

Abstract
Photochemical release (uncaging) of bioactive messengers with three-dimensional spatial resolution in light-scattering media would be greatly facilitated if the photolysis could be powered by pairs of IR photons rather than the customary single UV photons. The quadratic dependence on light intensity would confine the photolysis to the focus point of the laser, and the longer wavelengths would be much less affected by scattering. However, previous caged messengers have had very small cross sections for two-photon excitation in the IR region. We now show that brominated 7-hydroxycoumarin-4-ylmethyl esters and carbamates efficiently release carboxylates and amines on photolysis, with one- and two-photon cross sections up to one or two orders of magnitude better than previously available. These advantages are demonstrated on neurons in brain slices from rat cortex and hippocampus excited by glutamate uncaged from N-(6-bromo-7-hydroxycoumarin-4-ylmethoxycarbonyl)-l-glutamate (Bhc-glu). Conventional UV photolysis of Bhc-glu requires less than one-fifth the intensities needed by one of the best previous caged glutamates, γ-(α-carboxy-2-nitrobenzyl)-l-glutamate (CNB-glu). Two-photon photolysis with raster-scanned femtosecond IR pulses gives the first three-dimensionally resolved maps of the glutamate sensitivity of neurons in intact slices. Bhc-glu and analogs should allow more efficient and three-dimensionally localized uncaging and photocleavage, not only in cell biology and neurobiology but also in many technological applications.
Photolabile protecting groups have become a mainstay of organic synthesis (1), biotechnology, and cell biology (2, 3), because cleavage by light is a very mild deprotection step that usually is orthogonal to other experimental manipulations. The outstanding spatial and temporal precision with which light can be controlled enables diverse applications such as photolithographic construction of complex peptide and oligonucleotide arrays (4) or physiological release (uncaging) of bioactive substances in cells and tissues (2, 3). The ultimate in three-dimensional spatial precision would be obtained if the photochemistry were initiated not by the usual absorbance of a single UV photon but by excitation with two or more coincident IR photons of equivalent total energy (5–8). Such multiphoton excitation requires extremely high local intensities, typically obtained by focusing a femtosecond pulsed IR laser with a high-numerical-aperture lens and becomes insignificant away from the point of focus (Fig. 1). This nonlinear optical phenomenon is uniquely capable of noninvasively localizing the photochemistry to any given spot in three dimensions and will therefore be especially valuable in mapping biochemical sensitivities in complex tissues such as the brain (9, 10). Unfortunately, none of the commonly used photolabile protecting groups such as substituted 2-nitrobenzyls, 3-nitrophenyls, benzoins, and phenacyls (2, 11) have been reported to have sufficient photosensitivity for two-photon excitation without tissue damage. Photosensitivity is quantified as the uncaging action cross section δu, which is the product of the two-photon absorbance cross section δa and the uncaging quantum yield Qu2 (12). Ideally, δu should exceed 0.1 Goeppert–Mayer (GM), where 1 GM is defined as 10−50 cm4⋅s per photon. The most popular groups, such as 3,4-dimethoxy-6-nitrobenzyl, fall considerably short of this criterion. For example, DM-nitrophen, a Ca2+ chelator based on 3,4-dimethoxy-6-nitrobenzyl, has a maximum δu of 0.01 GM at 720 nm (12). The only existing caged biological messenger with adequate two-photon sensitivity is a Ca2+ complex of an azide-substituted chelator (δu = 1.4 GM at 700 nm), whose specific photochemistry (12, 13) would not be readily generalizable to release any other important messengers. A partial substitute for a true two-photon-excited photolysis is to use conventional UV with molecules bearing two separate caging groups, both of which have to be photolyzed to release biological activity (2, 10). However, this approach requires that the target molecule be blockable at either of two positions and that the extent of photolysis remains low. Also, the feasible depth of uncaging inside tissue will be limited by the much stronger absorbance and scattering of UV compared with IR wavelengths. We now report that carboxylate, carbamate, and phosphate esters of brominated 6-hydroxycoumarin-4-ylmethanol have the requisite action cross sections (δu ≈ 1 GM), photolysis kinetics, synthetic accessibility, water solubility, and stability in the dark to be a reasonably general solution to the problem of true two-photon uncaging of biologically important acids and amines. In addition, their cross sections for one-photon uncaging with UV at 365 nm or longer wavelengths are about an order of magnitude better than that of most previous caged compounds.
Figure 1.

(A) Scheme depicting the increased three-dimensional localization of uncaging with pairs of IR photons compared with single UV photons. The double cones of violet and red respectively symbolize beams of UV and IR, focused near the cell body of a schematized pyramidal neuron. Circles and triangles respectively represent the caging group and the bioactive molecule being caged. (B) Structures of caged molecules characterized in this study, showing the predominant state of ionization at pH 7.2.
MATERIALS AND METHODS
Organic Synthesis.
General. Chemicals and solvents were used directly as received unless otherwise noted. Acetonitrile, chloroform, and dichloromethane were dried over 4A molecular sieves. Proton magnetic resonance spectra (1H NMR) were recorded on a Gemini 200 MHz spectrometer (Varian) and reported in ppm (δ) downfield from tetramethylsilane with residual solvent peaks as the internal standards. UV spectra were recorded on a Cary 3E UV-Visible spectrophotometer (Varian). Fluorescence spectra were recorded on a Fluorolog spectrofluorometer (Spex Industries, Edison, NJ). Except where otherwise noted, KMops buffer consisted of 100 mM KCl/10 mM Mops titrated to pH 7.2 with KOH. Mass spectra were recorded on a electrospray mass spectrometer (5989B, Hewlett Packard). Thin layer and column chromatography were performed on precoated silica gel 60F-254 plates and 230–400 mesh silica gel 60, respectively (EM Separations Technology, Gibbstown, NJ). All manipulations of compounds sensitive to near-UV light were performed under an orange safety lamp.
Syntheses of the coumarins (Fig. 2) relied on condensation of ethyl 4-chloroacetoacetate with halogenated resorcinols (10b, 10c) in analogy to the known reaction with the unsubstituted parent resorcinol (10a) (14). The alternative of bromination after coumarin formation was hard to control short of tribromination, which yielded 11d. Acetate and glutamate esters (4–8) were prepared by direct displacement of chloride from 11a–11d. The preferred glutamate carbamate (9) was prepared by hydrolysis of the 4-chloromethyl side chain to hydroxymethyl in 13 (14), coupling to 4-nitrophenyl chloroformate to give 14, subsequent reaction with tert-butyl-protected glutamate to yield 15, and final acidolysis of the tert-butyls.
Figure 2.

Synthetic scheme for the new compounds prepared in this study. (a) Ethyl 4-chloroacetoacetate, H2SO4, rt, 6 d; (b) Br2, AcOH, rt, 1h; (c) DBU, AcOH, benzene, reflux, 1.5 h; (d) DBU, N-(tert-butoxycarbonyl)-α-tert-butyl glutamate, benzene, reflux, 1 h; (e) CF3CO2H, CH2Cl2, rt, 1 d; (f) H2O, reflux, 14 h; (g) i. DMAP, 4-nitrophenyl chloroformate, CH3CN, rt, 7 h. ii DMAP, di-tert-butyl glutamate hydrochloric acid salt, rt, 23 h; (h) CF3CO2H, CH2Cl2, rt, 2 d.
6-Bromo-4-chloromethyl-7-hydroxycoumarin (11c).
A solution of 936.1 mg (4.953 mmol) of 4-bromoresorcinol (10c), concentrated sulfuric acid (5 ml), and ethyl 4-chloroacetoacetate (1 ml, 7.4 mmol) was stirred for 6 days at room temperature. The reaction mixture was poured into ice water and stirred for 2 h to give a fine precipitate. The precipitate was collected by filtration, washed with cold water, and dried under vacuum over P2O5 to yield 847.6 mg (2.928 mmol, 59.1% yield) of 11c as a solid. 1H NMR (CD3OD) δ 7.96 (1H, s), 6.86 (1H, s), 6.43 (1H, s), 4.83 (2H, s).
6-Bromo-7-hydroxycoumarin-4-ylmethyl acetate (6).
A mixture of 67.1 mg (0.232 mmol) of 11c, dry benzene (2 ml), 175 μl (0.927 mmol) of 1,8-diazabicyclo[5.4.0]undec-7-ene, and 40 μl (0.70 mmol) of acetic acid was refluxed for 1.5 h. The reaction mixture was allowed to cool to room temperature, diluted with chloroform (10 ml), quenched with 1 N HCl (1 ml), and separated. The organic layer was dried over MgSO4 and evaporated to yield the crude product. Purification with column chromatography (10 g of SiO2, 4.7% methanol/chloroform) gave 43.1 mg (0.138 mmol, 59.4% yield) of 6 as an oil. 1H NMR (CDCl3 + 5% CD3OD) δ 7.56 (1H, s), 6.80 (1H, s), 6.21 (1H, s), 5.14 (2H, s), 2.11 (3H, s); MS (negative ion) 311.0 and 313.1; UV (KMops) λmax (ɛ) 370 nm (15,000 M−1·cm−1).
6-Chloro-4-chloromethyl-7-hydroxycoumarin (11b).
This compound was prepared from 4-chlororesorcinol (10b) by the same method as 11c to yield 2.50 g (99% yield) of 11b as a solid. 1H NMR (CD3OD) δ 7.53 (1H, s), 6.80 (1H, s), 6.29 (1H, s), 4.52 (2H, s).
6-Chloro-7-hydroxycoumarin-4-ylmethyl acetate (5).
This compound was prepared from 11b by the same method as 6 to yield 75.4 mg (0.281 mmol, 28% yield) of 5 as an oil. 1H NMR (CDCl3 + 5%CD3OD) δ 7.43 (1H, s), 6.85 (1H, s), 6.25 (1H, s), 5.16 (2H, s), 3.30 (1H, s), 2.14 (3H, s); MS (negative ion) 267.2 and 269.1; UV (KMops) λmax (ɛ) 370 nm (16,000 M−1·cm−1).
3,6,8-Tribromo-4-chloromethyl-7-hydroxycoumarin.
(11d). To a suspension of 212.6 mg (1.01 mmol) of 4-chloromethyl-7-hydroxycoumarin (11a) in acetic acid (2 ml) was added 0.2 ml (3.9 mmol) of bromine. The mixture first became a brown solution and then a yellow-orange slurry. After 1 h at room temperature, the reaction mixture was poured into ice water (70 ml) and allowed to stand at room temperature for 2 h. The yellow precipitate was collected by filtration, washed thoroughly with cold water, and dried over P2O5 under vacuum to give 477.2 mg of crude 11d as an orange solid, which was used for the next reaction without further purification. 1H NMR (CD3OD) δ 8.14 (1H, s), 4.83 (2H, s).
3,6,8-Tribromo-7-hydroxycoumarin-4-ylmethyl acetate (7).
This compound was prepared from 11d by the same method as 6 to yield 34.5 mg (73.3 μmol, 30% yield) of 7 as an oil. 1H NMR (CDCl3 + 5%CD3OD) δ 7.88 (1H, s), 5.42 (2H, s), 2.11 (3H, s); MS (negative ion) 466.9, 468.9, 470.9, and 472.9; UV (KMops) λmax (ɛ) 397 nm (15,900 M−1·cm−1).
N-(tert-Butoxycarbonyl)-l-glutamic acid α-tert-butyl γ-(6-bromo-7-hydroxycoumarin-4-yl)methyl ester (12).
A mixture of 56.0 mg (0.193 mmol) of 6-bromo-4-chloromethyl-7-hydroxycoumarin (11c), dry benzene (3 ml), 83 μl (0.44 mmol) of 1,8-diazabicyclo[5.4.0]undec-7-ene and 66.4 mg (0.219 mmol) of N-(tert-butoxycarbonyl)-l-glutamic acid α-tert-butyl ester was refluxed for 1 h. The reaction mixture was allowed to cool to room temperature, diluted with chloroform, quenched with 15% citric acid, and separated. The organic layer was dried (MgSO4) and evaporated to yield the crude product. Purification with column chromatography (10 g of SiO2, 2% methanol/chloroform) gave 33.8 mg (0.061 mmol, 31.4% yield) of 12 as an oil. 1H NMR (CDCl3) δ 7.63 (1H, s), 7.03 (1H, s), 6.36 (1H, s), 5.23 (2H, s), 4.25 (1H, m), 5.14 (1H, d, J = 8 Hz), 2.6–2.5 (2H, m), 2.24 (1H, m), 1.96 (1H, m), 1.48 (9H, s), 1.44 (9H, s); MS (negative ion) 554.0, 556.0.
l-Glutamic acid γ-(6-bromo-7-hydroxycoumarin-4-yl)methyl ester (8).
A solution of 31.2 mg (0.0561 mmol) of 12 in dry CH2Cl2 (3 ml) and CF3COOH (1 ml) was stirred for 1 day at room temperature and evaporated to dryness to give 23.2 mg of 8 (81% yield if trifluoroacetate salt). It could be purified by chromatography on Sephadex LH-20, eluting with H2O, or on silica gel 60, eluting with chloroform/methanol/acetic acid 1:1:0.1 by volume. 1H NMR (CD3OD) δ 7.84 (1H, s), 6.87 (1H, s), 6.30 (1H, s), 5.38 (2H, s), 4.09 (1H, dd, J = 6.5 and 7.0 Hz), 2.81–2.70 (2H, m), 2.28 (1H, ddt, J = 14, 6.5, and 7.2 Hz), 2.25 (1H, ddt, J = 14, 7.0, and 7.2 Hz); MS (positive ion) 400.0 and 401.9; UV (KMops) λmax (ɛ) 369 nm (19,550 M−1·cm−1).
6-Bromo-7-hydroxy-4-hydroxymethylcoumarin (13).
A suspension of 291.7 mg (1.01 mmol) of 11c in water (50 ml) was refluxed for 14 h. The resulting solution was cooled and evaporated to give 268.7 mg (0.99 mmol, 98% yield) of 13 as a solid, pure enough to use in the next step without further purification. 1H NMR (CD3OD) δ 7.83 (1H, s), 6.86 (1H, s), 6.40 (1H, s), 4.78 (2H, s).
N-(6-Bromo-7-hydroxycoumarin-4-yl)methoxycarbonyl-l-glutamic acid di(tert-butyl) ester (14).
To a stirred solution of 61.1 mg (0.225 mmol) of alcohol 13 in dry acetonitrile (5 ml) were added 56.9 mg (0.466 mmol) of 4-dimethylaminopyridine and 52.3 mg (0.252 mmol) of 4-nitrophenyl chloroformate simultaneously. Intermediate carbonate 14 was formed but not isolated. After stirring for 7 h at room temperature, another 58.1 mg (0.476 mmol) of 4-dimethylaminopyridine and 73.2 mg (0.247 mmol) of l-glutamic acid di(tert-butyl) ester hydrochloride were added. The reaction mixture was stirred at room temperature for 23 h, quenched with 15% citric acid, diluted with chloroform, and separated. The organic layer was dried (MgSO4) and evaporated to dryness to yield the crude product. Purification by column chromatography (15 g of SiO2, 33% ethyl acetate/hexane followed by 60% ethyl acetate/hexane) gave 47.0 mg (0.0845 mmol, 37.5% yield) of 15 as an oil and 29.0 mg (0.107 mmol, 47.5% recovery) of the starting alcohol 13. Compound 15 had 1H NMR (CDCl3) δ 7.63 (1H, s), 7.01 (1H, s), 6.36 (1H, s), 5.67 (1H, d, J = 8 Hz), 5.22 (2H, s), 4.28 (1H, m), 2.3–2.4 (2H, m), 2.15 (1H, m), 1.95 (1H, m), 1.48 (9H, s), 1.45 (9H, s); MS (negative ion) 553.9 and 555.9.
N-(6-Bromo-7-hydroxycoumarin-4-yl)methoxycarbonyl-l-glutamic acid (9), (Bhc-glu).
A solution of 16.9 mg (0.0304 mmol) of 15 in dry CH2Cl2 (3 ml) and CF3COOH (1 ml) was stirred for 2 days at room temperature and evaporated to dryness. Purification with column chromatography (8 g of SiO2, 25% methanol/chloroform containing 0.4% acetic acid) gave 12.7 mg (0.0286 mmol, 94.1% yield) of 9 as a yellow solid. 1H NMR (D2O) δ 7.72 (1H, s), 6.80 (1H, s), 6.26 (1H, s), 5.17 (2H, s), 3.96 (1H, m), 2.3–2.4 (2H, m), 2.10 (1H, m), 1.90 (1H, m); MS (positive ion) 466.1 and 468.2 (M+Na+), 482.1 and 483.9 (M+K+); UV (KMops) λmax (ɛ) 368 nm (17,470 M−1·cm−1).
Quantum Efficiencies for One-Photon Excitation and Rates of Hydrolysis in the Dark.
The photolysis quantum efficiencies for one-photon excitation were determined by irradiating a KMops-buffered solution of the substrate with 365-nm UV light from a mercury lamp (B-100; Spectronics, Westbury, NY). The duration of each irradiation period was controlled by an electronic shutter. Between each duration, a small aliquot (10 μl) of the solution was removed for analysis by reversed-phase HPLC with a BetaBasic-18 column (Keystone Scientific, Bellefonte, PA) eluted with an isocratic mixture of 40% acetonitrile and 60% water containing 0.1% trifluoracetic acid at a flow rate of 0.8 ml/min, using absorbance detection at 325 nm. Optical densities at 365 nm were kept around 0.1 so that inner-filtering of the irradiation and spatial gradients of concentrations could be neglected, and the progress curves were simple decaying exponentials. Quantum efficiencies were calculated as (Iσt90%)−1, where I is the irradiation intensity in einsteins⋅cm−2⋅s−1, σ is the decadic extinction coefficient in cm2⋅mol−1 (103 times ɛ, the usual extinction coefficient in M−1⋅cm−1), and t90% is the irradiation time in seconds for 90% conversion to product (15). The total UV intensity I was measured by using chemical actinometry with 6 mM potassium ferrioxalate in the same setup (16). Dark hydrolysis rates were measured similarly except without illumination.
Quantitative Analysis of Photolysis Product from Bhc-Glu (9).
Glutamate released from Bhc-Glu (9) on photolysis was quantified as its fluorescamine derivative by HPLC (17). For example, the production of glutamate from 100 μM 9 exposed to 365 nm irradiation was as follows: 30 s, 27.6% yield; 60 s, 52.7%; 90 s, 74.5%; 120 s, 98.7%. Therefore, glutamate was quantitatively released with the appropriate stoichiometry.
In Vitro Measurements of Uncaging Cross Sections.
Measurements of cross sections for two-photon uncaging were made in microcuvets with 10 × 1 × 1 mm illuminated dimensions and about 15 μl effective filling volume (26.10F-Q-10; Starna Cells, Atascadero, CA). Femtosecond IR pulses from a mode-locked titanium-sapphire laser (Tsunami pumped by a Millenium; both from Spectra-Physics) were aimed along the long axis of the sample chamber and focused in its center with a 25 mm focal length lens (06LXP003/076; Melles Griot, Irvine, CA) optimized for IR lasers. The pulse parameters were estimated by replacing the caged compound by a solution of fluorescein, a reference compound of known fluorescence quantum yield Qf2 (0.9) and two-photon absorbance crosssection δaF (30 and 38 GM at 740 and 800 nm respectively) (18). The time-averaged fluorescence 〈F(t)〉 collected by the detector is given by the equation (18)
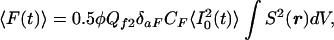 |
1 |
where φ is the collection efficiency of the detector, CF is the concentration of fluorescein, 〈I02(t)〉 is the mean squared light intensity, S(r) is a unitless spatial distribution function, and the integral is over the volume of the microcuvet. The fluorescence emitted at right angles to the laser beam was measured through a 535 ± 22.5 nm bandpass filter in front of a silicon photodiode radiometer (IL1700; International Light, Newburyport, MA). The collection efficiency φ was estimated as Ay/(4πR2n2), where A is the area of the detector (0.38 cm2), y is the fraction of the integrated emission spectrum transmitted by the interference filter (0.377, measured separately in a spectrofluorometer), R is the distance from the center of the cuvet to the detector (3.83 cm), and n is the refractive index of water (1.333). For photolysis of a caged compound or substrate, the number NP of molecules of product formed per unit time is
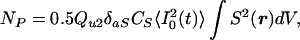 |
2 |
where Qu2, δaS, and CS are the the quantum efficiency for two-photon uncaging, absorbance cross section, and concentration of the substrate, respectively. NP was measured by HPLC sampling of the microcuvet as for one-photon photolysis. The uncaging action cross section δu (≡Qu2δaS) can therefore be estimated by combining the results of the fluorescence calibration and HPLC measurements of uncaging in the same apparatus:
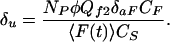 |
3 |
Similarly, when the substrate is also fluorescent, its two-photon fluorescence action cross section δfS (≡Qf2SδaS) can be estimated as
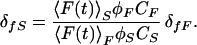 |
4 |
Here the subscripts S and F distinguish values for the substrate and for the fluorescein reference. If one assumes that one- and two-photon quantum efficiencies are the same, then δu/Qu1 and δfS/Qf1S should both equal δaS. In fact, δu/Qu1 is typically about twice δfS/Qf1S. At present we cannot say which values for δaS are more reliable, because the estimate of the geometric collection efficiency A/(4πR2n2) is quite crude and because Parma & Omenetto (19) have claimed that Qf1S is only 0.32Qf2S for the closely related 7-hydroxycoumarin. Therefore, the absolute magnitudes of the cross sections here are probably reliable to no better than a factor of two, although their relative magnitudes may be more accurate. Also, all cross sections are directly proportional to the assumed value of Qf2δaF for fluorescein.
One-Photon UV Uncaging on Brain Slices.
Coronal visual cortex slices from Long-Evans rats at postnatal days 26–28 were cut ≈400 μm thick by using an egg slicer-like device (20). Animals were anesthetized with a lethal dose of pentobarbital (100 mg/kg) and decapitated, and brains were rapidly placed in ice-cold artificial cerebrospinal fluid [containing (mM) NaCl (120), KCl (3.5), CaCl2 (2.5), MgCl2 (1.3), NaH2PO4 (1.25), NaHCO3 (26), d-glucose (10); saturated with 95% O2/5% CO2]. Slices were held submerged in an oxygenated chamber filled with artificial cerebrospinal fluid heated to 32–36°C. After at least a 1-h incubation, slices were transferred to a recording chamber where they were submerged in artificial cerebrospinal fluid at room temperature.
Whole-cell voltage clamp recordings were made from eight pyramidal neurons from layer II/III. Electrodes (4–8 MΩ resistance) were filled with a potassium gluconate-based intracellular solution containing (mM) potassium gluconate (170), Hepes (10), NaCl (10), MgCl2 (2), EGTA (1.33), CaCl2 (0.133), MgATP (3.5), GTP (1.0). Access resistance (<33 MΩ) was monitored continuously and all holding potentials were corrected for the liquid junctional potential. Cells were clamped at a constant potential of −60 mV during recording. Signals were amplified with an Axopatch 1D (Axon Instruments), low-pass filtered at 1 KHz, converted to digital signals, and recorded onto a computer with custom software.
Slices were bathed with artificial cerebrospinal fluid containing 50 μM of either Bhc-glu (4 cells) or CNB-glu (4 cells). Evoked currents were measured after uncaging glutamate with 10-ms pulses of UV light from an argon laser outfitted with UV optics and focused onto the specimen with a ×40 oil immersion objective as described (21). The UV light was applied from the bottom of the slice and focused to a depth of about 50 μm above the bottom surface. Recordings were made from neurons near the top of the slice. Light intensity was measured at the specimen plane with a calibrated photodiode. Neurons were stimulated repeatedly (0.1–0.033 Hz) at the same spot while the light intensity was systematically decreased or increased until there was no response or until the response appeared to saturate. Although a range of light intensities was tested for each cell, some cells were tested over a smaller set of intensities than others. The average amplitude was calculated for the light intensity that gave the largest responses for each cell during each trial, and this value was used to normalize all amplitude values for that cell. The largest responses were usually at the highest light intensity, but this was not always the case as some responses decreased at higher intensities.
To compare times to peak for currents generated from uncaging of CNB-glu vs. Bhc-glu, we determined the peak time for every response and pooled all of the values from each cell to obtain a mean value for that cell. The mean values for the four cells sampled with Bhc-glu were then compared with the values from the four cells sampled with CNB-glu.
Two-Photon IR Uncaging on Brain Slices.
Standard techniques were used to prepare 400-μm thick coronal brain slices from the hippocampus and cortex of 12–19 day postnatal rats. Slices were superfused at room temperature with oxygenated high-Mg/low-Ca saline containing (mM) NaCl (119), KCl (3), NaH2PO4 (1), CaCl2 (0.5), MgSO4 (4), NaHCO3 (25), and d-glucose (10). The patch pipette solution contained (mM) potassium methanesulfonate (135), Hepes (10), MgCl2 (0.3), Na2ATP (3), Na3GTP (0.3), and K2EGTA (0.5), pH adjusted to 7.30 with KOH. For some experiments, this solution also contained 0.1 mM K5-fura-2 (Molecular Probes). Whole-cell patch clamp recordings were made under visual control by using video contrast enhancement and under oblique illumination. Recordings were made in voltage clamp mode with Vh between −60 and −90 mV and accepted only when the holding current was less than 100 pA.
For delivery of caged glutamate, a 50-μm wide Kapton-coated fused-silica capillary tube (Polymicro Technologies, Phoenix, AZ) was positioned at the surface of the slice before recording, and its back end was immersed in a reservoir of local perfusion solution. Basic local perfusion solution contained (mM) NaCl (119), KCl (3), NaH2PO4 (1), CaCl2 (0.5), MgSO4 (4), Hepes (25), and d-glucose (16), pH adjusted to 7.30 with NaOH. To this solution 0–0.5 mM Bhc-glu (confirmed by optical absorption) and sometimes 200 μM Trolox C (Fluka), 1 mM DTT (Sigma), or 50 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (Research Biochemicals) were added. In some mapping experiments, currents were enhanced by including 100 μM cyclothiazide (Research Biochemicals) to block receptor desensitization (22). To start the flow of local solution, 5–10 psi pressure was applied to the reservoir.
Two-photon uncaging and fluorescence imaging were done by using custom hardware and software. Pulses 100 fs long from a Ti:sapphire laser (Tsunami, Spectra-Physics) tuned to 780 nm were directed through a Zeiss ×63 water-immersion objective (numerical aperture 0.9). Power measurements were made at the back-aperture of the objective. For mapping experiments slow scan speeds (2 s per line) were used to allow low-resolution images to be acquired (32 × 32 pixels; Fig. 7b). In tests of the power dependence and for the involvement of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors (Fig. 5), currents were recorded during scans with fast line rates (5 ms per line, fast scan direction is vertical in Fig. 5) that did not permit temporal and hence spatial resolution along the scan lines. To verify cell responsiveness, glutamate was uncaged also by conventional, one-photon absorption by using a pulsed mercury arc lamp (23).
Figure 7.

Glutamate-evoked current mapped at higher resolution on the soma of a cortical neuron. Transmitted-light image acquired by using the scanning laser (a), glutamate-evoked current map (b), or fluorescence image (c) in the presence of Bhc-glu. The superfusion solution contained 0.5 mM Bhc-glu. Laser power for the uncaging image was 85 mW.
Figure 5.

Responses to two-photon uncaging of Bhc-glu. (a) Two-photon fluorescence image of a hippocampal neuron bathed in 0.5 mM Bhc-glu. (b) Whole-cell currents recorded in response to full-field raster scans like the one used for the fluorescence image (2 ms per line, 256 lines) with the beam power between 0 to 75 mW. (c) Dependence of peak current on beam power. The curves indicate fits by eye to second- and third-power functions. (d) Block of uncaging-evoked currents by a glutamate receptor antagonist (laser power 80 mW). Traces show responses at the same location in the absence and presence of 50 μM CNQX (6-cyano-7-nitroquinoxaline-2, 3-dione), an antagonist of non-N-methyl-d-aspartate receptors.
RESULTS
In Vitro Measurements.
Large two-photon cross sections are found in molecules with extended conjugation and high extinction coefficients for π-π* transitions. Of the caging groups commonly used in biology, nitrobenzyl groups without alkoxy substituents have so far shown unmeasurably low cross sections (19), so we did not attempt to measure an uncaging cross section δu for CNB-glu (1). The 3,4-dimethoxy-6-nitrobenzyl chromophore has the longest wavelengths and highest extinction coefficients for one-photon absorption and therefore would seem to have more promise. However, we were unable to detect any two-photon photosensitivity for the commercially available l-glutamic acid α-(3, 4-dimethoxy-6-nitrobenzyl) ester (2), because its reaction kinetics (Fig. 3) were entirely dominated by rapid spontaneous dark hydrolysis with a time constant of 19.8 h, presumably because of the electron-withdrawing -NH3+ on the α-carbon of the glutamate. A simpler ester without this background problem, 3,4-dimethoxy-6-nitrobenzyl acetate (3), showed δu = 0.03 and 0.01 GM at 740 and 800 nm, respectively (Table 1). These values are comparable to that measured for DM-nitrophen (0.01 GM at 720 nm) by a very different analytical procedure. We therefore turned to 7-hydroxycoumarin-4-ylmethyl as the most promising member of a newer generation of caging groups (24, 25), because the unattached 7-hydroxycoumarin chromophore has an acceptable cross section for two-photon excitation of fluorescence (19). 7-Hydroxycoumarin-4-ylmethyl acetate (4) proved to have much higher δu values (1.07 and 0.13 GM at 740 and 800 nm) for hydrolysis to acetic acid and 7-hydroxycoumarin-4-ylmethanol. We hypothesized that halogenation of the coumarin would further increase the two-photon cross sections by lowering the pKa of the chromophore below physiological pH and promoting formation of the more strongly absorbing anion. Also, heavy atoms would promote intersystem crossing to the triplet, which is believed to be the photochemically reactive state (T.F., M. Kanehara, and M. Iwamura, unpublished data). Compounds 5–7 were indeed mostly anionic at pH 7.2. Table 1 shows that their maximum δus increased in the order 6-chloro < 6-bromo < 3,6,8-tribromo substitution as predicted. Although 7 had the highest cross section at 800 nm, its photolysis was complicated by multiple secondary photoproducts, and useful derivatives proved relatively difficult to synthesize, whereas analogs of the monobromo compound 6 were much more tractable. Compounds 4–7 had half-lives for dark hydrolysis of 202, 228, 183, and 218 h, respectively. To prepare physiologically useful caged compounds, the 6-bromo-7-hydroxycoumarin-4-ylmethyl (Bhc) group was linked to l-glutamate at either the γ-carboxyl or the amino group via a carbamate linker (Fig. 2). The γ-ester (8) and the carbamate (9) had about the same δu, 0.9–1.0 and ≈0.4 GM at 740 and 800 nm, respectively (Table 1). The superior sensitivity to two-photon photolysis of 9 compared with prior caged glutamates such as 2 is qualitatively obvious (Fig. 3). The main disadvantage of 8 was its spontaneous hydrolysis. At pH 7.2, 24°C, the percentages of 8 remaining after 17, 40, 65, 91, and 119 h in the dark were 84, 63, 41, 21, and 8, respectively, whereas there was no detectable loss of 9 (>99% remaining) over 35 h. This superiority of glutamate carbamates over γ-esters and especially α-esters with respect to dark hydrolysis is similar to that already reported for α-carboxy-2-nitrobenzyl protecting groups (26). Background hydrolysis is often a serious drawback because free glutamate readily desensitizes glutamate receptors, so carbamate (9) is preferred.
Figure 3.

Time course of two-photon photolysis of Bhc-glu (9) at 740 nm (average power 530 mW exiting the cuvet, 172 fs pulse width), compared with breakdown of l-glutamic acid α-(3,4-dimethoxy-6-nitrobenzyl) ester (DMNB-glu, 2) with or without irradiation. ●, Percentage of Bhc-glu remaining from an initial concentration of 100 μM. Solid line, least-squares curve fit to a simple decaying exponential, which gave a time constant of 38 min. The initial rate of this photolysis run, combined with Eq. 3, gave a value of δu2 = 0.85 GM. The mean ± SD of 4 runs was 0.95 ± 0.21 GM. □, Percentage of DMNB-glu remaining under the same conditions. Dashed line, a decaying exponential of time constant 19.8 h, which had been separately measured over much longer times for the breakdown of DMNB-glu in the dark in 100 mM KMops, pH 7.2. Thus the pulsed IR laser beam does not detectably increase the rate of DMNB-glu breakdown.
Table 1.
Spectroscopic and photolytic characteristics of cage compounds
| Cmpd | λabs | ɛ(λabs) | ɛ(365) | ɛ(400) | Qu1 | ɛQu1(365) | Qf1 | λem | δf(740) | δf(800) | δu(740) | δu(800) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1* | 262 | 5,100 | 180 | 0.14 | 25 | ∼0 | ||||||
| 2 | 345 | 5,900* | 4,900 | 1,050 | 0.006 | 29 | ∼0 | † | † | |||
| 3 | 346 | 6,100 | 5,200 | 1,300 | 0.005 | 26 | ∼0 | 0.03 | 0.01 | |||
| 4 | 325 | 11,600 | 4,100 | 1,000 | 0.025 | 103 | 0.20 | 474 | 3.85 ± 0.31 | 1.26 ± 0.04 | 1.07 ± 0.05 | 0.13 ± 0.01 |
| 5 | 370 | 16,000 | 15,900 | 5,000 | 0.01 | 302 | 0.37 | 474 | 9.68 ± 0.32 | 3.22 ± 0.09 | 1.07 ± 0.07 | 0.34 ± 0.02 |
| 6 | 370 | 15,000 | 14,800 | 5,000 | 0.037 | 548 | 0.22 | 474 | 4.24 ± 0.12 | 1.41 ± 0.05 | 1.99 ± 0.09 | 0.42 ± 0.01 |
| 7 | 397 | 15,900 | 9,700 | 15,700 | 0.065 | 630 | 0.16 | 498 | 0.96 ± 0.05 | 3.1 ± 0.2 | ||
| 8 | 369 | 19,550 | 19,200 | 7,560 | 0.019 | 364 | 0.89 ± 0.24 | 0.42 ± 0.11 | ||||
| 9 | 368 | 17,470 | 17,300 | 5,400 | 0.019 | 329 | 0.38 | 474 | 2.2‡ | 0.95 ± 0.21 | 0.37 ±0.06 |
λabs, wavelength in nm of absorbance maximum, ɛ, extinction coefficient in M−1 cm−1 at wavelength indicated; Qu1, quantum efficiency for uncaging with one-photon excitation at 365 nm; Qf1, quantum efficiency for fluorescence with one-photon excitation; λem, wavelength in nm for fluorescence emission peak; δf, fluorescence action cross section in 10−50 cm4 s/photon for two-photon excitation at wavelength indicated (δf = δaQf2, where δa is the absorbance cross section and Qf2 is the quantum efficiency for fluorescence, both with two-photon excitation); δu, uncaging action cross section in GM (10−50 cm4 s/photon) for two-photon excitation at wavelength indicated (δu = δaQu2, where Qu2 is the quantum efficiency for uncaging with two-photon excitation). Measurements were made in KMops at 24–25°C. Error estimates are standard deviations.
*Values taken from the literature (30, 31); For compound 1, the entries listed under ɛ(365) and ɛQu1(365) actually refer to 360 nm.
δu could not be measured because breakdown was dominated by dark hydrolysis with a 19.8-h exponential time constant.
Value was measured at 780 nm on an independent apparatus.
One-Photon UV Uncaging in Brain Slices.
To compare the physiological effects of invoking currents in neurons from uncaging Bhc-glu (9) or CNB-glu (1) at the same concentration (50 μM), eight cells from layer II/III of rat visual cortex were directly stimulated by focusing 10-ms pulses of UV laser light (351–364 nm) below the cell body. Fig. 4A shows typical current traces from stimulating cells at increasing light intensities. The three upper traces are from a cell bathed in Bhc-glu. The responses were evoked by stimulation at UV intensities of 2, 3, and 4 mW at the slice, whereas similar sized current responses from a different cell bathed in CNB-glu (lower three traces) required intensities of 10, 16, and 21 mW, about five times higher intensity. The times from onset of shutter opening to the peak inward current were longer for the Bhc-glu responses compared with CNB-glu. The mean peak times for all responses from the 4 cells sampled with Bhc-glu ranged from 29.8 to 45.7 ms (mean ± SEM = 38.9 ± 3.6 ms, n = 4 cells) whereas the values for cells sampled with CNB-glu ranged from 14.1 to 25.8 ms (mean 18.3 ± 5.2 ms, n = 4). Carbamates are known to release glutamate more slowly than do γ-esters (26), probably because of the time required for loss of CO2 from the carbamic acid of glutamate rather than any intrinsic slowness of coumarin-4-ylmethyl hydrolysis. The mean normalized current for all of the cells as a function of pulse energy, plotted on a log scale, is shown in Fig. 4B. Normalization of each cell to its own average maximum response was required because the neurons can vary in their absolute sensitivity to glutamate, partly because the neurons were patched blindly, so that the exact position of the cell body relative to the focus point of the objective was not determined (see Materials and Methods). The form of the dose-response curve is expected to be complex because of the cooperativity of the glutamate receptor and the competition between its activation and desensitization (27). Furthermore, saturating activation of the receptor was not achievable with CNB-glu because the maximal laser power was limited. Nevertheless, the data in Fig. 4B confirm that Bhc-glu is physiologically about five-fold more UV-sensitive than CNB-glu. The superiority of Bhc-glu is not as marked as implied by the thirteen-fold ratio of ɛQu1(365 nm) values in Table 1, probably because the 351–364 nm wavelengths from the laser are better for CNB-glu and worse for Bhc-glu and because the slightly slower kinetics of the carbamate allow more receptor desensitization to limit the peak amplitude.
Figure 4.

Bhc-glu requires about five times less UV intensity than does CNB-glu to generate similar currents in pyramidal neurons from layers II/III of rat visual cortex. (A) Evoked current responses from uncaging 50 μM Bhc-glu at 2, 3, and 4 mW and 50 μM CNB-glu at increasing light intensities of 10, 16, and 21 mW. The horizontal bars above the traces indicate the 10 ms-long UV photolysis, 20 ms after the beginning of trace. Vertical scale, 50 pA. Horizontal scale, 50 ms. (B) Mean normalized current amplitudes (±SEM) from uncaging Bhc-glu (□, n = 4 cells) or CNB-glu (○, n = 4 cells) are plotted as a function of varying light intensities delivered to the slice, on a log scale. The numbers of cells tested at each light intensity are indicated by the numbers inside the symbols. To reduce intercell variation in response sizes, amplitudes for each cell were normalized to the average maximum response at the light intensity giving the maximal response for that cell.
Two-Photon IR Uncaging in Brain Slices.
Bhc-glu was applied to slices locally by pressure-ejection from a capillary tube. The compound’s presence could be monitored directly by using its fluorescence at low levels of two-photon excitation, which allowed cells to be seen in “negative” stain (Fig. 5a). During whole-cell recordings, application of Bhc-glu did not cause detectable changes in holding current (15 cells), showing that this compound lacks intrinsic agonist activity.
The stream of pulses from the Ti:sapphire laser was raster scanned across cell bodies and dendrites. In the absence of caged compound, scans caused no changes in holding current (15 cells). After application of Bhc-glu, scans evoked inward currents (Fig. 5b) whose amplitude was strongly dependent on beam power (Fig. 5c). The peak current amplitude increased as a supralinear function of beam power, mostly caused by the nonlinear power dependence expected for a two-photon process (5, 10), but also probably including some contribution from positive cooperativity of the AMPA-type glutamate receptor (27). When the antagonist CNQX (50 μM) was included in the local perfusion solution, light-evoked currents were reversibly abolished (Fig. 5d) indicating that evoked currents arose from the activation of non-N-methyl-d-aspartate glutamate receptors.
A central property of two-photon excitation is the elimination of out-of-focus background (5). Two-photon uncaging should therefore allow the production of glutamate only in the plane of focus. This was tested by using two-photon scanning photochemical microscopy (8). Cells were filled with the indicator dye fura-2 to visualize their dendrites (Fig. 6a) and scanned with uncaging illumination with the focal planes set at different depths (Fig. 6b). Changes in holding current were recorded as a function of the beam position and displayed as pixel intensity, generating a map of the response amplitude. Responses varied as a function of the focal plane and corresponded with the dendritic elements that were in focus under fluorescence viewing (Fig. 6b). Scans over the cell body gave ring-like current maps characteristic of optical sectioning (Fig. 7b).
Figure 6.

Mapping of glutamate-evoked currents. (a) Hippocampal pyramidal neuron filled with 100 μM fura-2 (image stack of 51 frames taken at 1-μm intervals, median filtered and maximum projected). (b) Maps (Left) of glutamate-evoked currents (current range is 33 pA between white and black) on dendrites taken at 4-μm depth intervals, with corresponding fluorescence images (projections of ±3 positions, right). The small image at left shows a higher-resolution scan. Laser power for uncaging images was 35 mW. Bar in a (20 μm) applies to all images. The superfusion solution contained 0.5 mM Bhc-glu and 100 μM cyclothiazide.
DISCUSSION
The two-photon uncaging cross sections (δu) for Bhc and analogously halogenated 7-hydroxycoumarin-4-ylmethyl groups are on the order of 1 GM, about two orders of magnitude larger than those previously measured for any photolabile protecting groups. If one assumes equality of one- and two-photon quantum efficiencies, their absorbance cross sections (δa) would be 20–50 GM, reasonably consistent with previous values for coumarin dyes (18, 19). Molecules with δa as high as several thousand GM have recently been reported, but they are very large, hydrophobic, and not yet harnessed to any useful photolytic reaction (28). The mechanism of Bhc photolysis has not yet been analyzed, although experiments on the related 7-methoxycoumarin-4-ylmethyl diethylphosphate indicate that the solvolysis occurs via the triplet state with a lifetime of about 1 μs (T.F., M. Kanehara, and M. Iwamura, unpublished data). The intermediacy of a triplet state is supported by the increase in photolysis quantum yields from 6-chloro to 6-bromo to 3,6,8-tribromo substitution (Table 1). The kinetics of glutamate release from Bhc-glu after a photolysis flash remain to be fully characterized, although the above triplet-state kinetics suggest that the initial release of the carbamic acid is unlikely to be rate-limiting. Instead, the slow step is probably the final decarboxylation of the carbamic acid to regenerate glutamate, which takes a few ms at pH 7.2 and is faster yet at more acidic pH (26). All glutamates that are caged via a carbamate linkage have to go through this additional step. In the present trials, we preferred the better hydrolytic stability in the dark of Bhc-glu (9) over the potentially faster kinetics of the γ-ester (8). If one were confident that the tissue could handle background release of glutamate, e.g., by active uptake, and if maximal temporal and spatial resolution were required, the γ-ester (8) might prove superior. A modified γ-ester combining both fast kinetics and resistance to hydrolysis in the dark would be optimal.
Bhc-glu is the first caged glutamate to allow nondestructive three-dimensional mapping of the glutamate responsivity over the surface of a neuron. In various previous attemps to map glutamate sensitivities by using nitrobenzyl-based compounds (K. Svoboda and W.D., unpublished data) with two-photon excitation, no significant current signals were seen. The intrinsic fluorescence of Bhc-glu is helpful for visualizing the delivery of the caged compound but can hinder simultaneous use of UV- or violet-excited fluorescent indicators. Further modification of the chromophores to accelerate intersystem crossing should reduce such fluorescence and increase the photolysis quantum yields. Although the present work focused on caged glutamates, we have also prepared the Bhc ester of cAMP, which is hydrolytically stable in the dark but efficiently photolyzed by two-photon excitation (T.F. and T.M.D., unpublished results). Therefore, the Bhc group can cage carboxylates, phosphates, and amines (via carbamates), the major acidic and basic functionalities of biological relevance.
The good performance of the above coumarin-based caging groups has important implications in both photochemistry and practical applications. Traditional caging groups are based on small chromophores absorbing only in the UV and often utilizing weak n–π* transitions. The present coumarins are conceptually derived from well known fluorescent dyes by attaching the leaving group via a -CH2- to a site that undergoes a large increase in electron density on excitation of the delocalized chromophore. Also, heavy atoms are added to promote intersystem crossing to the triplet state. We suspect that these design principles can be applied to other fluorescent dyes to create other caging groups of even longer wavelengths and larger 1- and 2-photon cross sections. In conventional 1-photon photolysis, longer wavelengths and greater photosensitivity should help minimize photodamage, decrease irradiation times, simplify apparatus, make semiconductor light sources feasible, and permit separately controllable photolyses of two or more protecting groups at different wavelengths. Two-photon photolysis should facilitate higher-resolution three-dimensional optical memories (29), spatially arrayed combinatorial libraries (4), photodynamic therapy, and deeper and less-invasive mapping of the local responses of complex tissues to neurotransmitters and messengers.
Acknowledgments
This work was supported by National Institutes of Health Grants NS27177 (R.Y.T.) and EY10742 (E.M.C.), a fellowship from the Japan Society for the Promotion of Science (T.F.), and a National Science Foundation Predoctoral Fellowship (J.L.D.).
ABBREVIATIONS
- Bhc-glu
N-(6-bromo-7-hydroxycoumarin-4-ylmethoxycarbonyl)-l-glutamate
- CNB-glu
l-glutamic acid γ-(α-carboxy-2-nitrobenzyl) ester
- GM
Goeppert-Mayer or 10−50 cm4⋅s per photon
References
- 1.Pillai, V. N. R. (1980) Synthesis, 1–26.
- 2.Adams S R, Tsien R Y. Annu Rev Physiol. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- 3.Marriott, G., ed. (1998) Methods Enzymol.291. [DOI] [PubMed]
- 4.Pirrung M C. Chem Rev. 1997;97:473–488. doi: 10.1021/cr960013o. [DOI] [PubMed] [Google Scholar]
- 5.Denk W, Strickler J H, Webb W W. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 6.Williams R M, Piston D W, Webb W W. FASEB J. 1994;8:804–813. doi: 10.1096/fasebj.8.11.8070629. [DOI] [PubMed] [Google Scholar]
- 7.Denk W, Piston D W, Webb W W. In: Handbook of Biological Confocal Microscopy. Pawley J B, editor. New York: Plenum; 1995. pp. 445–458. [Google Scholar]
- 8.Denk W. Proc Natl Acad Sci USA. 1994;91:6629–6633. doi: 10.1073/pnas.91.14.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Callaway E, Katz L. Proc Natl Acad Sci USA. 1993;90:7661–7665. doi: 10.1073/pnas.90.16.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pettit D L, Wang S S H, Gee K R, Augustine G J. Neuron. 1997;19:465–471. doi: 10.1016/s0896-6273(00)80361-x. [DOI] [PubMed] [Google Scholar]
- 11.Givens R S, Jung A, Park C H, Weber J, Bartlett W. J Am Chem Soc. 1997;119:8369–8370. [Google Scholar]
- 12.Brown E, Shear J B, Adams S R, Tsien R Y, Webb W. Biophys J. 1999;76:489–499. doi: 10.1016/S0006-3495(99)77217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams S R, Lev-Ram V, Tsien R Y. Chem Biol. 1997;4:867–878. doi: 10.1016/s1074-5521(97)90119-8. [DOI] [PubMed] [Google Scholar]
- 14.Zagotto G, Gia O, Baccichetti F, Uriarte E, Palumbo M. Photochem Photobiol. 1993;58:486–491. doi: 10.1111/j.1751-1097.1993.tb04919.x. [DOI] [PubMed] [Google Scholar]
- 15.Adams S R, Kao J P Y, Grynkiewicz G, Minta A, Tsien R Y. J Am Chem Soc. 1988;110:3212–3220. [Google Scholar]
- 16.Hatchard C G, Parker C A. Proc R Soc London Ser A. 1956;235:518–536. [Google Scholar]
- 17.Simon S M. Nat Biotechnol. 1996;14:1221. doi: 10.1038/nbt1096-1221. [DOI] [PubMed] [Google Scholar]
- 18.Xu C, Webb W W. J Opt Soc Am B. 1996;13:481–491. [Google Scholar]
- 19.Parma L, Omenetto N. Chem Phys Lett. 1978;54:541–543. [Google Scholar]
- 20.Katz L C. J Neurosci. 1987;7:1223–1249. doi: 10.1523/JNEUROSCI.07-04-01223.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawatari A, Callaway E M. Nature (London) 1996;380:442–446. doi: 10.1038/380442a0. [DOI] [PubMed] [Google Scholar]
- 22.Yamada K A, Tang C. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denk W. J Neurosci Methods. 1997;72:39–42. doi: 10.1016/s0165-0270(96)00153-7. [DOI] [PubMed] [Google Scholar]
- 24.Furuta T, Torigai H, Sugimoto M, Iwamura M. J Org Chem. 1995;60:3953–3956. [Google Scholar]
- 25.Furuta T, Iwamura M. Methods Enzymol. 1998;291:50–63. doi: 10.1016/s0076-6879(98)91006-0. [DOI] [PubMed] [Google Scholar]
- 26.Rossi F M, Margulis M, Tang C-M, Kao J P Y. J Biol Chem. 1997;272:32933–32939. doi: 10.1074/jbc.272.52.32933. [DOI] [PubMed] [Google Scholar]
- 27.Clements J D, Feltz A, Sahara Y, Westbrook G L. J Neurosci. 1998;18:119–127. doi: 10.1523/JNEUROSCI.18-01-00119.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albota M, Beljonne D, Brédas J-L, Ehrlich J E, Fu J-Y, Heikal A A, Hess S E, Kogej T, Levin M D, Marder S R, et al. Science. 1998;281:1653–1656. doi: 10.1126/science.281.5383.1653. [DOI] [PubMed] [Google Scholar]
- 29.Strickler J H, Webb W W. Opt Lett. 1991;16:1780–1782. doi: 10.1364/ol.16.001780. [DOI] [PubMed] [Google Scholar]
- 30.Haugland R P. Handbook of Fluorescent Probes and Research Chemicals. Eugene, OR: Molecular Probes; 1996. [Google Scholar]
- 31.Wieboldt R, Gee K R, Niu L, Ramesh D, Carpenter B. Proc Natl Acad Sci USA. 1994;91:8752–8756. doi: 10.1073/pnas.91.19.8752. [DOI] [PMC free article] [PubMed] [Google Scholar]