

Abstract
Strict coordination of the two motor domains of kinesin is required for driving the processive movement of organelles along microtubules. Glutamate 164 of the kinesin heavy chain was shown to be critical for kinesin function through in vivo genetics in Drosophila melanogaster. The mutant motor E164K exhibited reduced steady-state ATPase activity and higher affinity for both ATP and microtubules. Moreover, an alanine substitution at this position (E164A) caused similar defects. It became stalled on the microtubule and was unable to bind and hydrolyze ATP at the second motor domain. Glu164, which has been conserved through evolution, is located at the motor-microtubule interface close to key residues on helix α12 of β-tubulin. We explored further the contributions of Glu164 to motor function using several site-directed mutant proteins: E164K, E164N, E164D, E164Q, and D165A. The results indicate that the microtubule-E164K complex can only bind and hydrolyze one ATP. ATP with increased salt was able to dissociate a population of E164K motors from the microtubule but could not dissociate E164A. We tested the basis of the stabilized microtubule interaction with E164K, E164N, and E164A. The results provide new insights about the motor-microtubule interface and the pathway of communication for processive motility.
Kinesin uses the energy of ATP hydrolysis to generate unidirectional processive movement along microtubules (1-4). The key to understanding the processivity of kinesin is to define the transition states and structural requirements for the coordination of the two catalytic motor domains with each other and with the microtubule. The nucleotide binding motifs of kinesin show both structural and sequence homology with G-proteins, myosins, and other kinesin superfamily members in amino acid sequence and in structural organization (5-17). In addition, these diverse enzymes exhibit a conserved mechanism in which the nucleotide state at the active site is translated, through switch I and switch II, into a series of conformational changes that are critical for function (18-34) (reviewed in refs 17, 35, and 36).
The neck linker of conventional kinesin has been shown to be a key motion-transducing element (13, 34, 37-41). ATP binding promoted neck linker docking onto the catalytic core in the direction toward the plus end of the microtubule, and ATP hydrolysis returned the neck linker to a more mobile conformation. The neck linker docking site includes parts of the motor domain near loop L12 (microtubule binding face) and switch II helix α4, which is involved in the communication between the active site and microtubule binding interface. On the basis of these and other results, it was proposed that the energy from ATP binding drives neck linker docking onto the catalytic core and that the structural transition causes a plus-end-directed swing of the neck linker that positions the partner motor domain forward onto the next microtubule binding site. In the context of the hand-over-hand mechanism, the forward head would initially be in a nucleotide state where the neck linker is mobile and not docked along the catalytic core, while the neck linker on the rearward head is docked. This configuration allows both motor domains to be microtubule bound and separated by 8 nm (39). These results viewed with an alternating site mechanism of ATP hydrolysis, mechanical data of load-dependent ATP state, and structural studies provide a model for force generation (20-22, 24-26, 42-46). Furthermore, kinetic analyses of switch I mutants (32, 33) suggested that ATP hydrolysis on one head is required to lock the forward head onto the microtubule tightly before the rearward head can detach from the microtubule. This mechanism would ensure that one head is always bound tightly to the microtubule throughout the ATPase cycle (Figure 1), thereby resulting in processive movement.
FIGURE 1.
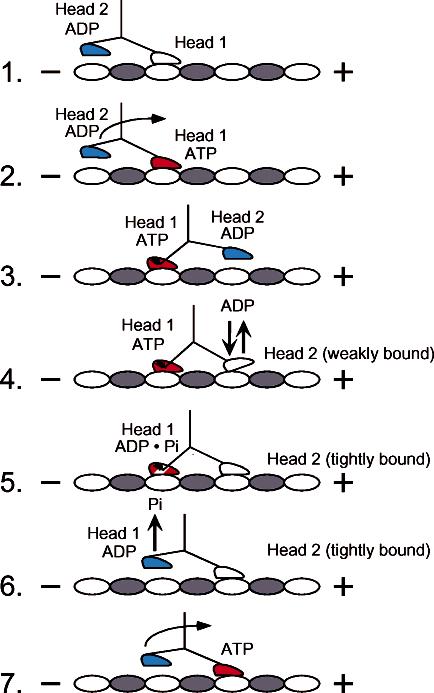
Alternating site model for kinesin stepping. The cycle begins as head 1 binds the microtubule with rapid ADP release. ATP binding at head 1 leads to the plus-end-directed motion of the neck linker to position head 2 forward at the next microtubule binding site. ATP binding at head 1 is sufficient to promote head 2 association with the microtubule followed by rapid ADP release. ATP hydrolysis at head 1 promotes tight binding of head 2 onto the microtubule, resulting in an intermediate with both heads strongly bound to the microtubule and with the neck linkers under mechanical strain. Phosphate is released, and the rearward head detaches from the microtubule. The active site of head 2 is now accessible for ATP binding, and the cycle is repeated.
We lack structural information about the microtubule-bound intermediates of kinesin during the ATPase cycle. Therefore, it is difficult to understand how the tight coupling of the ATPase and microtubule binding cycles within each motor domain and also between the two motor domains is governed. We have analyzed mutant dimeric kinesin motors to explore cooperativity between the two motor domains and to gain a better understanding of the motor-microtubule interface. Khc23 (E164K) was identified in a genetic screen for recessive lethal mutations in Drosophila melanogaster (47). This mutation disrupted axonal transport, and larval death occurred prior to emergence of any adult flies. In vitro, the E164K mutation caused striking defects in both ATP turnover and microtubule gliding, confirming the importance of Glu164 for kinesin function (47).
Glu164 is located at the boundary of β-strand 5a and loop L8b on the surface of the catalytic core of the kinesin heavy chain and is at the microtubule-motor interface (Figure 2) (48, 49). We examined this residue in the context of an alanine substitution (48). Our analysis revealed that the dimeric E164A motor stalled on the microtubule and was unable to detach from the microtubule. Moreover, only one site of the dimerized motor domains was able to bind and hydrolyze ATP. In an effort to dissect the details of the defect that led to this stalling, we evaluated a series of site-directed mutants at Glu164. The mutant motors analyzed were dimeric and included E164K, E164Q, E164N, and E164D as well as D165A, D165N, and D165E. On the basis of the results, we discuss the structural requirements and transitions that are essential for alternating site catalysis and transient interactions at the motor-microtubule interface.
FIGURE 2.

Docking model of the microtubule,kinesin complex. The potential interactions of Drosophila Glu164 (rat Glu158) at the β-sheet 5a/loop 8b junction with microtubule amino acids were explored by docking the coordinates of monomeric kinesin 2KIN (9) and α,β-tubulin (53) into the electron density map of microtubule·kinesin complexes obtained by cryo-EM (54). The kinesin motor domain is shown in red with R-tubulin in green. The β-tubulin subunit is shown in cyan with Taxol (yellow) bound, and the plus end of the microtubule is to the right. The neck linker (magenta) follows kinesin helix R6 and is shown docked onto the catalytic core and pointed toward the plus end of the microtubule. Note that Glu415, Met416, Glu417, and Phe418 are β-tubulin residues on helix α12 in close proximity to kinesin Glu164. Arg292 (human R284, rat R286) of kinesin helix α5 has been implicated in interactions with β-tubulin helix β12 as well as Glu164 (48, 49). The lower panels present the stereoview.
EXPERIMENTAL PROCEDURES
Protein Purification. The kinesin mutants were constructed by introducing single amino acid changes in the K401-wt1 plasmid, pET5b-K401 (50), using the Chameleon double-stranded site-directed mutagenesis protocol (Stratagene, Inc., La Jolla, CA). The mutations were verified by DNA sequencing, and the protein was expressed in Escherichia coli BL21(DE3)pLysS. K401-wt and K401 mutants were expressed, purified, and characterized as described previously (47, 50). The protein concentration was determined using the Bio-Rad protein assay with IgG as a protein standard. Although the kinesin motors are dimeric, the concentrations reported represent single motor domain or ATP binding site concentrations based on active site measurements (48).
Experimental Conditions. All experiments reported were performed in ATPase buffer (20 mM HEPES, pH 7.2, with KOH, 5 mM magnesium acetate, 0.1 mM EGTA, 0.1 mM EDTA, 50 mM potassium acetate, and 1 mM DTT) at 25 °C. The concentrations reported are the final concentrations after mixing.
Microtubule Preparation for Experiments. On the day of each experiment, purified tubulin was cold depolymerized, clarified, and assembled with Taxol. We report the tubulin concentration of the microtubules, which were stabilized with 20 μM Taxol, as the concentration after mixing.
Steady-State ATPase Assays. ATPase assays were performed at 25 °C in ATPase buffer at 50 mM potassium acetate by following the hydrolysis of [α-32P]ATP as described previously (51). The steady-state ATPase data as a function of ATP concentration were fit to the Michaelis-Menten equation to determine the kcat and the Km,ATP. The microtubule concentration dependence required the data be fit to the quadratic equation
| (1) |
where E represents the enzyme kinesin, E0) 0.5 μM kinesin, Mt is tubulin, and K0.5,Mt is the concentration of tubulin as microtubules required to provide one-half the maximal velocity.
Stopped-Flow Experiments. The pre-steady-state kinetics of mantATP binding (data not shown), microtubule association, ADP release from one or both heads, and detachment from microtubules were measured using a KinTek stopped-flow instrument (SF-2002; KinTek Corp., Austin, TX) at 25°C in ATPase buffer. N-Methylanthraniloyl fluorescence (mantATP and mantADP) was excited at 360 nm (mercury arc lamp) and detected at 450 nm using a 400 nm cutoff filter. MantATP binding data were fit to the linear function
| (2) |
where kobs is the rate constant from the exponential phase of the fluorescence enhancement, k+1 defines the second-order rate constant for mantATP binding, and koff corresponds to the observed rate constant of mantATP dissociation as determined by the y-intercept (Scheme 1). The association of the mutant motor with microtubules and dissociation of the Mt·E164K complex were monitored by changes in turbidity at 340 nm. The association data were fit to the linear function
| (3) |
where kobs is the rate of the initial exponential phase, k+5 defines the second-order rate constant for microtubule association, and k-5 corresponds to the observed rate constant of motor dissociation as determined by the y-intercept (Scheme 1).
Scheme 1.

For the ATP-promoted dissociation kinetics (Figure 6), the stopped-flow instrument was set to show the data during the first 1.4-2 ms of mixing. This experimental design facilitated the normalization of the transients to all begin at the same point on the y-axis. The normalization process mathematically adjusted the entire transient either up or down on the y-axis for direct comparison of each mutant. The amplitude and rate data for each transient were based on the fit of the data to two exponential functions and obtained prior to normalization.
FIGURE 6.

Dissociation kinetics of E164 mutants. A preformed Mt·E164 mutant complex (3 μM mutant, 2.9 μM tubulin, 20 μM Taxol) was rapidly mixed in the stopped-flow instrument with 1 mM MgATP plus 100 mM KCl, and a change in turbidity was monitored. Each transient was normalized to start at 0.14 V. There is an initial rapid drop in each transient during the first 3 ms that is attributed to mixing. Therefore, to fit the data and obtain amplitude information, the dissociation signal was considered to begin at 0.125 V. Note the variability in the amplitude of the transients, representing differences in motor detachment from the microtubule.
Acid Quench and Pulse-Chase Experiments. Pre-steady-state experiments to determine the kinetics of ATP binding and hydrolysis were performed with a rapid chemical quench-flow instrument (RQF-3; KinTek Corp., Austin, TX) at 25°C in ATPase buffer as described previously (48, 51, 52). For each time point, a preformed Mt·kinesin complex (5 μM kinesin, 18 μM tubulin, 20 μM Taxol) was reacted with [α-32P]ATP for times ranging from 5 to 400 ms. To measure ATP hydrolysis, the reaction mixture was quenched with 5 M formic acid and expelled from the instrument, and radiolabeled product was separated from substrate by thinlayer chromatography. The concentration of product was plotted as a function of time, and the data were fit to the burst equation
| (4) |
where A is the amplitude of the exponential burst phase, representing the formation of [α-32P]ADP·Pi at the active site during the first ATP turnover, kb is the rate constant of the pre-steady-state burst phase, kss is the rate constant of the linear phase and when divided by the enzyme concentration corresponds to steady-state turnover, and t is the time in seconds.
To investigate the reduced burst amplitude, pulse-chase experiments were performed such that the Mt·E164A,[α-32P]ATP reaction mixture was chased with 5 mM MgATP for 0.9 s and then quenched by 5 M formic acid. The chase allows enzyme-bound [α-32P]ATP to be converted to [α-32P]-ADP·Pi, yet any substrate unbound or bound loosely to the active site would be diluted by the excess unlabeled MgATP chase.
Molecular Docking. The atomic coordinates of the rat kinesin monomer 2KIN (9) and of α,β-tubulin (53) were computationally docked into the cryo-EM 3-D reconstruction of microtubules decorated with monomeric rat kinesin (54) using the software package O (55). Figure 2 was composed using Bobscript (56).
Sequence Analysis. The α-tubulin and β-tubulin sequences were obtained from NCBI Entrez and aligned with ClustalX v1.81(57) using the default parameters. The GENEDOC alignment editor (http://www.psc.edu/biomed/genedoc) was used to view and edit the alignments. The kinesin sequence alignments were obtained from the Kinesin Homepage (http://www.proweb.org/∼kinesin//).
RESULTS
Pre-Steady-State Kinetics of ATP Hydrolysis. To test the effects of the E164K mutation on ATP hydrolysis, we used a rapid chemical quench approach. Previous acid quench experiments for E164A proved to be informative, revealing fast ATP hydrolysis with only 13% of the available sites participating (48). Figure 3A shows the time course for ATP hydrolysis by E164K at five different ATP concentrations. Each transient is biphasic, with an initial fast exponential rate of ADP·Pi formation during the first ATP turnover (the burst), followed by a slower rate of product formation, which corresponds to steady-state turnover (the linear phase). The observation of an exponential burst of product formation is indicative that a step after ATP hydrolysis is rate-limiting for E164K as seen previously for K401-wt and E164A (48, 52). Figure 3B shows the burst rate plotted as a function of [α-32P]MgATP concentration. The fit of these data to a hyperbola provides the maximum rate constant of the burst phase at 187 s-1 and a Kd,ATP at 27 μM, which is comparable to the Km,ATP obtained from steady-state experiments. At high ATP concentrations, ATP binding becomes faster than ATP hydrolysis; therefore, this experiment provided the rate constant for ATP hydrolysis, k+2 187 s-1 (Scheme 1, Table 1). ATP hydrolysis for E164K is approximately double that of K401-wt, and the Kd,ATP reveals tighter binding of ATP by E164K than observed for K401-wt. Because ATP binding and ATP hydrolysis are so fast for E164K, the burst phase for the high ATP concentration transients was not well defined. The minimum time point attainable in the quench flow instrument is 3 ms. Therefore, the limitation of the instrument limits the precision of the rates that can be reported for both ATP hydrolysis (k+2, Figure 3) and the ATP-dependent conformational change (k+1’) detected by the pulse-chase experiments (Figure 5; see below). These rate constants must be viewed as estimates.
FIGURE 3.

Acid quench kinetics of ATP hydrolysis. A preformed Mt·E164K complex (5 μM E164K, 18 μM tubulin) was rapidly mixed with varying concentrations of [α-32P]MgATP in a chemical quench-flow instrument. (A) Transients for ATP hydrolysis in the presence of 10 μM (●), 20 μM (▲), 30 μM (○), 50 μM (△), and 200 μM (×) [α-32P]MgATP. The data were fit to eq 4. Only the first 100 ms of each transient is shown to expand the time domain of the initial, exponential burst phase. (B) The rate constants of the pre-steady-state burst phase determined for each transient in panel A were plotted as a function of [α-32P]MgATP concentration. The fit of the data to a hyperbola provides the maximum rate constant for the burst phase, kb) 187 ± 12 s-1 and Kd,ATP) 27 ± 7 μM. (C) The amplitudes of the pre-steady-state burst phase determined from each transient in panel A were plotted as a function of [α-32P]MgATP concentration. The data were fit to a hyperbola with 1.04 ± 0.2 μM as the maximum burst amplitude and Kd,ATP ) 82 ± 41 μM. Panels B and C include additional data obtained from experiments not shown in (A).
Table 1.
Microtubule-Kinesin Constantsa
| K401-wt simulationg | K401-wt | E164A | E164K | E164N | E164Q | E164D | D165A | |
|---|---|---|---|---|---|---|---|---|
| mantATP bindingb | ||||||||
| k+1 (μM-1 s-1) | 2 | 1.1b | 2.2 ± 0.1 | 1.3 ± 0.03 | ||||
| koff (s-1) | 9.8 | 20.6 ± 3.4 | 4.4 ± 0.7 | |||||
| acid quenchc | ||||||||
| k+2 (s-1) | 100 | 93c | 368 ± 20.4 | 187 ± 12 | ||||
| Kd,ATP (μM) | 87 | 16.5 ± 5 | 27 ± 7 | |||||
| amp | 0.55/site | 0.13/site | 0.21/site | |||||
| Kd,ATP (μM) | 59.1 ± 32.4 | 82 ± 41 | ||||||
| pulse−chasec | ||||||||
| k+1′ and k-1 (s-1) | 120 (k-1) | 239c | 521 ± 43.1 | 286 ± 31 | ||||
| Kd,ATP (μM) | 70 | 44.2 ± 12.6 | 19 ± 7 | |||||
| amp | 0.73/site | 0.34/site | 0.32/site | |||||
| Kd,ATP (μM) | 52.3 ± 12.2 | 40 ± 8 | ||||||
| ATP-promoted microtubule dissociationd | ||||||||
| k+3 (s-1) | 50 | 12-14d | 17.5 ± 0.5 | 37.4 ± 0.7 | 35.6 ± 3.1 | 47.4 ± 3.6 | 13.1 ± 0.6 | |
| K0.5,ATP (μM) | 5.7 ± 0.8 | 23.8 ± 2.6 | 39.1 ± 14.4 | 98.5 ± 23.9 | 22.3 ± 5.5 | |||
| dissociation | none | ∼63% | ∼34% | full | full | full | ||
| Pi releasee(s-1) | >150 | 13e | ||||||
| microtubule associationd | ||||||||
| k+5(μM-1 s-1) | 11 | 10-20d | 9.2 ± 0.6 | 16.5 ± 1.6 | 3.5 ± 0.24 | 4.8 ± 0.12 | 9.2 ± 0.6 | 14.04 ± 0.77 |
| koff(s-1) | 0.37 ± 3.3 | 19.4 ± 8.9 | 11.7 ± 1.4 | 8.7 ± 0.69 | 0.17 ± 3.5 | 3.2 ± 4.4 | ||
| mantADP release, both headsf | ||||||||
| release (s-1) | 300 | >200f | 71.6 ± 3.2 | 54.9 ± 1.8 | 81.9 ± 8.2 | 102.8 ± 11.3 | 120.3 ± 5.8 | 131.3 ± 20.8 |
| K0.5,Mt (μM) | 15 | 4.1 ± 0.7 | 5.5 ± 0.8 | 4.5 ± 1.8 | 20.5 ± 5.5 | 8.6 ± 1.4 | 18.9 ± 6.9 | |
| mantADP release, head 2f | ||||||||
| k+6 (s-1) (ATP) | 200 | >100f | 48.0 ± 1.3 | 32.8 ± 0.7 | 71.5 ± 1.7 | 133.02 ± 4.1 | 169.3 ± 5.9 | 75.6 ± 1.1 |
| K0.5,ATP(μM) | 11.7 ± 1.7 | 8.1 ± 0.96 | 13.8 ± 1.7 | 57.8 ± 6.2 | 78.3 ± 9.4 | 32.0 ± 2.5 | ||
| k+6(s-1) (ADP) | 6.6f | 35.0 ± 0.8 | 22.1 ± 0.7 | 24.3 ± 1.8 | 11.1 ± 0.85 | 11.1 ± 0.4 | 8.4 ± 0.68 | |
| K0.5,ADP (μM) | 24.4 ± 2.6 | 49.0 ± 7.1 | 126.2 ± 29.7 | 154.6 ± 35.7 | 68.6 ± 8.1 | 45.5 ± 17.7 | ||
| kcat (s-1) | 20.6 ± 0.9 | 4.9 ± 0.5 | 6.8 ± 0.6 | 7.8 ± 0.61 | 14.5 ± 1.5 | 13.5 ± 1.1 | 12.2 ± 0.73 | |
| Km,ATP (μM) | 94.4 ± 5.9 | 21.1 ± 2.6 | 21.3 ± 2.8 | 38.9 ± 3.07 | 73.3 ± 5.1 | 58.9 ± 5.5 | 110.4 ± 5.4 | |
| K0.5,Mt (μ) | 1.0 ± 0.05 | 0.42 ± 0.09 | 0.4 ± 0.2 | 0.26 ± 0.085 | 0.33 ± 0.02 | 0.77 ± 0.08 | 0.55 ± 0.115 |
Conditions: 20 mM HEPES, pH 7.2, with KOH, 5 mM magnesium acetate, 0.1 mM EGTA, 0.1 mM EDTA, 50 mM potassium acetate, and 1 mM DTT at 25 °C. Microtubule dissociation (see footnote d) and phosphate release (see footnote e) kinetics were determined at a final salt concentration of 150 mM.
MantATP binding (22).
FIGURE 5.

Pulse-chase kinetics of ATP binding. A preformed Mt·E164K complex (5 μM E164K, 18 μM tubulin) was rapidly mixed with varying concentrations of [α-32P]MgATP in a chemical quench-flow instrument, and the reaction times were varied from 5 to 400 ms followed by a 5 mM MgATP chase. The data were fit to eq 4. (A) Transients for ATP binding in the presence of 10 μM (●), 20 μM (▲), 30 μM (○), 50 μM (△), and 200 μM (×) [α-32P]-MgATP. (B) The rate constants of the pre-steady-state burst phase determined from each transient in (A) were plotted as a function of [α-32P]MgATP concentration. The fit of the data to a hyperbola provides the maximum rate constant, kb) 286 ± 31 s-1 and Kd,ATP) 19 ± 7 μM. (C) The amplitudes of the pre-steady-state burst phase determined from each transient in (A) were plotted as a function of [α-32P]MgATP concentration. The data were fit to a hyperbola with 1.6 ±0.1 μM as the maximum burst amplitude and Kd,ATP) 40 ±8 μM.
The burst amplitude, which represents the concentration of the Mt·K·ADP·Pi intermediate formed during the first ATP turnover, was plotted as a function of ATP concentration (Figure 3C). The maximum burst amplitude at 1.04 μM was significantly less than the concentration of 5 μM E164K active sites. Although this observation may represent a significant off rate for ATP as observed for K401-wt, this interpretation appears unlikely because of the tight binding of ATP. E164A also displayed tighter ATP binding and showed a burst amplitude significantly less than that of the enzyme concentration. For E164A, the low burst amplitude was attributed to only one motor domain of the dimer hydrolyzing ATP. Our next experiments were directed to determine the mechanistic basis of the reduced burst amplitude detected for E164K.
Pulse-Chase Kinetics. The pre-steady-state kinetics of ATP binding were investigated to explore the reduced burst amplitude in the acid quench experiments. A preformed Mt·E164K complex was rapidly mixed with [α-32P]MgATP in the chemical quench-flow instrument, followed by a 5 mM MgATP chase or an acid quench for direct comparison of ATP binding and ATP hydrolysis (Figure 4). In the chase experiment, the nonradiolabeled MgATP chase will drive the reaction toward the Mt·K·ADP·Pi intermediate. Any [α-32P]MgATP tightly bound at the active site will partition toward hydrolysis, and any [α-32P]MgATP weakly bound will dissociate and become diluted by the excess nonradiolabeled MgATP chase. The time courses for ATP binding and hydrolysis were determined for two different [α-32P]-MgATP concentrations. These results show that ATP binding was much faster than ATP hydrolysis. Moreover, ATP binding was sufficiently tight to allow for accumulation of a stable Mt·K*·ATP intermediate prior to the onset of ATP hydrolysis (Scheme 1). These results indicate that ATP binding for E164K is different from that observed for K401-wt, where the transients for ATP binding and hydrolysis were similar due to partitioning of the Mt·K·ATP intermediate (Figure 1 of ref 52). For K401-wt, there was a fast off rate for ATP (k-1) relative to ATP hydrolysis (k+2) such that the full burst amplitude could only be achieved at exceptionally high concentrations of ATP. These results indicate that ATP binding for E164K occurs without a significant off rate for ATP.
FIGURE 4.

Pre-steady-state kinetics of ATP binding and hydrolysis. A preformed Mt·E164K complex (5 μM E164K, 18 μM tubulin) was rapidly mixed with [α-32P]MgATP followed by either an acid quench or a 5 mM MgATP chase. The reaction times varied from 5 to 400 ms with the first 100 ms shown. The concentrations reported represent final concentrations. The time courses for ATP binding (▲) and ATP hydrolysis (●) were determined for two different [α-32P]MgATP concentrations (A, 20 μM; B, 50 μM).
Pulse-Chase Kinetics of ATP Binding. To explore further the reduced burst amplitude, pulse-chase experiments were performed as a function of ATP concentration. The cold MgATP chase drives the reaction toward product formation; therefore, at high [α-32P]MgATP concentrations all active sites should be saturated with radiolabeled ATP and detected after the pulse-chase as [α-32P]ADP. A preformed Mt·E164K complex (5 μM E164K, 18 μM tubulin) was rapidly mixed in the chemical quench-flow instrument with varying concentrations of [α-32P]MgATP, chased with 5 mM MgATP, and then quenched with 5 M formic acid. Figure 5A shows the transients for five ATP concentrations. Figure 5B presents the burst rate data plotted as a function of [α-32P]MgATP concentration. The fit of the data provides a maximum rate constant, k +1′,at286s -1 with the Kd,ATP at 19 μM. The burst rate is slightly greater than that of K401-wt at 239 s-1 (Table 1), and its observation indicates that the E164K mutant can make the structural transition to the M,K*,ATP intermediate that proceeds directly to ATP hydrolysis (Scheme 1). This rapid isomerization has been assumed to represent ATP-dependent neck linker docking reported by Rice et al. (37). An isomerization at 200-300 s-1 has also been detected by mantATP binding kinetics (23), by fluorescence resonance energy transfer (41), and by molecular force clamp (42). The Kd,ATP derived from the fit of the data in Figure 4B also indicates that E164K binds ATP with higher affinity than K401-wt (Table 1).
Figure 5C shows the burst amplitude plotted as a function of ATP concentration. These data provided a maximum burst amplitude of 1.6 μM, which is less than the concentration of active sites (5 μM E164K). These results indicate that ∼32% of the available sites bind radiolabeled ATP and go forward to ATP hydrolysis. Because ATP binding appears to be tight, these data suggest that there is a population of sites unavailable for ATP binding as was the case for the E164A mutant (48).
ATP-Promoted Dissociation Kinetics of the Mt·Mutant Complex. Both E164A and E164K appear to exhibit similar kinetic profiles for ATP binding and ATP hydrolysis, and the steady-state kinetic parameters are also similar (Table 1). This was surprising considering the difference in the side chains of alanine and lysine. Because E164A becomes stalled on the microtubule after the first ATP turnover, we pursued experiments to evaluate whether E164K as well as the other E164 mutants showed ATP-dependent detachment from the microtubule. The Mt·kinesin complex was mixed in the stopped-flow apparatus with MgATP plus KCl to provide final concentrations of 1 mM MgATP, 100 mM KCl, and 50 mM potassium acetate from the buffer. Because of kinesin’s high processivity, the additional salt was required to obtain an optical signal for dissociation (20, 23). The higher salt does not affect the first ATP turnover but weakens rebinding of the detached motor to the microtubule (20).
In Figure 6, the dissociation kinetics for each of our Glu164 site-directed mutants is shown in comparison to K401-wt. In this experiment, a decrease in turbidity is interpreted as release of the motor from the microtubule; therefore, the amplitude associated with the kinetics reflects the ability of the motor to detach from the microtubule. E164Q and E164D show amplitudes more similar to K401-wt. However, the amplitude of the E164K transient is ∼ 63% of K401-wt. The amplitude of the E164N transient is ∼ 34% of that of K401-wt, and E164A does not readily dissociate from the microtubule as observed previously (48). Although E164A and E164K appear to have similar ATP binding and hydrolysis kinetics, their dissociation transients are very different, which may be due to the additional salt included in the ATP syringe. These data indicate that E164K can detach from the microtubule, yet E164A at the same conditions cannot detach from the microtubule.
The intermediate amplitudes obtained for E164N and E164K are more difficult to interpret because the results suggest that each population is not homogeneous in its response to MgATP. There are at least three likely possibilities. First, there is a significant fraction of inactive motors. This hypothesis appears unlikely because the active site titrations indicated that the protein purified was fully active. Second, there is an equilibrium between the attached and detached states such that ATP promotes dissociation, but the motor rebinds immediately to the microtubule and does not step forward. This equilibrium effect would result in a decrease in the amplitude observed. We have evaluated an equilibrium effect by performing the same experiment but at higher concentrations of the Mt·E164K (or Mt·E164N) complex. The experiment was repeated with the Mt·motor complexes at 3 μM motor plus 2.9 μM Mts and at 6 μM motor plus 6 μM Mts (data not shown). The transients were compared to K401-wt at the same conditions. The rate of dissociation remained the same for the mutant motors and K401-wt regardless of protein concentration. The transients for the mutant motors were compared directly to K401-wt, and the amplitude of each was obtained relative to the amplitude of K401-wt. There was no difference in the percent dissociation regardless of motor concentration in the experiment, suggesting that an equilibrium effect was not the cause of a subpopulation of motors remaining attached to the microtubule. The third hypothesis is based on kinesin’s high processivity. The ATP-promoted dissociation experiments are performed with additional salt included in the ATP syringe to weaken rebinding to the microtubule as kinesin takes its next step. A significant fraction of the K401-wt motors (>90%) do detach at these conditions. However, dissociation is not 100% complete. We propose that the intermediate amplitudes observed for E164K and E164N reflect differential stepping or run lengths of individual motors within the population. Some detach after the first turnover, yet others may continue stepping along the microtubule lattice for additional ATP turnovers. Others may stall on the microtubule. Evidence in support of this hypothesis is the apparent slow phase from 0.5 to 2 s seen in each of the transients (Figure 6). The results though do indicate that, for E164A, E164N, and E164K, a significant fraction of the motors remains attached to the microtubule at the conditions that result in K401-wt detachment.
MantADP Dissociation Kinetics To Investigate Cooperativity. Another major defect revealed by the E164A kinetic analysis was the loss of cooperativity between the motor domains. This defect may result because the active site can no longer distinguish ATP from ADP, or the active site cannot communicate its nucleotide state to the other motor domain, and/or the communication pathway between the microtubule and active site was disrupted. To evaluate the possible loss of motor domain cooperativity in E164K, a series of mantADP dissociation experiments were performed (Figures 7 and 8). We examined the rate of mantADP release from the high-affinity site of the kinesin dimer. The asymmetry of sites is established when kinesin binds the microtubule (species 1, Figure 1). One motor domain is bound tightly to the microtubule and free of nucleotide, and the other motor domain is tethered or weakly bound to the microtubule but is able to accumulate mantADP at its active site (24). An equilibrium mixture was prepared where 6 μM E164K was incubated with 3 μM mantADP and 15 μM microtubules. Under these conditions, only one mantADP is expected to be bound per kinesin dimer. MantADP release is then activated by rapidly mixing the complex with either ATP (Figure 7) or ADP (Figure 8). The ATP (or ADP) should bind to the microtubule-bound motor domain, driving the second head to bind to the microtubule and release its mantADP (see Figure 1, steps 1-4) (21, 24, 25, 45).
FIGURE 7.

MantADP release from head 2 initiated by ATP. The preformed Mt·E164K complex (6 μM E164K, 3 μM mantADP, 15 μM tubulin) was rapidly mixed in the stopped-flow instrument with varying concentrations of ATP (3.125-500 μM). (A) Transients are shown for the following concentrations of MgATP: 3.125, 6.25, 12.5, 25, and 100 μM (from the top to the bottom transient). Fluorescence signals were normalized to begin at 6.42 V for each MgATP concentration. The smooth lines represent the fit of the data to a single exponential function. (B) The observed exponential rate constants of the MgATP-dependent fluorescence change increased as a function of MgATP concentration. The data were fit to a hyperbola which defines the maximum rate of mantADP release from the second head activated by ATP to be 32.8 ± 0.7 s-1.
FIGURE 8.

MantADP release from head 2 initiated by ADP. (A) The preformed Mt±E164K complex (6 μM E164K, 3 μM mant-ADP, 15 μM tubulin) was rapidly mixed in the stopped-flow instrument with varying concentrations of MgADP (10-1000 μM). Transients are shown for the following concentrations of MgADP: 12.5, 25, 50, and 100 μM (from the top to the bottom trace). Fluorescence signals were normalized to begin at 5.5 V for each MgADP concentration. The smooth lines represent the fit of the data to two exponential functions. (B) The initial exponential rates of the MgADP-dependent fluorescence change increased as a function of MgADP concentration. The fit of the data to a hyperbola provided the maximum rate constant of mantADP release from the second head, kobs) 22.1 ± 0.7 s-1. (C) The preformed Mt·E164K complex (6 μM E164K, 3 μM mantADP) was rapidly mixed in the stopped flow with 1 mM MgATP (purple, bottom transient), MgADP (green), MgATPγS (red), or MgAMP-PNP (blue). Fluorescence signals were normalized to begin at 1 V, and the smooth lines represent the fit of the data to a single exponential function. The rate of mantADP release from the high-affinity site (head 2) was 33.9 ±0.5 s-1 initiated by MgATP, 19.9 ±0.3 s-1 by MgADP, 19.9 ±0.3 s-1 by MgATPγS, and 17.1 ±0.3 s-1 by MgAMP-PNP.
Figure 7 shows representative transients from five different ATP concentrations with the exponential decrease in fluorescence correlated with mantADP release from the active site. The maximum rate of mantADP release from the second head is 33 s-1 when initiated by ATP. This experiment was then repeated, but E164K motor domain binding to the microtubule was initiated by ADP (Figure 8). The maximum rate of mantADP release was 22 s-1. We also investigated the effect of initiation by the nucleotide analogues ATPγS (slowly hydrolyzable) and AMP-PNP (nonhydrolyzable) (Figure 8C). These data show that the rate of mantADP release in the presence of the analogues, ATPγS at 20 s-1 and AMP-PNP at 17 s-1, is similar to that of ADP (Table 1).
The rate of mantADP release from the high-affinity site in the presence of ATP (33 s-1) was much slower than that observed previously for K401-wt (>100 s-1). However, the release of mantADP from the second head in the presence of ADP at 19-22 s-1 was much faster than that previously observed for K401-wt at 6.6 s-1. Although ADP does stimulate mantADP release, we assume that ADP cannot drive the structural transitions to form species 4 and 5 and generate force (Figure 1).
Although ATP, ATP analogues, and ADP can elicit mantADP release from the second head of the E164K dimer, the rates and amplitudes are similar rather than different as observed for K401-wt. These results indicate that γ-phosphate recognition and/or motor domain communication is (are) also aberrant as observed for E164A (Table 1). Although E164N shows a similar kinetic profile as E164A and E164K, the data suggest that the cooperativity defect is not as severe with the asparagine mutant as observed for E164A and E164K. The mantADP dissociation kinetics for E164Q, E164D, and D165A are more similar to K401-wt (Table 1).
DISCUSSION
Our goal was to identify and understand the structural transitions that are driven by ATP turnover at each site of the kinesin dimer for processive motion. However, it is difficult to design experiments that report from each motor domain of the kinesin dimer to address motor domain communication. In addition, the most important conformational changes driven by kinesin occur on the microtubule rather than in the detached state. Because the experimental insights we seek are inaccessible by traditional experiments, we have used a structure-function approach to evaluate the mechanistic and structural basis of kinesin cooperativity.
Mechanistic Comparison of E164K and E164A. Previously, we showed that the alanine mutant, E164A, exhibited tight ATP binding and fast ATP hydrolysis (48). However, the kinetics for the Mt·E164A complex indicated that only one of its two motor domains was able to bind and hydrolyze ATP. Furthermore, ADP release was slowed upon microtubule binding, and there was a loss of communication between the two motor domains. Our interpretation of the data was that the E164A mutant was stalled on the microtubule, and the structural transitions to allow ATP binding on head 2 were prevented (step 7, Figure 1).
The data presented here demonstrate that the E164K mutant has a kinetic profile similar to E164A despite the difference in side chain length and charge. The kinetics for E164K reveal tighter ATP binding and fast ATP hydrolysis (Figure 3, Table 1). ADP release is also slowed in E164K (Figure 7). We propose that ATP hydrolysis on head 1 occurs prior to ADP release from head 2; therefore, head 2 becomes tightly bound to the microtubule, slowing ADP release from its active site (species 5, Figure 1). In addition, the higher microtubule affinity may alter the conformation of the collision complex and slow mantADP release. E164K also shows loss of cooperativity, which was detected by the mantADP release kinetics from head 2 (Figures 7 and 8, Table 1). This experiment requires nucleotide recognition, i.e., the active site of head 1 be able to discriminate ATP from ADP (species 2, Figure 1) and the ATP state once recognized be transmitted via helix α4 for neck linker docking onto the catalytic core (species 3). Lastly, the structural transitions that result must be able to propel head 2 forward to the next microtubule binding site, followed by collision with the microtubule and rapid ADP release (species 4). These transitions must involve communication from the microtubule binding interface (α5-L12-α4, L11) to switch II-switch I (α4-L11-α3a). For E164K, mantADP release from head 2 was much slower when initiated by ATP in comparison to K401-wt (Table 1). However, when ADP was used to signal microtubule association, the mantADP release kinetics were similar to those initiated by ATP. For K401-wt, ATP leads to rapid mantADP release from head 2 at >100 s-1, and ADP results in slow release of mantADP at 5-6 s-1. Therefore, the mantADP release kinetics for both E164A and E164K indicate that cooperativity was disrupted by both the alanine and lysine substitutions. The aberrant kinetics may be due to altered contacts with nucleotide in the switch I-switch II region of the motor that senses the γ-phosphate; or alternatively, if the nucleotide state were recognized, then the communication pathway to the other motor domain and microtubule is affected. The observations that ATP binding was tighter and ATP hydrolysis more rapid imply that the aberrant mantADP release kinetics did not result strictly from a loss of communication but that amino acid interactions with the nucleotide must be altered within the active site also.
The pulse-chase kinetics for E164K reveal a burst amplitude that is <50% of the sites (Figure 5), suggesting that, like E164A, E164K cannot bind and hydrolyze ATP at the forward head (species 7, Figure 1). We propose that both mutants stall on the microtubule as species 5 (Figure 1), in part because of stabilizing interactions with the microtubule (Figure 2). For E164K, the Mt·K complex may be stabilized through electrostatic interactions with Glu415 and Glu417 on β-tubulin helix α12. For E164A, the microtubule-kinesin interface may be stabilized through hydrophobic interactions of alanine with Phe418 and Met416 on β-tubulin helix α12. The dissociation kinetics for the mutants support this interpretation (Figure 6).
Dissociation of Microtubule-E164 Mutant Complexes. Although the kinetics reveal a similar profile for E164A and E164K, the ATP-dependent dissociation kinetics are quite different. For this experiment, additional salt was added to the ATP syringe to weaken the affinity for the microtubule when the motor attempts to rebind the microtubule after detachment. E164A did not show dissociation, and this result is expected if stabilizing hydrophobic interactions were involved. The E164K transient reflected an amplitude that was ∼63% of K401-wt (Figure 6, Table 1), suggesting that the stabilizing electrostatic interactions with the microtubule were now disrupted by the additional salt. Furthermore, the amplitude of the E164N transient shows only 34% dissociation, yet E164Q and E164D are more similar to K401-wt. The microtubule association experiments indicated that all mutants were able to bind microtubules (Table 1). The dissociation and association kinetics for E164D were similar to K401-wt kinetics, illustrating the importance of the negative charge at amino acid 164 in mediating microtubule interactions. We propose that as kinesin transitions to its weakly bound state on the microtubule, its detachment is facilitated by the repulsive interactions of Glu415 and Glu417 on β-tubulin helix α12 (Figure 2). The aberrant dissociation kinetics for E164A, E164N, and E164K are consistent with this interpretation.
Characterization of E164 Mutants. To further examine the mechanistic reasons for the variability in kinetics based on side chain size and charge at this residue, we looked at steady-state turnover, mantADP release from both heads, and mantADP release from head 2 for E164N, E164Q, E164D, and neighboring residue D165A (Table 1, Figure 2). All of the E164 mutants as well as D165A displayed a slower kcat and higher affinity for microtubules, though E164Q and E164D exhibit rates closer to that of K401-wt. The Km,ATP suggests tighter binding of ATP for the mutants with E164Q and E164D closest to wild type. The K0.5,Mt suggests higher microtubule affinity for these mutants, but the K0.5,Mt for E164D is near wild type, reaffirming that the negative charge is important in maintaining proper microtubule interactions.
The rate of mantADP release measured from both heads was slower in the Glu164 mutants with E164D more similar to K401-wt. For E164A and E164K, ADP and ATP can activate mantADP release from head 2 with similar rates, suggesting aberrant cooperativity between motor domains. It seems that cooperativity between the motor domains approaches wild-type rates in E164N, E164Q, and E164D because ADP initiates a slower rate of mantADP release from head 2 than ATP in these mutants. The two extremes in side chain length (alanine and lysine) clearly disrupt the active site response to nucleotide perhaps through interactions with helix β5 or stabilizing interactions with the microtubule lattice through β-tubulin helix α12 (Figure 2). The asparagine, glutamine, and aspartic acid are closer in size to glutamic acid and may restore some of the structural contacts needed for γ-phosphate recognition and nucleotide communication.
Importance of Interactions with Kinesin Helix α5. E164A, E164K, and E164N show more similar kinetic trends with varying degrees of severity in the defect. Although E164Q and E164D have some defects, their kinetics were more similar to those of wild-type kinesin. However, maintenance of the negative charge (E164D) seems important for microtubule interactions, and preserving the side chain length (E164Q) also restored activity to near wild-type activity. From this analysis, we conclude that both side chain charge and size are important for Glu164 function.
Woehlke et al. reported that the human R284A2 mutant (Drosophila Glu292, Figure 2) also slows ATP turnover 2-fold as was seen with our E164K and E164A mutants (49). This residue resides on kinesin helix α5. It is also in close proximity to β-tubulin helix 12, and Arg292 has been implicated in interactions with Glu164 (48). The Drosophila T291M mutation (58) also on helix α5 resulted in a dimeric kinesin motor that showed the same communication defect as E164A and E164K, suggesting that these mutations do affect the path between the microtubule and the active site through helix α5. In addition to its link to the active site, helix α5 is involved in microtubule interactions. Therefore, Glu164 through its interaction with kinesin helix α5 may directly affect both the active site and the microtubule interface.
Importance of the Glu164 Position on the Microtubule Binding Face. Glu164 points directly at the microtubule binding surface, which may account for the defects we see in microtubule dissociation and association. However, the residue beside Glu164, Asp165, points away from the microtubule. D165A (Table 1), D165N, and D165E (data not shown) all show slightly slower ATP turnover. They exhibit wild-type dissociation from the microtubule and near wildtype kinetics of mantADP release from both heads and head 2 (Table 1). Although Asp165 is beside the critical Glu164, it is not as functionally significant for kinesin mechanochemistry based on our mechanistic analysis. This makes sense given that Asp165 is not positioned to interact with either α- or β-tubulin nor to interact with residues on helix α5 (Figure 2).
The importance of Glu164 to kinesin function is also reinforced by the fact that it is a highly conserved residue— identical in all KHC, kinesin II (KRP85/95), and Unc104/KIF1 kinesins. These kinesins have been implicated in plus-end-directed transport of membranous organelles or large proteinaceous rafts for kinesin II intraflagellar transport, and both conventional kinesin and Unc104 exhibit processive motility (59-61). Interestingly, this glutamate as well as neighboring amino acids is not conserved in the BimC/Eg5 subfamily even though these kinesins also have an N-terminal motor domain and exhibit plus-end-directed microtubule movements (62, 63). BimC/Eg5 kinesins have been implicated in spindle dynamics and have been shown to be essential for bipolar spindle formation (64). It may be that conventional kinesin, kinesin II, and Unc104 kinesins share a common ATPase mechanism to generate force, and Glu164 plays a pivotal role for their cargo transport function.
The analysis of microtubule-kinesin complexes by cry-oEM and 3-D reconstruction show Glu164 in close proximity to Glu415, Met416, Glu417, and Phe418 on helix α12 of β-tubulin (Figure 2). The cryoEM does not have the resolution required to identify these β-tubulin amino acids as being structurally relevant to Glu164 on the basis of hydrogen bond length or van der Waals distances. However, the kinetic results presented here and the high similarity in β-tubulin sequences warrant experiments to test this hypothesis directly. The EMEF motif is identical in the 25 β-tubulins of D. melano-gaster, Homo sapiens, Sus scrofa (pig), Mus musculus, Rattus norvegicus, Cricetulus griseus (Chinese hamster), Gallus gallus (chicken), Xenopus laevis, Caenorhabditis elegans, Chlamydomonas reinhardtii, and Schizosaccharomyces pombe. In Saccharomyces cerevisiae, there is a single β-tubulin gene, and its amino acid sequence is ELEF where a leucine is substituted for the methionine. Huffaker and his colleagues generated a series of β-tubulin mutants in S. cerevisiae (65). One, tub2-454, contained alanine substitutions for the glutamate residues of ELEF, resulting in an ALAF motif on β-tubulin helix 12. This mutant was recessive lethal; however, the cells did contain microtubule arrays, and ∼50% of the cells were able to assemble bipolar spindle arrays. This type of analysis indicates that the EMEF motif on β-tubulin helix α12 is important for microtubule function. For our studies, we hypothesize that key β-tubulin residues on helix α12 stabilized interactions with kinesins E164A, E164K, and E164N such that the kinesin ATPase cycle and the structural pathways for communication were disrupted.
This combined approach of genetics and biochemistry has proven to be powerful in the elucidation of the mechanistic contribution of specific amino acids to kinesin function. Two of our alleles with mutations on the microtubule binding surface of the motor (T291M and E164K) have dramatic effects on ATP binding and hydrolysis. Thr291 appears to be part of a conserved hydrophobic pocket that may be critical both in the positioning of loop L12 for microtubule binding and in maintaining the structural dynamics of the active site. Disruption of this pocket by the T291M mutation resulted in failure to distinguish ADP from ATP and cooperativity defects for mantADP release (58). In contrast to the E164 mutants, T291M exhibited weak ATP binding and weak microtubule affinity.
In summary, these results illustrate the intimate coupling of the ATP and microtubule binding sites. They also provide insight into the elegant structural transitions that must take place to coordinate the motor domains, both with each other and with the microtubule.
ACKNOWLEDGMENT
We thank Brain W. Robertson, Troy Krzysiak, Alycia Bittner, and Dr. Eric Polinko (University of Pittsburgh) for contributions to this study and Dr. Steven S. Rosenfeld, University of Alabama at Birmingham, for thoughtful comments during preparation of the manuscript. The authors sincerely thank the anonymous reviewers for their critical comments on the manuscript and for suggesting alternative interpretations of the dissociation kinetics.
Footnotes
This work was supported by National Institutes of Health Grants GM54154 (S.P.G.) and GM46295 (W.M.S.), National Institute of Arthritis and Musculoskeletal and Skin Diseases Career Development Award K02-AR47841 (S.P.G.), a predoctoral fellowship from the American Heart Association, Indiana Affiliate, Inc. (K.M.B.), and an American Heart Association Established Investigatorship with funds contributed in part by the AHA Indiana Affiliate, Inc. (W.M.S.).
- KHC
- kinesin heavy chain
- K401
- KHC fragment containing the N-terminal 401 amino acids
- K401-wt
- wild-type K401
- AMP-PNP
- 5’-adenylyl β,γ-imidodiphosphate
- ATPγS
- adenosine 5’-[γ-thio]triphosphate
- Mt
- microtubule
- mantADP
- 2’(3’)-O-(N-methyl-anthraniloyl)adenosine 5’-diphosphate
- EM
- electron microscopy.
Human R284, D. melanogaster R292, rat R286, Neurospora crassa R289.
REFERENCES
- 1.Vale RD, Milligan RA. The way things move: Looking under the hood of molecular motor proteins. Science. 2000;288:88–95. doi: 10.1126/science.288.5463.88. [DOI] [PubMed] [Google Scholar]
- 2.Schief WR, Howard J. Conformational changes during kinesin motility. Curr. Opin. Cell Biol. 2001;13:19–28. doi: 10.1016/s0955-0674(00)00169-1. [DOI] [PubMed] [Google Scholar]
- 3.Schliwa M, Woehlke G. Molecular motors. Nature. 2003;422:759–765. doi: 10.1038/nature01601. [DOI] [PubMed] [Google Scholar]
- 4.Mandelkow E, Johnson KA. The structural and mechanochemical cycle of kinesin. Trends Bochem. Sci. 1998;23:429–433. doi: 10.1016/s0968-0004(98)01278-x. [DOI] [PubMed] [Google Scholar]
- 5.Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sablin EP, Kull FJ, Cooke R, Vale RD, Fletterick RJ. Crystal structure of the motor domain of the kinesin-related motor ncd. Nature. 1996;380:555–559. doi: 10.1038/380555a0. [DOI] [PubMed] [Google Scholar]
- 7.Sablin EP, Case RB, Dai SC, Hart CL, Ruby A, Vale RD, Fletterick RJ. Direction determination in the minus-end-directed kinesin motor ncd. Nature. 1998;395:813–816. doi: 10.1038/27463. [DOI] [PubMed] [Google Scholar]
- 8.Kozielski F, Sack S, Marx A, Thormählen M, Schönbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. Phe crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 9.Sack S, Müller A, Marx M, Thormählen M, Mandelkow E-M, Brady ST, Mandelkow E. X-ray structure of motor and neck domains from rat brain kinesin. Biochemistry. 1997;36:16155–16165. doi: 10.1021/bi9722498. [DOI] [PubMed] [Google Scholar]
- 10.Song Y-H, Marx A, Müller J, Woehlke G, Schliwa M, Krebs A, Hoenger A, Mandelkow E. Structure of a fast kinesin: Implications for ATPase mechanism and interactions with microtubules. EMBO J. 2001;20:6213–6225. doi: 10.1093/emboj/20.22.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulick AM, Song H, Endow SA, Rayment I. X-ray crystal structure of the yeast Kar3 motor domain complexed with Mg.ADP to 2.3 Å resolution. Biochemistry. 1998;37:1769–1776. doi: 10.1021/bi972504o. [DOI] [PubMed] [Google Scholar]
- 12.Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, Hirokawa N. Switch-based mechanism of kinesin motors. Nature. 2001;411:439–445. doi: 10.1038/35078000. [DOI] [PubMed] [Google Scholar]
- 13.Sindelar CV, Budny MJ, Rice S, Naber N, Fletterick R, Cooke R. Two conformations in the human kinesin power stroke defined by X-ray crystallography and EPR spectroscopy. Nat. Struct. Biol. 2002;9:844–848. doi: 10.1038/nsb852. [DOI] [PubMed] [Google Scholar]
- 14.Turner J, Anderson R, Guo J, Beraud C, Fletterick R, Sakowicz R. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J. Biol. Chem. 2001;276:25496–25502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 15.Kull FJ, Vale RD, Fletterick RJ. The case for a common ancestor: kinesin and myosin motor proteins and G proteins. J. Muscle Res. Cell Motil. 1998;19:877–886. doi: 10.1023/a:1005489907021. [DOI] [PubMed] [Google Scholar]
- 16.Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 17.Song Y-H, Marx A, Mandelkow E. Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 2003;1994;91:6865–6869. doi: 10.1073/pnas.91.15.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hackney DD. Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 1994;91:6865–6869. doi: 10.1073/pnas.91.15.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furch M, Fujita-Becker S, Geeves MA, Holmes KC, Manstein DJ. Role of the salt-bridge between switch-1 and switch-2 of Dictyostelium myosin. J. Mol. Biol. 1999;290:797–809. doi: 10.1006/jmbi.1999.2921. [DOI] [PubMed] [Google Scholar]
- 20.Gilbert SP, Webb MR, Brune M, Johnson KA. Pathway of processive ATP hydrolysis by kinesin. Nature. 1995;373:671–676. doi: 10.1038/373671a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilbert SP, Moyer ML, Johnson KA. Alternating site mechanism of the kinesin ATPase. Biochemistry. 1998;37:792–799. doi: 10.1021/bi971117b. [DOI] [PubMed] [Google Scholar]
- 22.Moyer ML, Gilbert SP, Johnson KA. Pathway of ATP hydrolysis by monomeric and dimeric kinesin. Biochemistry. 1998;37:800–813. doi: 10.1021/bi9711184. [DOI] [PubMed] [Google Scholar]
- 23.Ma Y-Z, Taylor EW. Mechanism of microtubule kinesin ATPase. Biochemistry. 1995;34:13242–13251. doi: 10.1021/bi00040a040. [DOI] [PubMed] [Google Scholar]
- 24.Ma Y-Z, Taylor EW. Interacting head mechanism of microtubule-kinesin ATPase. J. Biol. Chem. 1997;272:724–730. doi: 10.1074/jbc.272.2.724. [DOI] [PubMed] [Google Scholar]
- 25.Crevel I, Carter N, Schliwa M, Cross RA. Coupled chemical and mechanical reaction steps in a processive Neurospora kinesin. EMBO J. 1999;18:5863–5872. doi: 10.1093/emboj/18.21.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenfeld SS, Xing J, Jefferson GM, Cheung HC, King PH. Measuring kinesin’s first step. J. Biol. Chem. 2002;277:36731–36739. doi: 10.1074/jbc.M205261200. [DOI] [PubMed] [Google Scholar]
- 27.Xing J, Wriggers W, Jefferson GM, Stein R, Cheung HC, Rosenfeld SS. Kinesin has three nucleotide-dependent conformationss—Implications for strain-dependent release. J. Biol. Chem. 2000;275:35413–35423. doi: 10.1074/jbc.M004232200. [DOI] [PubMed] [Google Scholar]
- 28.Yun M, Zhang XH, Park CG, Park HW, Endow SA. A structural pathway for activation of the kinesin motor ATPase. EMBO J. 2001;20:2611–2618. doi: 10.1093/emboj/20.11.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wriggers W, Schulten K. Nucleotide-dependent movements of the kinesin motor domain predicted by simulated annealing. Biophys. J. 1998;75:646–661. doi: 10.1016/S0006-3495(98)77555-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naber N, Minehardt TJ, Rice S, Chen X, Grammer J, Matuska M, Vale RD, Kollman PA, Car R, Yount RG, Cooke R, Pate E. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 2003;300:798–801. doi: 10.1126/science.1082374. [DOI] [PubMed] [Google Scholar]
- 31.Naber N, Rice S, Matuska M, Vale RD, Cooke R, Pate E. EPR spectroscopy shows a microtubule-dependent conformational change in the kinesin switch 1 domain. Biophys. J. 2003;84:3190–3196. doi: 10.1016/S0006-3495(03)70043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farrell CM, Mackey AT, Klumpp LM, Gilbert SP. The role of ATP hydrolysis for kinesin processivity. J. Biol. Chem. 2002;277:17079–17087. doi: 10.1074/jbc.M108793200. [DOI] [PubMed] [Google Scholar]
- 33.Klumpp LM, Mackey AT, Farrell CM, Rosenberg JM, Gilbert SP. A kinesin switch I arginine to lysine mutation rescues microtubule function. J. Biol. Chem. 2003;278:39059–39067. doi: 10.1074/jbc.M304250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asenjo AB, Krohn N, Sosa H. Configuration of the two kinesin motor domains during ATP hydrolysis. Nat. Struct. Biol. 2003;10:836–842. doi: 10.1038/nsb984. [DOI] [PubMed] [Google Scholar]
- 35.Sablin EP, Fletterick RJ. Nucleotide switches in molecular motors: structural analysis of kinesins and myosins. Curr. Opin. Struct. Biol. 2001;11:716–724. doi: 10.1016/s0959-440x(01)00265-2. [DOI] [PubMed] [Google Scholar]
- 36.Kull FJ, Endow SA. Kinesin: switch I & II and the motor mechanism. J. Cell Sci. 2002;115:15–23. doi: 10.1242/jcs.115.1.15. [DOI] [PubMed] [Google Scholar]
- 37.Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 38.Rice S, Cui Y, Sindelar C, Naber N, Matuska M, Vale R, Cooke R. Thermodynamic properties of the Kinesin neck-region docking to the catalytic core. Biophys. J. 2003;84:1844–1854. doi: 10.1016/S0006-3495(03)74992-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skiniotis G, Surrey T, Altmann S, Gross H, Song YH, Mandelkow E, Hoenger A. Nucleotide-induced conformations in the neck region of dimeric kinesin. EMBO J. 2003;22:1518–1528. doi: 10.1093/emboj/cdg164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kallipolitou A, Deluca D, Majdic U, Lakämper S, Cross RA, Meyhöfer E, Moroder L, Schliwa M, Woehlke G. Unusual properties of the fungal conventional kinesin neck domain from Neurospora crassa. EMBO J. 2001;20:6226–6235. doi: 10.1093/emboj/20.22.6226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenfeld SS, Jefferson GM, King PH. ATP reorients the neck linker of kinesin in two sequential steps. J. Biol. Chem. 2001;276:40167–40174. doi: 10.1074/jbc.M103899200. [DOI] [PubMed] [Google Scholar]
- 42.Schnitzer MJ, Visscher K, Block SM. Force production by single kinesin motors. Nat. Cell Biol. 2000;2:718–723. doi: 10.1038/35036345. [DOI] [PubMed] [Google Scholar]
- 43.Kaseda K, Higuchi H, Hirose K. Coordination of kinesin’s two heads studied with mutant heterodimers. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16058–16063. doi: 10.1073/pnas.252409199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Block SM, Asbury CL, Shaevitz JW, Lang MJ. Probing the kinesin reaction cycle with a 2D optical force clamp. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2351–2356. doi: 10.1073/pnas.0436709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hackney DD. Pathway of ADP-stimulated ADP release and dissociation of tethered kinesin from microtubules. Implications for the extent of processivity. Biochemistry. 2002;41:4437–4446. doi: 10.1021/bi0159229. [DOI] [PubMed] [Google Scholar]
- 46.Rosenfeld SS, Fordyce PM, Jeffereson GM, King PH, Block SM. Stepping and stretching: How kinesin uses internal strain to walk processively. J. Biol. Chem. 2003;278:18550–18556. doi: 10.1074/jbc.M300849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brendza KM, Rose DJ, Gilbert SP, Saxton WM. Lethal kinesin mutations reveal amino acids important for ATPase activation and structural coupling. J. Biol. Chem. 1999;274:31506–31514. doi: 10.1074/jbc.274.44.31506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klumpp LM, Brendza KM, Rosenberg JM, Hoenger A, Gilbert SP. Motor domain mutation traps kinesin as a microtubule rigor complex. Biochemistry. 2003;42:2595–2606. doi: 10.1021/bi026715r. [DOI] [PubMed] [Google Scholar]
- 49.Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD. Microtubule interaction site of the kinesin motor. Cell. 1997;90:207–216. doi: 10.1016/s0092-8674(00)80329-3. [DOI] [PubMed] [Google Scholar]
- 50.Gilbert SP, Johnson KA. Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. Biochemistry. 1993;32:4677–4684. doi: 10.1021/bi00068a028. [DOI] [PubMed] [Google Scholar]
- 51.Gilbert SP, Mackey AT. Kinetics: A tool to study molecular motors. Methods. 2000;22:337–354. doi: 10.1006/meth.2000.1086. [DOI] [PubMed] [Google Scholar]
- 52.Gilbert SP, Johnson KA. Pre-steady-state kinetics of the microtubule-kinesin ATPase. Biochemistry. 1994;33:1951–1960. doi: 10.1021/bi00173a044. [DOI] [PubMed] [Google Scholar]
- 53.Nogales E, Wolf SG, Downing KH. Structure of α,β tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 54.Hoenger A, Thormählen M, Diaz-Avalos R, Doerhoefer M, Goldie KN, Müller J, Mandelkow E. A new look at the microtubule binding patterns of dimeric kinesins. J. Mol. Biol. 2000;297:1087–1103. doi: 10.1006/jmbi.2000.3627. [DOI] [PubMed] [Google Scholar]
- 55.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 56.Esnouf RM. An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graphics Modell. 1997;15:132–133. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 57.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brendza KM, Sontag CA, Saxton WM, Gilbert SP. A kinesin mutation that uncouples motor domains and desensitizes the γ-phosphate sensor. J. Biol. Chem. 2000;275:22187–22195. doi: 10.1074/jbc.M001124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–158. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- 60.Block SM, Goldstein LSB, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 61.Tomishige M, Klopfenstein DR, Vale RD. Conversion of Unc104/KIF1A kinesin into a processive motor after dimerization. Science. 2002;297:2263–2267. doi: 10.1126/science.1073386. [DOI] [PubMed] [Google Scholar]
- 62.Sawin KE, LeGuellec K, Philippe M, Mitchison TJ. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature. 1992;359:540–543. doi: 10.1038/359540a0. [DOI] [PubMed] [Google Scholar]
- 63.Fox LA, Sawin KE, Sale WS. Kinesin-related proteins in eukaryotic flagella. J. Cell Science. 1994;107:1545–1550. doi: 10.1242/jcs.107.6.1545. [DOI] [PubMed] [Google Scholar]
- 64.Enos AP, Morris NR. Mutation of a gene that encodes a kinesin-like protein blocks nuclear division in A. nidulans. Cell. 1990;60:1019–1027. doi: 10.1016/0092-8674(90)90350-n. [DOI] [PubMed] [Google Scholar]
- 65.Reijo RA, Cooper EM, Beagle GJ, Huffaker TC. Systematic mutational analysis of the yeast β-tubulin gene. Mol. Biol. Cell. 1994;5:29–43. doi: 10.1091/mbc.5.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]