

Abstract
Synthetic hydrogels have been molecularly engineered to mimic the invasive characteristics of native provisional extracellular matrices: a combination of integrin-binding sites and substrates for matrix metalloproteinases (MMP) was required to render the networks degradable and invasive by cells via cell-secreted MMPs. Degradation of gels was engineered starting from a characterization of the degradation kinetics (kcat and Km) of synthetic MMP substrates in the soluble form and after crosslinking into a 3D hydrogel network. Primary human fibroblasts were demonstrated to proteolytically invade these networks, a process that depended on MMP substrate activity, adhesion ligand concentration, and network crosslinking density. Gels used to deliver recombinant human bone morphogenetic protein-2 to the site of critical defects in rat cranium were completely infiltrated by cells and remodeled into bony tissue within 4 wk at a dose of 5 μg per defect. Bone regeneration was also shown to depend on the proteolytic sensitivity of the matrices. These hydrogels may be useful in tissue engineering and cell biology as alternatives for naturally occurring extracellular matrix-derived materials such as fibrin or collagen.
Keywords: extracellular matrix‖biomaterials‖proteolytic degradation
Advances in the field of tissue engineering are connected to the performance of biomaterials that help in guiding tissue formation or regeneration. Design principles in biomaterials have typically been influenced by the function of the extracellular matrix (ECM). Because the ECM has been demonstrated to play a key role in signal transduction (1–3), the development of materials that can specifically and molecularly interact with cells has become an emerging area of research (4, 5). The regulation of cell behavior through receptor-mediated adhesion [e.g., by functionalizing materials with integrin-binding oligopeptides (6, 7)] and by growth factors (8) has thus been targeted.
We and others have been focusing on the development of synthetic materials that are targeted to assist tissue regeneration (9–11). Placed at the site of a defect, such materials should actively and temporarily participate in the regeneration process by providing a platform on which cell-triggered remodeling could occur. Consequently, these matrices must display some key characteristics of the provisional ECM. One of the critical initial functions of the fibrin-rich network that fills tissue defects after trauma, almost regardless of the injury site, lies in its ability to foster the invasion of inflammatory cells. These cells then initiate the remodeling process by partially degrading the matrix and by secreting molecular signals for attraction and differentiation of other cell types such as fibroblasts that build up new ECM (12). Because the provisional ECM presents itself in such situations often as a biophysical barrier to these cells, invasion and remodeling depend on the action of cell-secreted proteases enabling cell migration by clearing of a path (13, 14). Thereby, matrix metalloproteinases (MMPs) have been implicated as key players (15, 16). Secreted as inactive proenzymes and activated near the cell surface or expressed at the surface in activated form as membrane-anchored MMPs (MT-MMP), these enzymes can cleave virtually all constituents of the ECM at specific sites. Besides their role in migration, they affect other cell functions such as proliferation and apoptosis (17).
The objective of this work is to develop synthetic materials that can assist tissue regeneration by mimicking the MMP-mediated invasion of the natural provisional matrix. Because 3D migration through dense ECMs involves a complex integration of both adhesion (18) and proteolytic degradation (14, 19) events, these synthetic materials must display, as a minimum, molecular signals for cell adhesion and sites for proteolytic breakdown. This was accomplished by crosslinking linear oligopeptide substrates for MMPs into 3D networks on reaction with multiarm end-functionalized poly(ethylene glycol) (PEG) macromers (20). Together with integrin-binding domains (here, RGDSP) attached in a pendant fashion to hydrogels, cells were anticipated to be able to migrate within these networks by MMP- and integrin-dependent mechanisms (21).
To function effectively as a cell-ingrowth matrix in vivo, the degradation behavior by MMPs and the cell invasion behavior of these artificial provisional matrices, respectively, must be well understood and optimized. In the present study, we therefore examined how the molecular composition of the material (variables: adhesiveness, sensitivity to MMPs, and network architecture) influenced its 3D cellular invasion. By using both in vitro and in vivo assays, we demonstrate that the proteolytic degradation of these hydrogel networks can be rationally controlled on multiple levels.
Materials and Methods
Determination of Degradation Kinetics of Soluble Peptides by MMP-1 Using Michaelis–Menten Analysis.
The kinetic parameters of substrate hydrolysis (Km and kcat) were measured by using a fluorometric assay that has been published (22). Briefly, MMP-1 (40 nM, kind gift from H. Nagase, Imperial College of Science, Technology and Medicine, London) was incubated with individual substrates at 30°C in buffer solution followed by reaction with fluorescamine and detection of the fluorescence (λexcitation = 387 nm, λemission = 480 nm) on a Perkin–Elmer spectrofluorometer. Initial reaction velocities under first-order conditions ([S]o ≪ Km) were obtained from plots of fluorescence versus time, by using only data points corresponding to <40% hydrolysis. The slope from these plots was divided by the fluorescence change corresponding to complete hydrolysis and then multiplied by the substrate concentration to obtain initial velocity in units of micromolar per second. Kinetic parameters (Km and kcat) were determined by Lineweaver–Burk analysis, i.e., transforming the data to double reciprocal plots (1/[S] vs. 1/vi).
Hydrogel Synthesis.
In a typical gel preparation, 4arm-PEG-tetravinyl sulfone (20 kg/mol) (base materials and functionalization are published as Supporting Materials and Methods in supporting information on the PNAS web site, www.pnas.org) was dissolved in triethanolamine-buffered saline (0.3 M, pH 8.0) to give a 10% (wt/vol) solution. The integrin-binding peptide Ac-GCGYGRGDSPG (in the same buffer) was added to this solution. After 10 min, triethanolamine-buffered saline containing a bis-cysteine peptide with a highly MMP-sensitive sequence [e.g., Ac-GCRD-GPQG↓IWGQ-DRCG; herein referred to as MMP(W)]X, the X indicating participation in a crosslinked network) a moderately MMP-sensitive sequence [e.g., Ac-GCRD-GPQG↓IAGQ-DRCG; MMP(A)X], or an MMP-insensitive sequence [Ac-GCRD-GDQGIAGF-DRCG; (DF)X] was added. Gelation occurred within a few minutes; however, the crosslinking reaction was continued for ≈30 min at 37°C. Abbreviations for peptides and PEG macromers used throughout the work can be found at the bottom of Table 1.
Table 1.
Hydrolysis of peptide substrates by MMP-1 in soluble and gel form
| Peptides/gels | kcat, s−1 | Km, μM | kcat/Km, M−1⋅s−1 |
|---|---|---|---|
| MMP(W) | 0.54 ± 0.11* | 620 ± 160* | 870 ± 170* |
| MMP(W)x | 0.51 ± 0.10* | 290 ± 30* | 1,760 ± 280* |
| mPEG750-MMP(W)x | 0.64 ± 0.13† | 300 ± 60† | 2,130 ± 430† |
| mPEG5000-MMP(W)x | 0.83 ± 0.17† | 860 ± 170† | 970 ± 190† |
| MMP(A)x | 0.17 ± 0.03† | 430 ± 90† | 400 ± 80† |
| (DF)x | ND | ND | ND |
| 4PEG20-MMP(W)x | 1.21 | ND | ND |
| 4PEG20-MMP(A)x | 0.40 | ND | ND |
ND, not determined. MMP(W) corresponds to the peptide Ac-GPQG↓IWGQ-NH2; MMP(W)x is Ac-GCRD-GPQG↓IWGQ-DRCG-NH2; MMP(A)x is Ac-GCRD-GPQG↓IAGQ-DRCG-NH2; (DF)x is Ac-GCRD-GDQGIAGF-DRCG-NH2⋅mPEG750; and mPEG5000 refers to attached PEG chains of molecular mass 750 and 5,000 Da, respectively. 4PEG20-MMP(W)x corresponds to hydrogel networks synthesized by 4arm-PEG-vinyl sulfone of molecular weight 20 kg/mol and the above bis-Cys-peptide MMP(W)x.
Standard deviation (n = 3).
On the basis of the largest relative error of above measurements, MMP(W) and MMP(W)x.
Cell Culture and 3D Cell Invasion.
Human foreskin fibroblasts (HFFs) (neonatal normal human dermal fibroblast, Clonetics, San Diego) were cultured according to ref. 10. For invasion experiments, HFFs were used at passages 10–14. A previously described cell invasion assay (9) was used to quantify cell-induced proteolytic activity of the gels. Cell-loaded fibrin clots (3 × 107 cells per ml) were embedded within 10 μl of hydrogel discs by placing clusters into precursor solution before gelation (n = 9–12 per condition) and samples cultured in serum-containing DMEM for up to 20 d. Cells were imaged by inverted phase–contrast microscopy and confocal scanning laser microscopy according to the methods described in Supporting Materials and Methods.
Bone Regeneration in the Rat Cranium and Analysis of Healing.
Eight-millimeter-diameter craniotomy defects were created with a trephine in a dental handpiece, carefully avoiding dural perforation. The surgical area was flushed with saline to remove bone debris and a preformed PEG-based gel (≈100-μl swollen volume) with physically entrapped BMP-2 (5 μg) was placed within the defect. BMP-2 was prepared as described previously (23). Sprague–Dawley albino rats (84–92 d old, female) were killed by CO2 asphyxiation 4 wk after implantation (n = six per treatment). Craniotomy sites were recovered from the skull and fixed in ethanol. Specimens were radiographed, x-ray image photographed, scanned, and then assessed for radioopacity within a standard 8-mm-diameter circle superimposed over the defect site by using nih image software (1.62f). Samples were histologically processed according to a previously described method (24).
Statistics.
Statistical analysis was performed by using statview 4.5 (Abacus Concepts, Berkeley, CA) by unpaired nonparametric Mann–Whitney tests at a 95% confidence level. Mean values and standard deviations are shown.
Results
Characterization of Soluble MMP Substrates as Network Building Blocks.
Bioactive networks were created by reacting molecular building blocks bearing either structural (PEG) or biological (oligopeptides) function (Fig. 1). When bis-cysteine peptides have reacted with PEG end-groups on both sides, they become part of a crosslinked network structure. As a result, lysis of these building blocks will lead to degradation of the entire network when they contain (for example) substrates for MMPs, and the gels are subjected to the action of these enzymes.
Figure 1.

A Michael-type addition reaction between vinyl sulfone-functionalized multiarm PEGs and mono-cysteine adhesion peptides (step 1, in high stoichiometric deficit) or bis-cysteine MMP substrate peptides (step 2, to come up to stoichiometric equivalence) was used to form gels from aqueous solutions in the presence of cells. These elastic networks were designed to locally respond to local protease activity at the cell surface (step 3).
The linear oligopeptide MMP substrate GPQG↓IAGQ derived from the α1(I) collagen chain of calf and chick as well as human (25) and its mutated version GPQG↓IWGQ were chosen as MMP-sensitive building blocks. Single substitutions of this sequence, such as the exchange of Ala with Trp, led to wide variations in degradation kinetics as summarized in (26). Many MMP family members can cleave these octapeptides at the Gly-Ile bond. Charged amino acids (Arg and Asp) and Cys were added on both sides of these sequences to increase water solubility and to enable reaction with conjugated unsaturated moieties such as vinyl sulfones, respectively. As a negative control, the human α1 type IV collagen sequence GDQGIAGF was chosen and was described to be insensitive to collagenase degradation (27).
Individual kinetic parameters for MMP-1 (as a representative member of the MMP family) hydrolysis of the above peptides with and without grafted PEG chains on both sides were determined by Michaelis–Menten analysis. Substrates were PEGylated for analysis in a local environment similar to that within a crosslinked network, in which the PEG might sterically alter the formation of the substrate–enzyme complex and thus change the overall enzymatic activity of the system. Substrate degradation was followed by fluorescamine reaction, and kinetic parameters kcat and Km were obtained by using Lineweaver–Burk plots. All double-reciprocal plots were linear over the entire range of substrate concentrations studied, indicating that Michaelis–Menten kinetics was obeyed, as shown in the example of Fig. 2. Modifications of the short MMP substrate GPQG↓IWGQ, by addition of charged amino acids and Cys or by PEGylation, both changed the kinetic parameters (Table 1). The bis-Cys-peptide had a significantly higher Km than its shorter version. The substitution of Ala by Trp caused a change of both Km (decrease by 30%) and kcat (increase by 300%). The enzymatic activity, denoted as kcat/Km, differed by a factor of almost 5. The degradation rate of the peptide (DF)X was below measurable values. PEGylation of the peptides changed the degradation characteristics depending on the PEG molecular mass: increasing the chain length from 750 to 5,000 Da led to an increase of Km, likely due to steric hindrance. The proteolytic sensitivity (kcat) of MMP-1 for a specific substrate was only slightly dependent on the presence of PEG chains flanking the substrate.
Figure 2.

Kinetic analysis of MMP-1 degradation of two oligopeptides, GCRD-GPQG↓IAGQ-DRCG (●) and GCRD-GPQG↓IWGQ-DRCG (□), bearing MMP substrates flanked by charged amino acids and a Cys on both sides. Degradation was followed fluorometrically by the reaction of the free amine group with fluorescamine. Lineweaver–Burk plots yielded the corresponding kinetic parameters kcat and Km, for the enzymatic reaction.
Hydrogel Degradation Is Independent of Km.
After the study in soluble models as described above, we examined the degradation of these substrates by MMP-1 when crosslinked into a 3D network. Quantitative analysis of peptide bonds by fluorescamine reaction yielded a linear increase of the fluorescent signal with time for both peptides (Fig. 3A). Apparently, the enzyme is acting under vmax conditions, implying the degradation rate to be zero-order. Hence, the kinetics did not depend on the substrate concentration in the hydrogel for these two substrate/MMP-1 pairs. Because the substrate concentration within the gel (3.67 mM) was much higher than Km for the previously determined peptides (0.3–0.9 mM), the Michaelis–Menten conditions were not fulfilled, and the zero-order hydrolysis rate was to be expected. Under zero-order reaction conditions, the enzyme is fully saturated with substrate and kcat in the gel phase was calculated according to
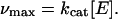 |
1 |
In comparison to the substrates in the soluble phase, kcat was found to be higher when crosslinked into a solid network (Table 1 Lower): 1.21 s−1 compared with 0.5 s−1 for soluble MMP(W)x, and 0.40 s−1 compared with 0.17 s−1 for soluble MMP(A)x, respectively. Relative to each other, kcat values of both sequences did not change when compared in the soluble and gel phase.
Figure 3.

Degradation kinetics of hydrogels respond to the MMP activity of the incorporated substrates: □, MMP(W)X; ●, MMP(A)X; ▵, (DF)X. Kinetics was measured by a quantitative fluorescamine assay yielding a linear increase of the fluorescent signal with time for both sample types (A) and by conducting swelling measurements (i.e., the volume increase of the gel samples) (B).
As cleavage of individual peptide bonds by MMP-1 occurred (Fig. 3A), the overall crosslink density of the network was decreased, perturbing the balance between hydration of PEG chains and elastic forces exerted by the immobilized crosslinks. Gels responded macroscopically by acquiring a new equilibrium between these forces, manifesting itself in an increase in volume during degradation (Fig. 3B), first swelling and then passing through a point of dissolution into high molecular weight soluble products (28). The observed increase in swelling over time, rather than degradation at the material surface, implied that bonds were cleaved homogeneously throughout the entire network. Hence, the degradation of these networks by MMP-1 occurred predominantly by bulk degradation rather than by a surface-erosion mechanism. The individual MMP substrates within the gel are cleaved according to the zero-order kinetic equation
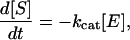 |
2 |
and hence,
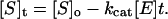 |
3 |
[S]o is the concentration of substrate within the intact gel. The limit where degradation of substrates has advanced sufficiently, such that the gel can no longer form a network, can be calculated by using a statistical network formation model such as the one derived by Macosko and Miller (29) in a reversed sense (for more detailed information, see Supporting Materials and Methods). Following this approach and combining with Eq. 3, the time at gel breakdown (tc) can be predicted:
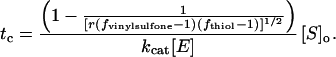 |
4 |
Hence, tc is a function of the network architecture (fvinylsulfone, fthiol as the functionalities of the precursor components and r as the molar ratio of vinyl sulfone to thiol groups; the latter is equal to 1 in this stoichiometrically balanced system), the substrate concentration (i.e., the crosslinking density, as seen through [S]o), the kinetic constant (kcat), and the enzyme concentration [E]. Eq. 4 predicts a tc for 4PEG20-MMP(W)x of 8.9 h (with kcat = 1.21 s−1 and [E] = 40 nM) and 27 h for 4PEG20-MMP(A)x (kcat = 0.4 s−1), respectively. For the former gels, the experiment yielded degradation times of ≈11 h, for the latter ≈24 h. Because the underlying theoretical model to arrive to Eq. 4 was based on ideal networks, i.e., gels having no defects such as pendant chains or entanglements, the match of theoretical and experimental values appears reasonable.
Cell Invasion: Degradation of Gels by Cell-Derived MMPs.
HFFs entrapped in fibrin and polymerized as clusters within the gels grew out radially from clusters into matrices of 4PEG20-MMP(W)X-RGD, at an RGD ligand density of 85 μM in the swollen gel (Fig. 4A). In contrast to the spread and polygonally shaped cells on the surface of these materials, invading fibroblasts typically assumed spindle-like shapes. Confocal laser microscopy with double staining of cell membranes and nuclei revealed that the migration of these fibroblasts occurred in a cohort manner (30) (Fig. 4B). Hence, the cells moved en masse by keeping cell–cell contacts clearly visible by the lining up of single cell nuclei (stained with 4′,6-diamidino-2-phenylindole) into cord-like structures. Invasion distances increased approximately linearly with culture time (Fig. 4C) reaching levels up to >1 mm within 1 week (corresponding to an invasion rate of ≈7 μm/hr).
Figure 4.

(A) Fibroblasts radially invaded the adhesive and MMP-sensitive synthetic hydrogel matrix (bar = 250 μm). (B) Migration of spindle-like-shaped fibroblasts occurred in a cohort manner (bar = 150 μm). (C) Cell invasion distances increased approximately linear with culture time. (D) Cell invasion rate depended on the proteolytic activity of the incorporated peptide substrates. (E) Migration rate depended on adhesion ligand density, i.e., the concentration of RGD-containing peptide sites fixed throughout the three-dimensional material, in a biphasic manner. (F) Crosslink density of hydrogels influenced cell invasion dramatically.
Cell Invasion Depends on the MMP Sensitivity of the Networks.
As anticipated from the biochemical measurements on soluble peptides and gels described above, cellular invasion responded to the enzymatic sensitivity of peptides (Fig. 4D), at constant RGD ligand density and network structure. Gels containing the peptide MMP(W)X (kcat = 1.21 s−1) allowed significantly higher 3D cell invasion rates compared with MMP(A)X (kcat = 0.4 s−1). When the peptide linker was MMP-insensitive, i.e., (DF)X, little invasion was measured (on average ≈100 μm in 1 wk, corresponding to 0.6 μm/hr). Thus, 3D migration was predominantly controlled by MMP-mediated gel degradation. The addition of a broad-spectrum MMP inhibitor (GM6001, Chemicon, 25 μM) to the medium produced similar inhibition. The remaining migration could be due to the action of other proteases or the presence of microscopic network defects.
Cell Invasion Rate Depends on RGD Ligand Density in a Bimodal Manner.
The RGD ligand concentration (tested at eight different concentrations ranging from 2.5 to 340 μM) mediated cell invasion in a biphasic manner (Fig. 4E) with the incorporation of 42.5 and 85 μM RGD significantly enhancing the level of HFF outgrowth (P < 0.05, compared with 10 and 140 μM, respectively). Matrices that were not functionalized with adhesion ligands or that contained low ligand concentrations (2.5 and 5 μM) did not support cell invasion at all, because cells, due to the weak cell–matrix interaction, were not able to exert sufficient traction for forward movement. A ligand density of 10 μM (i.e., the concentration of RGD peptides fixed three-dimensionally within the gel network) was found to be the minimal concentration necessary for HFF invasion.
Network Crosslink Density Is a Critical Regulator of Cell Invasion.
Cell invasion into synthetic gels also depended on the structure of the networks (Fig. 4F). At a constant ligand density of 85 μM (achieved by adjusting the RGD concentration in the precursor solution by correcting for the difference in swelling of the matrices) and for the same linking peptide MMP(W)X, invasion rates were found to dramatically decrease with crosslink density. Lowering the PEG molecular mass from 20 to 15 kDa or the volumetric swelling ratio Q from ≈72 to 47, respectively, decreased the invasion rate by almost a factor of 4. More highly crosslinked networks 4PEG10-MMP(W)X-RGD (Q ≈ 40) completely impaired invasion.
Bone Healing in the Rat Cranium.
In an exploration of the relations from molecular design to performance in vivo, synthetic hydrogels were applied as carriers for BMP-2 to mediate repair of critical size defects in rat calvaria. BMP-2 was noncovalently incorporated during gel crosslinking. We have previously demonstrated that BMP-2 within such materials is kept precipitated by the presence of PEG and is not released over periods up to 5 d when not exposed to MMPs; MMP exposure triggers degradation of the network, which allows BMP-2 dissolution and release (21). Synthetic gels of the compositions 4PEG20-MMP(W)X-RGD loaded with 5 μg BMP-2 per implant of ≈100 μl were completely infiltrated by cells and showed intramembranous bone formation after 4 wk of implantation (Fig. 5C). In all of the samples treated with this material, complete bridging of the defect was observed. In no animals were examples of chronic inflammation observable; indeed, when cell invasion-enabling materials were used, the materials were either completely or nearly resorbed by this time point, and even when materials were used that did not permit cell invasion, inflammation was not apparent.
Figure 5.

Healing of critical size rat calvarial defects 4 wk after implantation. The ability of various matrices to promote healing was assessed by radiography (A–C Left) and histology (A–C Right). The healing outcome of the excised samples is reported as percentage of the total area of the original defect covered by mineralized tissue. Healing in vivo depended on the enzymatic sensitivity of the incorporated substrate: gels that were not susceptible to degradation by MMPs [A, the gel (DF)X] or moderately sensitive [B, the gel MMP-(A)X] showed significantly less bone formation than was observed in materials that were highly susceptible to invasion [C, the gel MMP-(W)X]. This relationship was also observed quantitatively by radiography [D, P < 0.01 for both (DF)X and MMP-(A)X relative to MMP-(W)X].
Bone Regeneration Depends on the MMP Sensitivity of the Networks.
Proteolytic sensitivity of the matrices was critical for remodeling and bone formation. Gels that were previously shown to be insensitive to MMP-mediated cell invasion [4PEG20-(DF)X-RGD] showed significantly less healing as quantified by radiography (Fig. 5 A and D). The slower degrading peptide MMP(A)X (kcat = 0.4 s−1) led to significantly less cell infiltration (Fig. 5B) and bone formation close to the surface; healing scored by radiography was not statistically different. Thus, the healing response in vivo depended on the enzymatic sensitivity of the incorporated substrate and correlated well with the invasive behavior of fibroblasts presented in the previous in vitro results.
Discussion
A New Class of Bioactive Materials for Tissue Regeneration.
MMPs have been implicated in many physiologic and pathologic processes of ECM turnover. Tissue regeneration is one of the examples where the primary function of these enzymes is to break down the proteins of the provisional ECM barrier to permit cell migration. Here we demonstrate that synthetic materials can be created that are cell-adhesive and susceptible to cell-secreted MMPs, enabling cells to invade these networks. In the example of a critical size defect model of calvarial bone, we have shown that such material characteristics can be exploited in conducting bone regeneration. In contrast to traditional synthetic biomaterials, which are either permanent or degrade via chemical pathways (such as nonenzymatic hydrolysis of ester bonds), the cell-mediated mechanism of matrix breakdown and invasion, respectively, occurred in the biomimetic matrices in temporal and spatial synchrony with endogenous bone regeneration. MMP sensitivity was found to be critical for this process. These hydrogels may be useful in such applications as alternatives for naturally occurring ECM-derived materials such as fibrin or collagen, which require difficult purification procedures and carry the risks of immunogenicity and disease transmission.
Not only did we demonstrate the potential of such materials in a therapeutic context, but we have also shown that the synthetic strategy of material fabrication allowed for engineering its properties such that more fundamental aspects of cell–matrix interactions could be systematically investigated. To optimize in vivo cell infiltration, in vitro model systems were used toward a better understanding of the mechanisms involved in MMP-mediated matrix degradation, without the additional complexities of the in vivo situation.
Hydrogel Degradation by Cell-Derived MMPs.
Primary human fibroblasts were able to invade these completely artificial matrices. However, only a finely orchestrated combination of network components allowed these cells to do so. Due to the homogeneous microstructure with mesh sizes (indicative of the distance between crosslinks) that are one to two orders of magnitude smaller than cellular structures (not including network defects), cell movement depended critically on the action of cell-secreted MMPs. Invasion rates responded to the proteolytic activity of the incorporated peptides. A quantitative correlation between the gel degradation kinetics by MMP-1 as assessed by fluorescamine or swelling measurements and the cell invasion rate is not possible at this point, because many other MMPs are secreted by these cells within gels (predominantly the gelatinases MMP-2, MMP-9, and MMP-3; M.P.L. and G. P. Raeber, unpublished results) that will be able to cleave the substrates with different kinetics. However, based on the data published previously for these sequences (26), it appears that cleavage of the Trp-containing substrate is more efficient by all MMP members that have been tested (MMP-1, -2, -3, -7, -8, and -9). Thus, we cannot conclude at the moment which particular MMP or MMPs are involved in cellular invasion in this model, but we can conclude at least that one or more of the MMPs are involved. An MMP-independent pathway for cellular invasion within collagen matrices in vitro has been observed (31), and it would appear that such pathways are not present in the gel materials reported herein. This difference is likely due to the very small mesh size of these synthetic networks, one to two orders of magnitude smaller than the typical length scale of cellular processes (20), and is the subject of continued study (G. P. Raeber, M.P.L., and J.A.H., unpublished observations).
The influence of adhesion ligand concentration on cellular invasion was also analyzed. Similar to previously published (both experimental and theoretical) reports for cell migration on surfaces (18, 32) and within 3D matrices (33–36), a biphasic dependence of migration on ligand density was found. In contrast to cell migration across surfaces, 3D cell migration is more complex, as it involves additional cellular strategies to overcome the biophysical resistance of the matrix (37). The latter may include cell shape changes or proteolytic degradation, depending on the type of cell and matrix. The complexity of studying 3D cell migration as a function of a single network parameter, such as concentration of adhesion ligands, is delicate, because it can be done only with matrices that allow controlling one variable independent of any others. In this work, migration was studied as a function ligand concentration in an RGD concentration range that did not affect the network crosslink density and therefore matrix microstructure or MMP substrate concentration. However, to conclude that the observed migration behavior depended solely on ligand density, we had to assume that the level of cell-secreted MMPs was not changed on variation of the ligand concentration, an assumption that needs to be experimentally tested, because it is known that MMP regulation can also occur by integrin binding [e.g., by αvβ3, the main RGD-binding integrin (38)].
The crosslinking density of the networks was shown to dramatically alter cell invasion behavior. Interestingly, this response was not linear with PEG structure (crosslink density), because relatively dense networks (composed of 4arm-PEG-10kDa) completely blocked invasion. It appears thus that the level of active MMPs produced by the cells responded to the network microarchitecture, a hypothesis that remains to be further investigated.
Cell invasion was always observed to be linear over time. If diffusion of active MMPs and degradation away from the cell front would be significant, a change (presumably an increase) in invasion rate over time should be expected (assuming MMP levels to be constant). Noteworthy, in all experiments the gels were intact even after culture periods up to 1 month, implying that the cell-triggered proteolysis was precisely localized to the cell periphery. Either the presence of membrane-anchored MMPs (MT-MMPs) (14), a localization of MMP-2 to the cell surface by interaction with RGD-binding integrin αvβ3 (39), or a tightly regulated balance between active MMPs and their natural inhibitors (TIMPs) could be responsible for such a localized degradation behavior in the synthetic matrix.
We have demonstrated that synthetic materials can be created that mimic MMP-mediated invasion of natural provisional ECM. These networks have a defined molecular architecture. Their synthesis scheme allows for an enormous flexibility and versatility in design. The biological character can be created on a passive PEG background (responsible for the physicochemical characteristics) de novo by incorporating any desired combination of biologically active peptides or proteins bearing reduced thiols. The concentration of the ligands can be controlled, and, although limited, the structural and biological properties of the networks can be designed independent from each other. Thus we believe that such networks, apart from their therapeutic potential, could become a useful tool in studying more fundamental aspects of cell–matrix interactions.
Supplementary Material
Acknowledgments
We thank Dr. J. C. Schense (Kuros Therapeutics, Zurich) for help with the animal work, G. P. Raeber (Institute for Biomedical Engineering) for help with the confocal laser-scanning microscopy, and Drs. M. Höchli and T. Bächi of the Electron Microscopy Laboratory at the University of Zurich for confocal scanning laser microscopy. This work was funded by grants from the Swiss National Science Foundation (to J.A.H.) and the National Institutes of Health (Grants CA 77402 and CA 98799 to G.B.F.).
Abbreviations
- ECM
extracellular matrix
- MMP
matrix metalloproteinases
- PEG
polyethylene glycol
- HFF
human foreskin fibroblast
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Howe A, Aplin A E, Alahari S K, Juliano R L. Curr Opin Cell Biol. 1998;10:220–231. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz M A, Baron V. Curr Opin Cell Biol. 1999;11:197–202. doi: 10.1016/s0955-0674(99)80026-x. [DOI] [PubMed] [Google Scholar]
- 3.Streuli C. Curr Opin Cell Biol. 1999;11:634–640. doi: 10.1016/s0955-0674(99)00026-5. [DOI] [PubMed] [Google Scholar]
- 4.Hubbell J A. Curr Opin Biotechnol. 1999;10:123–129. doi: 10.1016/s0958-1669(99)80021-4. [DOI] [PubMed] [Google Scholar]
- 5.Griffith L G. Ann NY Acad Sci. 2002;961:83–95. doi: 10.1111/j.1749-6632.2002.tb03056.x. [DOI] [PubMed] [Google Scholar]
- 6.Ruoslahti E. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 7.Hubbell J A. In: Principles of Tissue Engineering. Lanza R P, Langer R, Vacanti J, editors. San Diego: Academic; 2000. pp. 237–250. [Google Scholar]
- 8.Whitaker M J, Quirk R A, Howdle S M, Shakesheff K M. J Pharm Pharmacol. 2001;53:1427–1437. doi: 10.1211/0022357011777963. [DOI] [PubMed] [Google Scholar]
- 9.Pratt A B, Hubbell J A. Cell-Responsive Synthetic Biomaterials Formed in Situ. Pasadena, CA: California Institute of Technology; 2001. [Google Scholar]
- 10.Halstenberg S, Panitch A, Rizzi S, Hall H, Hubbell J A. Biomacromolecules. 2002;3:710–723. doi: 10.1021/bm015629o. [DOI] [PubMed] [Google Scholar]
- 11.Mann B K, Gobin A S, Tsai A T, Schmedlen R H, West J L. Biomaterials. 2001;22:3045–3051. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 12.Clark R A F. Molecular and Cellular Biology of Wound Repair. New York: Plenum; 1996. [Google Scholar]
- 13.Werb Z. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 14.Murphy G, Gavrilovic J. Curr Opin Cell Biol. 1999;11:614–621. doi: 10.1016/s0955-0674(99)00022-8. [DOI] [PubMed] [Google Scholar]
- 15.Woessner J F, Nagase H. Matrix Metalloproteinases and TIMPs. New York: Oxford Univ. Press; 2000. [Google Scholar]
- 16.Sternlicht M D, Werb Z. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vu T H, Werb Z. Genes Dev. 2000;14:2123–2133. doi: 10.1101/gad.815400. [DOI] [PubMed] [Google Scholar]
- 18.Lauffenburger D A, Horwitz A F. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 19.Ivaska J, Heino J. Cell Mol Life Sci. 2000;57:16–24. doi: 10.1007/s000180050496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lutolf, M. P. & Hubbell, J. A. (2003) Biomacromolecules, in press. [DOI] [PubMed]
- 21. Lutolf, M. P., Weber, F. E., Schmoekel, H. G., Schense, J. C., Kohler, T., Müller, R. & Hubbell, J. A. (2003) Nat. Biotechnol., in press. [DOI] [PubMed]
- 22.Lauer-Fields J L, Tuzinski K A, Shimokawa K, Nagase H, Fields G B. J Biol Chem. 2000;275:13282–13290. doi: 10.1074/jbc.275.18.13282. [DOI] [PubMed] [Google Scholar]
- 23.Weber F E, Eyrich G, Graetz K W, Thomas R M, Maly F E, Sailer H F. Biochem Biophys Res Commun. 2001;268:554–558. doi: 10.1006/bbrc.2001.5408. [DOI] [PubMed] [Google Scholar]
- 24.Weber F E, Eyrich G, Graetz K W, Maly F E, Sailer H F. Int J Oral Maxillofac Surg. 2002;31:60–65. doi: 10.1054/ijom.2001.0154. [DOI] [PubMed] [Google Scholar]
- 25.Aimes R T, Quigley J P. J Biol Chem. 1995;270:5872–5876. doi: 10.1074/jbc.270.11.5872. [DOI] [PubMed] [Google Scholar]
- 26.Nagase H, Fields G B. Biopolymers. 1996;40:399–416. doi: 10.1002/(SICI)1097-0282(1996)40:4%3C399::AID-BIP5%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 27.Babel W, Glanville R W. Eur J Biochem. 1984;143:545–556. doi: 10.1111/j.1432-1033.1984.tb08404.x. [DOI] [PubMed] [Google Scholar]
- 28.Metters A T, Bowman C N, Anseth K S. J Phys Chem B. 2000;104:7043–7049. [Google Scholar]
- 29.Macosko C W, Miller D R. Macromolecules. 1976;9:199–206. doi: 10.1021/ma60050a003. [DOI] [PubMed] [Google Scholar]
- 30.Nabeshima K, Inoue T, Shimao Y, Sameshima T. Pathol Int. 2002;52:255–264. doi: 10.1046/j.1440-1827.2002.01343.x. [DOI] [PubMed] [Google Scholar]
- 31.Wolf K, Mazo I, Leung H, Engelke K, Von Andrian U H, Deryugina E I, Strongin A Y, Brocker E B, Friedl P. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palecek S P, Loftus J C, Ginsberg M H, Lauffenburger D A, Horwitz A F. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- 33.Kuntz R M, Saltzmann W M. Biophys J. 1997;72:1472–1480. doi: 10.1016/S0006-3495(97)78793-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schense J C, Hubbell J A. J Biol Chem. 2000;275:6813–6818. doi: 10.1074/jbc.275.10.6813. [DOI] [PubMed] [Google Scholar]
- 35.Gobin A S, West J L. FASEB J. 2002;16:751–753. doi: 10.1096/fj.01-0759fje. [DOI] [PubMed] [Google Scholar]
- 36.Burgess B T, Myles J L, Dickinson R B. Annu Biomed Eng. 2000;28:110–118. doi: 10.1114/1.259. [DOI] [PubMed] [Google Scholar]
- 37.Friedl P, Broecker E-B. Cell Mol Life Sci. 2000;57:41–64. doi: 10.1007/s000180050498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Rodriguez D, Petitclerc E, Kim J J, Hangai M, Yuen S M, Davis G E, Brooks P C. J Cell Biol. 2001;154:1069–1079. doi: 10.1083/jcb.200103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brooks P C, Stromblad S, Sanders L C, von Schalscha T L, Aimes R T, Stetler-Stevenson W G, Quigley J P, Cheresh D A. Cell. 1996;85:683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.