plasma antioxidant parameters, as evaluated from analysis using HPLC coupled with electrochemical detection, are summarized in Table 2. No significant changes in the antioxidant parameters were observed during the tenure of the experiment, or between the start and end of each march. A slight tendency of an increase, which was not statistically significant, in the total concentration of AA, and the dehydro-ascorbic acid could be identified. OSAA, a bona fide parameter reflecting the plasma antioxidant status (33), also did not vary during the experiment. Plasma levels of UA increased by 25% during the 50-km march and by 37% during the 80-km march. It should be noted that plasma UA had dropped markedly between the two marches (≈2 weeks), but did not reach a normal basal level before the second march (Table 2) and apparently represented the intense level of daily training between the marches.
Table 2.
Plasma antioxidants parameters and oxidative stress of the subjects, as measured by HPLC
| 50 km
|
80 km
|
|||
|---|---|---|---|---|
| Before | After | Before | After | |
| UA, mg/liter | 77 ± 3 | 96 ± 3 | 90 ± 5 | 123 ± 4 |
| AA, mg/liter | 12.1 ± 0.8 | 13.3 ± 0.9 | 13.3 ± 0.9 | 14.4 ± 1.0 |
| TAA, mg/liter | 13.2 ± 0.9 | 14.6 ± 0.1 | 15.0 ± 1.0 | 15.9 ± 1.1 |
| DHAA, mg/liter | 1.2 ± 0.19 | 1.34 ± 0.21 | 1.33 ± 0.18 | 1.41 ± 0.19 |
| OSAA, % | 8.8 ± 1.0 | 8.9 ± 1.1 | 8.7 ± 1.1 | 9.4 ± 1.3 |
Two-tailed paired t test. Data are given as values ± SE. The differences between [UA] “Before” and “After” each march are significant (P < 0.0001). Other changes are nonsignificant. The differences of [UA] values between the beginning of the 80-km march and the beginning of the 50-km march are significant (P < 0.02). UA, AA, TAA (total ascorbic acid), and DHAA (dehydro-ascorbate) were measured by HPLC coupled with electrochemical detection, and values of the OSAA were calculated, before and after marches of 50 and 80 km, which were separated by 2 weeks. OSAA is defined as the ratio between DHAA and TAA.
The plasma levels of CPK and AST (Table 3) could serve as more direct indicators of the severity of exercise and its effect on the tissues. A dramatic rise in CPK levels (≈10-fold) was observed between the start and the end levels for each march. Also, the CPK level before the first march was at least 3 times higher than the normal value among healthy population. Also, like the changes in UA levels (Table 2), CPK values did not return to their basal levels before the second march (Table 3).
Table 3.
Plasma levels of CPK and AST
| 50 km
|
80 km
|
|||||
|---|---|---|---|---|---|---|
| Before | After | P | Before | After | P | |
| CPK | 331 ± 40 | 4600 ± 529 | <0.0001 | 865 ± 330 | 8118 ± 1390 | <0.0002 |
| AST | 35 ± 2 | 134 ± 10 | <0.0001 | 48 ± 11 | 206 ± 33 | <0.0001 |
Two-tailed paired t test. Data are given as values ± SE. The changes of plasma CPK values between the beginning of the 50-km march and the beginning of the 80-km march are significant (P < 0.02). The plasma concentrations of CPK and AST after 50- and 80-km marches were determined. The 80-km march took place 2 weeks after the 50-km march.
The values of other biochemical parameters are summarized in Tables 3 and 4. Plasma AST, an indicator for liver function, increased by ≈4-fold. Bilirubin, creatinine, urea, and glucose also increased significantly. Urea values at the start of the second march were significantly higher than those in the first march.
Table 4.
Additional plasma parameters
| 50 km
|
80 km
|
|||||
|---|---|---|---|---|---|---|
| Before | After | P | Before | After | P | |
| Bilirubin | 0.39 ± 0.05 | 1.09 ± 0.08 | <0.0001 | 0.51 ± 0.08 | 1.71 ± 0.24 | <0.0005 |
| Creatinine | 0.99 ± 0.02 | 1.2 ± 0.03 | <0.0001 | 1.04 ± 0.03 | 1.36 ± 0.05 | <0.0001 |
| Urea | 32 ± 1 | 51 ± 2 | <0.0001 | 39 ± 2 | 62 ± 5 | <0.001 |
| Glucose | 92 ± 3 | 113 ± 5 | <0.0003 | 86 ± 5 | 110 ± 5 | <0.005 |
Two-tailed paired t test. Data are given as values ± SE. The difference of [urea] values between the beginning of the 80-km march and the beginning of the 50-km march is significant (P < 0.008).
There was no correlation between UA and CPK values. A correlation coefficient of 0.888 and 0.814 was found between AST and CPK values after 50 and 80 km, respectively (Fig. 1).
Figure 1.

Correlation between AST and CPK values. The correlation was calculated for the changes in the plasma values of AST and CPK after the 50- and 80-km marches (R250 = 0.888; R280 = 0.814).
PCC was measured as an indicator of protein oxidation (35). A significant decrease in the PCC between the start and the end of both marches was clearly observed (P < 0.001 and P < 0.04; two-tailed t test) (Fig. 1). Additionally, the PCC level at the start of the 80-km march was lower than that at the beginning of the 50-km march. Because only 16 subjects had finished the second march, the analysis was conducted in two modes. In the first, the PCC values at the end of the march were compared with the values of the same 16 subjects at the start of the march (paired analysis), not considering the start PCC values of those who did not complete the march. In the second mode, the average PCC value at the end of each march was compared with the average value of the entire group at the start. No difference was observed when either mode of analysis was used, including the degree of significance for the differences between the start and end PCC values.
Discussion
The experimental group was enrolled in a 6-month, 5 days/week, intensive physical activity program. The subjects shared a uniform lifestyle, balanced nutrition, and physical fitness. After 4 months of training, they reached a level of fitness allowing the two marches, of 50 and 80 km, as described. As far as we are able to judge, the physical effort of this group was more demanding than other reported studies. Indeed, the high levels of plasma CPK activity reflect the intensity of the fitness program (Table 3).
The characteristics of experimental groups reported in the literature vary markedly from one another in their degree of intensity, duration of exercise, fitness level of the subjects, diet regimens, and degree of compliance. Thus, the validity of a comparison between the studies should be cautiously and critically handled.
Various indicators have been used for the characteriztion of the oxidative stress exerted on cells and tissues. Here we have used the levels of plasma AA, UA, OSAA, and PCC, as well as the degree of leakge of CPK and AST into the circulation.
PCC has been often used as a bona fide indicator of oxidative stress (35). Caution should be exercised when evaluating its meaning. The turnover time for protein carbonyls is of many hours to days, unlike thiobarbituric acid reactive substances that turn over within minutes (36, 37). Indeed, the decrease in the PCC level, during each march (Fig. 2), might be a consequence of an activation of a mechanism that removes the oxidatively modified proteins from the circulation, or alternatively, activation of an antioxidant mechanism that removes the ROS. As the decrease in PCC during the first march was higher than that during the second march, it is tempting to speculate that the activation of the “removal mechanism” is stronger in the first hours of challenge. In a recent study, serum PCC level was monitored during and after coronary artery bypass operation. PCC increased during the early phase of reperfusion (2 min) and remained constant for hours. At 18 h, PCC level was markedly reduced to ≈50% of the value at early reperfusion (2 min to 4 h) (38). In another animal study it was indicated that muscle PCC level was unaffected by extensive exercise or age (39, 40).
Figure 2.

PCC. Oxidative modification of protein as reflected by the PCC marker in plasma of subjects immediately before and at the end of two marches of 50 and 80 km.
AA is considered an effective antioxidant in biological fluids (1, 2). AA is a reducing agent trapping a variety of free radicals, including the peroxyl radicals (41). Furthermore, it directly inhibits the propagation of lipid-peroxidation chain reactions in cellular membranes (42) and helps to regenerate vitamin E from the α-tocopheryl radical (43, 44). It was therefore anticipated that the AA level would decline during and after the marches, as was reported in some studies (45, 46). However, the plasma AA did not change from the start to the end of each march. Likewise, no difference was observed between the two marches, in spite the 2-week lapse between them (Table 2). This is in accord with other reports showing no change or even an increase in AA level (2, 23, 24, 28, 29, 42, 47–50). Other reports on intense but short-duration exercise have shown a marked decrease in AA and red blood cell glutathione levels (51).
The ratio between the concentration of the oxidized form of AA and total ascorbic acid is a simple but informative parameter that reflects the recent oxidative history of the system, as well as its antioxidant capacity to combat future challenge. OSAA has been shown to reflect changes in the antioxidant status of human subjects (33). No change in OSAA was recorded in our study (Table 2). This is in accord with other studies of the antioxidant capacity after severe exercise (46, 52). By contrast, some other reports have demonstrated an unexpected increase in the antioxidant capacity of athletes after exercise (23, 48, 53).
UA serves as a free radical scavenger in vivo (54, 55). Plasma UA can trap peroxyl radicals in aqueous phase and contribute to the plasma antioxidant defense (56). During exercise, energy-rich purine phosphates are used and catabolized, resulting in accumulation of hypoxanthine, xanthine, and UA in tissues. The conversion of hypoxanthine to xanthine and UA is associated with the formation of toxic oxygen free radicals (4). Bilirubin was shown to efficiently scavenge peroxyl radicals in vitro and to act as a metal-binding species, thus functioning as a selective antioxidant (56).
The increase in plasma UA during each march (Table 2) is in accord with the proposed mechanism of enhanced catabolism of purine nucleotides. This increase, in turn, depends on the metabolic rate, and is associated with the production of free radicals (2, 4, 24, 54, 57, 58). This process is analogous to the processes during ischemia and reperfusion (59) (Fig. 3).
Figure 3.

Catabolism of purine nucleotides from ATP to UA. Exercise-caused stress that enhances the catabolism of ATP to hypoxanthine, by virtue of the enzyme xanthine oxidase, is further converted to UA. In the process, free radicals, including the toxic superoxide and hydroxyl radicals, are produced and cause muscle cell damage.
Our results are in contradiction with those of Robertson et al. (48), where blood UA was lower in high-training runners (80–147 km/week) than in low-training runners (16–43 km/week), and which was even lower than in sedentary subjects.
Morning serum urea has been suggested as a marker of the intensity of training in the previous day, in endurance athletes (60). Indeed, urea levels at the start and the end of the marches were different, and are in accord with this suggestion (Table 4). These results are inconsistent with those of Raastad and Hallen (61).
Plasma CPK levels increased dramatically during each march, reaching levels 40- to 80-fold higher than the typical value for healthy sedentary subjects (Table 3). This result is in accord with other studies (22, 48, 50, 52, 62). These increases presumably reflect persistent damage to the muscles, resulting in a loss of cell membrane integrity, and consequent leakage of proteins into the blood. Interorgan injurious effects are often observed after acute insult. Indeed, elevated AST levels (by 3.8- and 4.5-fold) were also observed (Table 3), reflecting injury to liver cells. Hence, there is a possibility for direct interaction between the antioxidant status and muscle and liver injury. Such direct or secondary impact on cell function of other organs following severe exercise needs further clarification.
The lack of direct correlation between UA and CPK values may reflect the changes in UA and CPK that are initiated at different effort levels.
Collectively, the results of the present communication combined with other reported data can be integrated into a working hypothesis on the interrelationships among exercise, antioxidant status, and cell injury (Fig. 4). We suggest that elevation of the respiration rate during the present study may have led to the generation of more ROS than the antioxidant systems can scavenge, and that the increase in muscle injury also reflects an increase in the generation of the ROS to levels greater than the antioxidants can handle. It is assumed that, during exercise, the level of AA, which is a charged molecule, decreased within the cells while scavenging ROS. Hence, AA did not infiltrate out from the tissue to the plasma, and no change in plasma AA level was found after the marches. At the same time, UA, bilirubin, creatinine, and urea, the catabolic products that accumulate in cells, leaked or were secreted out of the cells, and the plasma levels increased significantly. It is only during or after strenuous exercise that the integrity of the muscle cell is compromised, and allows the leakage of large molecules, CPK and AST, and allows also the leakage of charged molecules, such as AA. The amount of AA leakage into the plasma would depend on its intracellular level that had decreased as a result of combating ROS.
Figure 4.

A hypothetical model for exercise-induced cell damage. The process in muscle cell injury vs. the duration of severe exercise is proposed.
In summary, it is postulated that, after severe physical exercise, the exercise-associated changes in the plasma parameters, that are coupled to intracellular processes, could provide a better indication for the ability of subjects to withstand physical activity including highly strenuous exercise.
Acknowledgments
We appreciate the superb technical help of and fruitful discussions with Dr. Rodney Levine and Ms. Nancy Wehr (National Heart, Lung, and Blood Institute, National Institutes of Health), and Dr. Raphael N. Melmed (Hadassah University Hospital, Israel). This study was supported by a research grant from the Chief Medical Officer, Israeli Defense Forces, and the Dr. Moshe and Pepka Bergman Memorial Fund.
Abbreviations
- AA
reduced ascorbic acid
- UA
uric acid
- CPK
creatine phosphokinase
- AST
aspartate transaminase
- PCC
protein carbonyl content
- ROS
reactive oxygen species
References
- 1.Sies H, Stahl W, Sundquist A R. Ann NY Acad Sci. 1992;669:7–20. doi: 10.1111/j.1749-6632.1992.tb17085.x. [DOI] [PubMed] [Google Scholar]
- 2.Rokitzki L, Logemann E, Sargedos A N, Murphy M, Wetzel Roth W, Keul J. Acta Physiol Scand. 1994;151:149–158. doi: 10.1111/j.1748-1716.1994.tb09732.x. [DOI] [PubMed] [Google Scholar]
- 3.Davies K J, Quintanilha A T, Brooks G A, Packer L. Biochem Biophys Res Commun. 1982;107:1198–1205. doi: 10.1016/s0006-291x(82)80124-1. [DOI] [PubMed] [Google Scholar]
- 4.Sjodin B, Westing Y H, Apple F S. Sports Med. 1990;10:236–254. doi: 10.2165/00007256-199010040-00003. [DOI] [PubMed] [Google Scholar]
- 5.Witt E H, Reznick A Z, Viguie C A, Starke-Reed P, Packer L. J Nutr. 1992;122:766–773. doi: 10.1093/jn/122.suppl_3.766. [DOI] [PubMed] [Google Scholar]
- 6.Alessio H M. Med Sci Sports Exercise. 1993;25:218–224. [PubMed] [Google Scholar]
- 7.Aruoma O I. J Nutr Biochem. 1994;5:370–381. [Google Scholar]
- 8.Ji L L. Free Radical Biol Med. 1995;18:1079–1086. doi: 10.1016/0891-5849(94)00212-3. [DOI] [PubMed] [Google Scholar]
- 9.Boveris A, Chance B. Biochem J. 1973;134:707–718. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson M. In: Handbook of Oxidants and Antioxidants in Exercise. Sen C K, Packer L, Hännier O, editors. Amsterdam: Elsevier; 2000. pp. 57–68. [Google Scholar]
- 11.Shi Y, Niculescu R, Wang D, Patel S, Davenpeck K, Zalewski A. Arterioscler Thromb Vasc Biol. 2001;21:739–745. doi: 10.1161/01.atv.21.5.739. [DOI] [PubMed] [Google Scholar]
- 12.Jones D A, Newham D A, Round J M, Tolfree S E J. J Physiol. 1986;375:435–448. doi: 10.1113/jphysiol.1986.sp016126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sen C K. J Appl Physiol. 1995;79:675–686. doi: 10.1152/jappl.1995.79.3.675. [DOI] [PubMed] [Google Scholar]
- 14.Inoue T, Mu Z, Sumikawa K, Adachi K, Okochi T. Jpn J Cancer Res. 1993;84:720–725. doi: 10.1111/j.1349-7006.1993.tb02035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meydani M, Evans W J, Handelman G, Biddle L, Fielding R A, Meydani S N, Burrill J, Flatarone M A, Blumberg J B, Cannon J G. Am J Physiol. 1993;264:R992–R998. doi: 10.1152/ajpregu.1993.264.5.R992. [DOI] [PubMed] [Google Scholar]
- 16.Reznick A Z, Witt E H, Matsumoto M, Packer L. Biochem Biophys Res Commun. 1992;189:801–806. doi: 10.1016/0006-291x(92)92273-z. [DOI] [PubMed] [Google Scholar]
- 17.Rajguru S U, Yeargans G S, Seidler N W. Life Sci. 1994;54:149–157. doi: 10.1016/0024-3205(94)00584-2. [DOI] [PubMed] [Google Scholar]
- 18.Saxton J M, Donnely A E, Roper H P. Eur J Appl Physiol. 1994;68:189–193. doi: 10.1007/BF00376765. [DOI] [PubMed] [Google Scholar]
- 19.Ames B N, Shigenaga M K, Hagen T M. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gutteridge J M C. Free Radical Res Commun. 1993;19:141–158. doi: 10.3109/10715769309111598. [DOI] [PubMed] [Google Scholar]
- 21.Halliwell B, Gutteridge J M. Arch Biochem Biophys. 1990;280:1–8. doi: 10.1016/0003-9861(90)90510-6. [DOI] [PubMed] [Google Scholar]
- 22.Kanter M M, Lesmes G R, Nequin N D, Kaminsky L A, Saeger J M. Ann Sports Med. 1986;3:39–41. [Google Scholar]
- 23.Child R B, Wilkinson D M, Fallowfield J L, Donnely A E. Med Sci Sports Exercise. 1998;30:1603–1607. doi: 10.1097/00005768-199811000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Liu M L, Bergholm R, Makimattila S, Lahdenpera S, Valkonen M, Hilden H, Yki-Jarvinen H, Taskinen M R. Am J Physiol. 1999;276:E1083–E1091. doi: 10.1152/ajpendo.1999.276.6.E1083. [DOI] [PubMed] [Google Scholar]
- 25.Sies H. Am J Med. 1991;91:31S–38S. doi: 10.1016/0002-9343(91)90281-2. [DOI] [PubMed] [Google Scholar]
- 26.Natta C, Tatum V, Chow C K. Ann NY Acad Sci. 1992;669:365–367. doi: 10.1111/j.1749-6632.1992.tb17125.x. [DOI] [PubMed] [Google Scholar]
- 27.Brent J, Rumack B. J Free Radical Biochem Clin Toxicol. 1993;31:139–171. doi: 10.3109/15563659309000383. [DOI] [PubMed] [Google Scholar]
- 28.Duthie G G, Robertson J D, Maughan R J, Morrice P C. Arch Biochem Biophys. 1990;282:78–83. doi: 10.1016/0003-9861(90)90089-h. [DOI] [PubMed] [Google Scholar]
- 29.Viguie C A, Frei B, Shigenaga M K, Ames B N, Packer L, Brooks G A. J Appl Physiol. 1993;75:566–572. doi: 10.1152/jappl.1993.75.2.566. [DOI] [PubMed] [Google Scholar]
- 30.Kanter M M. Exercise Sport Sci Rev. 1995;23:375–397. [PubMed] [Google Scholar]
- 31.Motchnik P, Frei B, Ames B N. Methods Enzymol. 1994;234:269–279. doi: 10.1016/0076-6879(94)34094-3. [DOI] [PubMed] [Google Scholar]
- 32.Chevion S, Berry E M, Kitrossky N, Kohen R. Free Radical Biol Med. 1997;22:411–421. doi: 10.1016/s0891-5849(96)00337-1. [DOI] [PubMed] [Google Scholar]
- 33.Chevion S. Ph.D. thesis. Jerusalem: The Hebrew University of Jerusalem; 1998. [Google Scholar]
- 34.Levine L, Williams J A, Stadtman E R, Shacter E. Methods Enzymol. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- 35.Chevion M, Berenshtein E, Stadtman E R. Free Radical Biol Med. 2000;33:S99–108. [PubMed] [Google Scholar]
- 36.Davies K J A, Goldberg A L. J Biol Chem. 1987;262:8220–8226. [PubMed] [Google Scholar]
- 37.Augustin W, Wiswedel I, Noack H, Reinheckel T, Reichelt O. Mol Cell Biochem. 1997;174:199–205. [PubMed] [Google Scholar]
- 38.Pantke U, Volk T, Schmutzler M, Kox W J, Sitte N, Grune T. Free Radical Biol Med. 1999;27:1080–1086. doi: 10.1016/s0891-5849(99)00144-6. [DOI] [PubMed] [Google Scholar]
- 39.Bejma J, Ji L L. J Appl Physiol. 1999;87:465–470. doi: 10.1152/jappl.1999.87.1.465. [DOI] [PubMed] [Google Scholar]
- 40.Radak Z, Kaneko T, Tahara S, Nakamoto H, Ohno H, Sasvari M, Nyakas C, Goto S. Free Radical Biol Med. 1999;27:69–74. doi: 10.1016/s0891-5849(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 41.Wefer H, Sies H. Eur J Biochem. 1988;174:766–773. doi: 10.1111/j.1432-1033.1988.tb14105.x. [DOI] [PubMed] [Google Scholar]
- 42.Hornsby P J, Crivello J F. Mol Cell Endocrinol. 1983;30:1–20. doi: 10.1016/0303-7207(83)90197-1. [DOI] [PubMed] [Google Scholar]
- 43.Gohil K, Packer L, Lumen B, Brooks G A, Terblanche S E. J Appl Physiol. 1986;60:1986–1991. doi: 10.1152/jappl.1986.60.6.1986. [DOI] [PubMed] [Google Scholar]
- 44.Kanter M M, Nolte L A, Holloszy J. J Appl Physiol. 1993;74:965–969. doi: 10.1152/jappl.1993.74.2.965. [DOI] [PubMed] [Google Scholar]
- 45.Goldfarb A H. Med Sci Sports Exercise. 1993;25:232–236. [PubMed] [Google Scholar]
- 46.Schroder H, Navarro E, Tramullas A, Mora J, Galiano D. Int J Sports Med. 2000;21:146–150. doi: 10.1055/s-2000-8870. [DOI] [PubMed] [Google Scholar]
- 47.Gleeson M, Robertson J, Maughan R J. Clin Sci. 1987;73:501–505. doi: 10.1042/cs0730501. [DOI] [PubMed] [Google Scholar]
- 48.Robertson J D, Maughan R J, Duthie G G, Morrice P C. Clin Sci. 1991;80:611–618. doi: 10.1042/cs0800611. [DOI] [PubMed] [Google Scholar]
- 49.Ginsburg G S, Agil A, O'Tool M, Rimm E, Douglas P S, Rifel N. J Am Med Assoc. 1996;276:221–225. [PubMed] [Google Scholar]
- 50.White T, Estrada M, Walker K A, Wisnia P, Filgueria G, Valdes F, Araneda O, Behn C, Martinez R. Comp Biochem Physiol. 2001;128:99–104. doi: 10.1016/s1095-6433(00)00286-5. [DOI] [PubMed] [Google Scholar]
- 51.Gohi K, Viguie C, Stanley W C, Brooks G A, Packer L. J Appl Physiol. 1988;64:115–119. doi: 10.1152/jappl.1988.64.1.115. [DOI] [PubMed] [Google Scholar]
- 52.Hinchcliff K W, Reinhart G A, Disilvestro R, Reynolds A, Blostein-Fujii A, Swenson R A. Am J Vet Res. 2000;61:512–517. doi: 10.2460/ajvr.2000.61.512. [DOI] [PubMed] [Google Scholar]
- 53.Snow D. In: Ascorbic Acid in Domestic Animals: Proceedings of the 2nd Symposium, October 1990, Kartaus Ittingen, Switzerland. Wenk C, Fenster R, Völker L, editors. 1990. pp. 221–242. [Google Scholar]
- 54.Hellsten Y, Tullson P C, Richer E A, Bangsbo J. Free Radical Biol Med. 1997;22:169–174. doi: 10.1016/s0891-5849(96)00286-9. [DOI] [PubMed] [Google Scholar]
- 55.Chevion S, Roberts M A, Chevion M. Free Radical Biol Med. 2000;28:860–870. doi: 10.1016/s0891-5849(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 56.Wayner D D M, Burton G W, Ingold K U, Barclay L C, Locke S J. Biochem Biophys Acta. 1987;924:408–419. doi: 10.1016/0304-4165(87)90155-3. [DOI] [PubMed] [Google Scholar]
- 57.Rasanen L, Wiitanen P A, Lilius E M, Hyyppa S, Witztum J L. Comp Biochem Physiol B. 1996;114:139–144. doi: 10.1016/0305-0491(96)00022-3. [DOI] [PubMed] [Google Scholar]
- 58.Mastaloudis A, Leonard S W, Traber M G. Free Radical Biol Med. 2001;31:911–922. doi: 10.1016/s0891-5849(01)00667-0. [DOI] [PubMed] [Google Scholar]
- 59.McCord J M. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 60.Bacharach D W, Petit M, Rundell K W. J Strength Con Res. 1996;10:105–108. [Google Scholar]
- 61.Raastad T, Hallen J. Eur J Appl Physiol. 2000;82:206–214. doi: 10.1007/s004210050673. [DOI] [PubMed] [Google Scholar]
- 62.Child R, Brown S, Day S, Donnely A E, Roper H P, Saxton J M. Clin Sci. 1999;96:105–115. [PubMed] [Google Scholar]