

Abstract
We previously reported that Bacillus thuringiensis strain 407 Cry 32− secretes a zinc-requiring metalloprotease, InhA2, that is essential for virulence in orally infected insects. Analysis of the inhA2-lacZ transcriptional fusion showed that inhA2 expression is repressed in a PlcR− background. Using DNase I footprinting experiments, we demonstrated that PlcR activates inhA2 transcription directly by binding to a DNA sequence showing a one-residue mismatch with the previously reported PlcR box. It was previously reported that PlcR is essential for B. thuringiensis virulence in oral infection by contributing to the synergistic properties of the spores on the insecticidal activity of the Cry1C protein. We used complementation experiments to investigate whether the PlcR− phenotype was due to the absence of InhA2. The results indicated that overexpression of inhA2 in the ΔplcR strain did not restore the wild-type phenotype. However, virulence was fully restored in the ΔinhA2 complemented mutant. Thus, inhA2 is the first example of a PlcR-regulated gene found to be directly involved in virulence. However, it is not sufficient for pathogenicity when the other members of the PlcR regulon are lacking. This suggests that InhA2 may act in concert with other PlcR-regulated gene products to provide virulence.
Bacillus thuringiensis and Bacillus cereus are two gram-positive spore-forming bacteria belonging to the cereus group, which also includes the human pathogen Bacillus anthracis and the nonpathogenic Bacillus mycoides. Molecular analysis showed that B. thuringiensis and B. cereus share the same genetic background (7, 8, 21). B. thuringiensis is well known for its entomopathogenic properties, partly due to the cytoplasmic crystallized δ-endotoxins (also termed Cry proteins) that are specifically active against insect larvae (35). B. cereus does not produce crystallized proteins and is an opportunistic human pathogen, causing food-borne gastroenteritis (23). In some rare cases, B. cereus is responsible for systemic and local infections such as endophthalmitis, periodontitis, meningitis, or pneumonia (4, 6).
Although these bacteria infect distinct hosts (insects versus mammals), they share some common pathogenic features. Indeed, the intrahemocoelic administration of low inocula of B. cereus or acrystalliferous B. thuringiensis strains to susceptible insect larvae leads to lethal septicemia (20, 32, 37, 39). Furthermore, in some insects, the presence of spores from both of these species, but not from other bacterial species, strongly increases the killing activity of B. thuringiensis crystals administrated via the oral route (10, 33). Finally, the opportunistic properties of B. thuringiensis have been demonstrated in mice by infection via nasal instillation of spores (22, 23). B. thuringiensis and B. cereus produce common potential virulence factors, which are thought to facilitate their development within the host. These factors include degradative enzymes and toxins (18, 19). A large number of B. thuringiensis genes, encoding potential virulence factors, are regulated by a pleiotropic transcriptional activator named PlcR (1, 25). The transcription of the plcR gene is both autoregulated (25) and under the control of the sporulation key factor Spo0A (26). Alignment of the promoter regions of about 15 PlcR-regulated genes from B. thuringiensis and B. cereus revealed the presence of a highly conserved palindromic sequence (TATGNAN4TNCATA), named the PlcR box (1, 30). This sequence is located in various positions upstream from the transcription start site and is essential for transcription (1). PlcR acts by binding to the PlcR box. This binding requires the product of a small gene (papR) that lies immediately downstream of plcR (36). papR is regulated by PlcR and encodes a quorum-sensing effector that controls the expression of the PlcR regulon in members of the B. cereus group. Analysis of the extracellular proteome in the B. cereus strain ATCC 14579 revealed that the disruption of plcR considerably reduced the amounts of up to 56 exported proteins (15). Moreover, the inactivation of plcR decreased the pathogenicity of B. cereus and B. thuringiensis in both insects and mice, suggesting that one or several PlcR-regulated genes are involved in pathogenicity (33).
We recently characterized a new B. thuringiensis virulence factor, InhA2 (13). InhA2 is a zinc metalloprotease that is highly homologous to the B. thuringiensis InhA, which was originally identified as an extracellular protease that specifically hydrolyzes the antibacterial peptides cecropins and attacins from the insect Hyalophora cecropia (9, 12). We showed that InhA2 plays a major role in potentiating the toxicity of Cry proteins in orally infected insects (13). The expression of inhA2 is induced at the onset of the stationary phase and is negatively regulated by Spo0A (13). Recent studies have shown that InhA2 is synthesized by B. cereus strain ATCC 14579 and is one of the B. cereus proteases strongly downregulated in ΔplcR mutants (15). However, the B. cereus inhA2 gene does not display the previously reported PlcR box consensus target upstream of its coding sequence and the mechanism by which PlcR affects InhA2 production has not been determined.
In the present study we investigated the involvement of the pleiotropic regulator PlcR in the control of inhA2 expression. Experiments with inhA2′-lacZ transcriptional fusions and DNase I footprinting demonstrated that B. thuringiensis inhA2 belongs to the PlcR regulon. We also assessed the virulence of the ΔplcR mutant after transcomplementation with inhA2 in orally infected Galleria mellonella. Our findings indicate that B. thuringiensis InhA2 is not sufficient by itself for providing virulence in the absence of PlcR-regulated genes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The sporogenic acrystalliferous B. thuringiensis strain 407 Cry− belonging to serotype 1 (27) and the 407 Cry− ΔplcR strain carrying a plcR disrupted gene (33) were used throughout the present study. The 407 Cry− ΔinhA2 strain carrying an inhA2 disrupted gene has been described previously (13). Escherichia coli K-12 strain TG1 [Δ(lac-proAB) supE thi hsdΔ5 (F′ traD36 proA+ proB+ lacIq lacZΔM15)] (14) was used as a host for the construction of plasmids and cloning experiments. E. coli strains SCS 110 [rpsL (Strr) thr leu endA thi-1 lacY galK galT ara tonA tsx dam dcm supE44 Δ(lac-proAB) (F′ traD36 proA+ proB+ lacIq lacZΔM15; Stratagene, La Jolla, Calif.] and ET12567 (F− dam-13::Tn9 dcm-6 hsdM hsdR recF143 zjj-202::Tn10 galK2 galT22 ara14 pacY1 xyl-5 leuB6 thi-1) were used to generate unmethylated plasmid DNA prior to B. thuringiensis transformation. Electroporation was used to transform E. coli (11) and B. thuringiensis (27).
E. coli and B. thuringiensis cells were routinely grown in Luria broth (LB) medium with vigorous agitation at 37 and 30°C, respectively. The following antibiotic concentrations were used for bacterial selection: ampicillin at 100 μg ml−1 (for E. coli) and kanamycin at 200 μg ml−1 and erythromycin at 10 μg ml−1 (for B. thuringiensis). Bacteria with the Lac+ phenotype were identified on LB plates containing X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) at 40 μg ml−1.
Spores of B. thuringiensis strains were obtained by culturing cells in 40 ml of sporulation-specific (HCT) medium (24) at 30°C for 3 days. Spores were harvested by centrifugation (5,000 × g for 10 min), washed with distilled water (twice, each time with 40 ml), and finally resuspended in 6 ml of sterile distilled water. The concentrations of the spore preparations were estimated by plating dilutions onto LB agar plates containing appropriate antibiotics.
The Cry1C toxin was prepared from the asporogenic strain 407 ΔsigK (5) transformed with pHT1C (34) as described by Gominet et al. (16).
DNA manipulation.
Plasmid DNA was extracted from E. coli and B. thuringiensis by a standard alkaline lysis procedure by using QIAprep spin columns (Qiagen), with the following modification in the first step of the lysis procedure for B. thuringiensis: incubation at 37°C for 1 h with 5 mg of chicken egg white lyzosyme (14,300 U/mg). Chromosomal DNA was extracted from B. thuringiensis cells harvested in mid-log phase as described previously (29). Restriction enzymes and T4 DNA ligase were used as recommended by the manufacturer (New England Biolabs). Oligonucleotide primers were synthesized by Genset (Paris, France). PCRs were performed in a GeneAmp PCR system 2400 thermal cycler (Perkin-Elmer). Amplified DNA fragments were purified by using the QIAquick PCR purification Kit (Qiagen) and separated on 0.7% agarose gels after digestion. Digested DNA fragments were extracted from agarose electrophoresis gels by using a centrifugal filter device (Ultrafree-DA; Amicon Laboratories).
Trans-complementation of ΔplcR and the ΔinhA2 mutant strains with inhA2.
A 473-bp HindIII/BamHI fragment containing the promoter region of the kanamycin resistance gene aphA3 was amplified by PCR by using the primers Km1 (5′-GAGGTGATAGGTAAG-3′) and Km2 (5′-CCCAAGAAGCTAATTATAAC-3′) and pDG783 DNA carrying the aphA3 gene from Enterococcus faecalis (38) as a template. PCRs were carried out in a volume of 100 μl containing a 200 μM concentration of deoxynucleoside triphosphates, 1.5 mM MgSO4, 50 pmol of each primer, 0.5 μg of DNA template, and 0.5 U of Pwo DNA polymerase (Roche Boheringer) in a 1× reaction buffer. The amplified DNA fragment was digested with the appropriate restriction enzymes and inserted between the HindIII/BamHI sites of the gram-positive-gram-negative shuttle plasmid, pHT315 (3). The resulting plasmid was designated pHT315ΩpaphA3. A 2,571-bp BamHI/EcoRI DNA fragment carrying a promoterless inhA2 gene (positions −46 and +126 with respect to the ATG and TAA codons of the inhA2 coding sequence, respectively) was obtained by PCR amplification by using the 407 Cry− chromosomal DNA as a template and oligonucleotides inhA2.1 (5′-CGCGGATCCCACCGATTTATCTG-3′) and inhA2.2 (5′-CCGGAATTCCTTTCCCCACATAATTTG-3′). The DNA fragment harboring the inhA2 coding sequence was digested and ligated downstream of the promoter region of aphA3 gene between the EcoRI/BamHI sites of pHT315ΩpaphA3. The ligation mixture was used to transform B. thuringiensis strain 407 Cry− ΔplcR and strain 407 Cry− ΔinhA2. The transformant clones harboring the recombinant pHT315ΩpaphA3-inhA2 were designated 407 Cry− [ΔplcR (pinhA2)] and 407 Cry− [ΔinhA2 (pinhA2)].
Construction of the inhA2′-lacZ transcriptional fusion.
The inhA2′-lacZ transcriptional fusion was constructed by cloning a 593-bp BamHI/PstI DNA fragment harboring the inhA2 promoter between the BamHI and PstI sites of pHT304-18′Z (2). The DNA fragment was generated by PCR amplification performed on 407 Cry− chromosomal DNA with the primers B6sqBt (5′-AAACTGCAGCCCAGCAAACGTAATTGCTTC-3′) and InB9 (5′-CGCGGATCCCTCTTTTGCTGGCGTTTCTGC −3′). The recombinant plasmid, designated pHT304ΩinhA2′-Z, was introduced into B. thuringiensis wild-type and ΔplcR mutant strains by electroporation. The transformants were named 407 Cry− (pHT304ΩinhA2′-Z) and 407 Cry− ΔplcR (pHT304ΩinhA2′-Z), respectively, and were resistant to erythromycin (10 μg ml−1).
β-Galactosidase assay.
Cells of B. thuringiensis strains harboring plasmid lacZ transcriptional fusions were cultured in LB medium in the absence of antibiotics at 30°C with vigorous shaking. β-Galactosidase specific activities were measured as described previously (29). Specific activities are expressed in units of β-galactosidase per milligram of protein (Miller units).
DNase I footprinting.
DNase I footprinting assays were performed by using purified PlcR as previously described (36). A+G Maxam and Gilbert reactions (28) were carried out on the appropriate 32P-labeled DNA fragments and loaded alongside the DNase I footprinting reactions. Gels were dried and analyzed by autoradiography.
Two-dimensional gel electrophoresis.
Two-dimensional gel electrophoresis was conducted as described previously (15). The culture supernatant was collected 2 h after the onset of stationary phase, centrifuged, and filtered. Proteins were precipitated by using the deoxycholic acid-trichloroacetic acid method (31), washed with ethanol ether (1:1), and dissolved in a urea-thiourea-CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate}-ampholine mixture. A total of 20 μg of proteins were loaded onto each IPG strip for the first dimension and isoelectrofocusing was performed for 35,000 V·h. The strips were then equilibrated first in urea-sodium dodecyl sulfate-Tris-dithiothreitol, followed by a second equilibration in urea-sodium dodecyl sulfate-Tris-acetamide. The second dimension was done on a 10 to 12.5% gradient acrylamide gel. Gels were then silver stained and scanned for image analysis. Proteins were identified by mass spectrometry after trypsin digestion or by N-terminal sequencing by using the Edman method. Mass spectrometry and N-terminal sequencing were performed by the Unité de Recherches en Biochimie et Structure des Protéines at INRA (Jouy-en-Josas, France).
Insects and force-feeding assays.
G. mellonella eggs were hatched at 30°C, and the larvae were reared on beeswax and pollen (Naturalim). Groups of 20 last-instar G. mellonella larvae, weighing ca. 200 mg, were force-fed with spore-crystal suspensions in sterile water (10 μl/larva) by using a 0.5-by-25-mm needle (Burckard Manufacturing) and a microinjector (Burckard). The larvae were kept in individual boxes containing beeswax and pollen at 25°C. A control group was fed with sterile water. Experiments were repeated three times, and mortality was recorded daily over a 3-day period.
Statistical analysis.
Means and standard errors of the means were calculated and plotted, and the statistical significance of the difference between groups was estimated by using the log linear model.
RESULTS
Effect of the plcR-null mutation on inhA2 expression.
In B. thuringiensis strain 407 Cry− grown in LB medium, inhA2 transcription is induced at the onset of stationary phase (13). Recent data showed that the closely related species B. cereus strain ATCC 14579 produces InhA2 and that the amount of this protease is significantly decreased in the isogenic strain carrying a disrupted plcR gene (15). These findings led us to check whether InhA2 was controlled by PlcR at the transcriptional level. The present study was carried out in B. thuringiensis strain 407 Cry− as the predicted amino acid sequence of B. cereus strain ATCC 14579 InhA2 (http://www.integratedgenomics.com/genomereleases.html#list0) is 99% identical to that of B. thuringiensis InhA2 (accession no. AF421888). Moreover, the nucleotide sequences of both gene promoter regions are identical (result not shown). We thus investigated the effect of the plcR-null mutation on B. thuringiensis inhA2 expression by analyzing a plasmid transcriptional fusion between the 480-bp DNA region extending upstream from the inhA2 start codon and the lacZ gene in pHT304-18′Z. The recombinant plasmid (pHT304ΩinhA2′-Z) was introduced into B. thuringiensis wild-type and ΔplcR mutant strains. Cells were cultured at 30°C in LB medium, and β-galactosidase production was monitored at different stages of bacterial growth between t−1 and t+3 (tn indicates the number of hours before [−] or after [+] the onset of the stationary phase). The inhA2′-lacZ fusion was not expressed when introduced into the ΔplcR mutant strain (β-galactosidase level of <10 Miller units), whereas its expression increased during the stationary phase when introduced into the wild-type strain (Fig. 1). These results indicate that PlcR positively controls inhA2 expression during bacterial growth.
FIG. 1.

PlcR is a positive regulator of B. thuringiensis inhA2 expression. Specific β-galactosidase activity (Miller units) of strain 407 Cry− (▪) and strain 407 Cry− ΔplcR (□) harboring the transcriptional inhA2′-lacZ fusion. The cells were grown in LB medium at 30°C.
PlcR directly activates inhA2 expression.
Analysis of the inhA2 promoter sequence revealed an atypical PlcR box (5′-CATGCAATTTTGCATA-3′) located 5 bp upstream of the previously identified −35 promoter sequence (Fig. 2A). This box displayed one mismatch compared to the reported PlcR binding consensus sequence (5′-TATGNAN4TNCATA-3′) (1). This mismatch concerned the first nucleotide of the PlcR box 5′, a thymine, which was substituted with a cytosine in the inhA2 gene. To investigate whether PlcR directly activates inhA2 gene expression by binding this putative PlcR box, we performed a DNase I footprinting experiment with purified PlcR and the inhA2 DNA promoter region extending 213 bp upstream and 112 bp downstream of the transcription start site (Fig. 2B). The footprinting assay showed that PlcR binds to the promoter region and that the area protected by this pleiotropic regulator exactly overlaps the putative PlcR box.
FIG. 2.

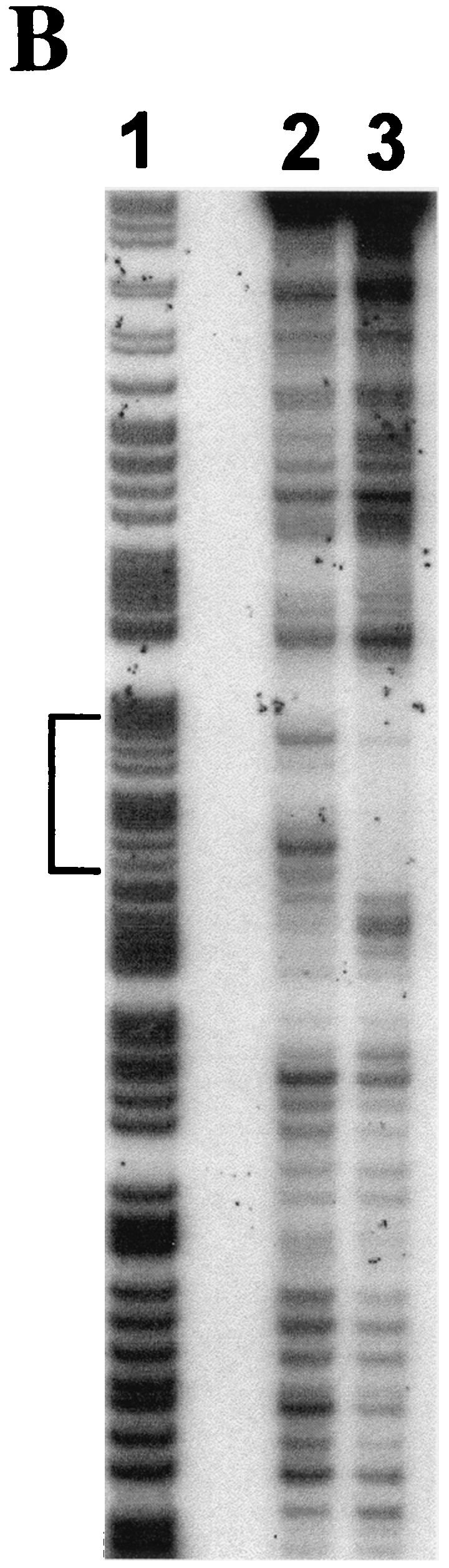
PlcR directly activates inhA2 transcription. Sequence of the inhA2 promoter region (A). The putative −10 and −35 boxes of the inhA2 promoter are boxed. The potential ribosome-binding site and the start codon (ATG) are underlined. The potential reverse Spo0A box and the PlcR box are shaded. The broken arrow indicates the transcription start site. Arrows indicate the positions of primers used in the DNase I footprinting experiments. DNase I footprinting analysis of PlcR binding to the inhA2 promoter region (B). Lane 1 indicates the A+G Maxam and Gilbert reactions of the labeled template strand of inhA2 promoter region. Lane 2, no PlcR; lane 3, 100 pmol of purified PlcR. Brackets indicate the region protected by PlcR.
Effect of InhA2 production on the pathogenicity of B. thuringiensis ΔplcR spores against orally infected G. mellonella larvae.
PlcR and InhA2 play a major role in the pathogenicity of B. thuringiensis against G. mellonella infected via the oral route (13, 33). We evaluated the synergistic effect of the spores on the insecticidal activity of the crystal protein Cry1C. Since the expression of inhA2 depends on PlcR, the loss of synergy with ΔplcR spores might be due to the absence of the InhA2 protein. To test this hypothesis, we measured the virulence of a ΔplcR strain expressing the inhA2 gene. A ΔplcR strain was complemented by introducing a plasmid harboring the inhA2 coding sequence cloned downstream of the constitutive aphA3 gene promoter. This recombinant plasmid was also introduced into the 407 Cry− ΔinhA2 mutant strain to verify that InhA2 complements the avirulent InhA2− phenotype. B. thuringiensis ΔplcR and ΔinhA2 mutant strains, carrying the inhA2 gene under the control of the aphA3 promoter, were referred to as 407 Cry− [ΔplcR (pinhA2)] and 407 Cry− [ΔinhA2 (pinhA2)], respectively. Two-dimensional electrophoresis was performed on extracellular proteins as described previously (15). Dense spots corresponding to InhA2 were observed in the wild-type strain (Fig. 3A). These spots were abolished in the ΔinhA2 strain (data not shown). In the ΔplcR strain, faint spots, migrating to the same pI and MW as InhA2, were detected (Fig. 3B). Mass spectrometry failed to identify these faint spots, which might be unrelated to InhA2. However, the presence of small amounts of InhA2 in the ΔplcR culture supernatant, even though the plasmid transcriptional fusion showed that inhA2 is not expressed in this mutant, may reflect a weak transcriptional activity of inhA2 in a chromosomal context. The mutant strains 407 Cry− [ΔplcR (pinhA2)] and 407 Cry− [ΔinhA2 (pinhA2)] secreted the InhA2 protease and the InhA2 spots for these strains were as dense as for the wild-type strain (Fig. 3C and D).
FIG. 3.

Two-dimensional gel electrophoresis of B. thuringiensis culture supernatants collected 2 h after the onset of the stationary phase. The gel area shown is the InhA/InhA2 zone, between isoelectric points 5.1 (left) and 5.9 (right) and in the molecular mass range from 70 to 90 kDa. (A) Strain 407 Cry−; (B) strain 407 Cry− ΔplcR; (C) strain 407 Cry− [ΔplcR (pinhA2)]; (D) strain 407 Cry− [ΔinhA2 (pinhA2)]. ColB, collagenase B; Ypr5, enhancin; NheB, component B of enterotoxin Nhe.
We next assessed the synergistic activity of B. thuringiensis 407 Cry−, 407 Cry− ΔplcR, 407 Cry− [ΔplcR (pinhA2)], and 407 Cry− [ΔinhA2 (pinhA2)] spores on the killing effect of the Cry1C toxin (Fig. 4). The ingestion of Cry1C-containing crystals alone (3.3 μg of protein/larva) or of spores alone (2 × 106 spores/larva) resulted in very low levels of mortality (<13% for the crystals and 0% for the spores from all B. thuringiensis strains). The coingestion, of the same concentrations of Cry1C-containing crystals and spores from the parental 407 Cry− strain resulted in a significant increase in mortality, demonstrating synergy. As reported previously (13), spores from the ΔinhA2 mutant were not synergistic. However, the 407 Cry− [ΔinhA2 (pinhA2)] spores completely recovered the wild-type virulent phenotype, thus demonstrating that the complemented strain was functional. In contrast, the 407 Cry− [ΔplcR (pinhA2)] spores were as ineffective as the parental ΔplcR mutant strain to provide a synergistic or even a cumulative effect on Cry1C insecticidal activity. These results indicate that the effect of plcR inactivation on synergy is not reversed by the presence of the InhA2 metalloprotease in bacterial cells.
FIG. 4.

Contribution of InhA2 to the virulence of B. thuringiensis against insects. Last-instar G. mellonella larvae were force-fed with spores alone (2 × 106 spores/larva), crystals alone (3.3 μg/larva) or spore-crystal mixtures. For all of the strains, spores alone caused no mortality. Vertical bars indicate the standard error of the mean. The results were obtained from three pooled independent experiments.
DISCUSSION
We previously reported that B. thuringiensis strain 407 Cry− possesses two related genes, inhA and inhA2, encoding putative zinc-requiring metalloproteases (13). inhA and inhA2 are inversely regulated by the sporulation factor Spo0A. inhA appears to be positively regulated by Spo0A and thus to be overproduced in sporulation-specific medium (17), whereas inhA2 is repressed by Spo0A and needs a relatively rich medium (such as LB) to be expressed (13). In the present study, we showed that the expression of inhA2 depends on the pleiotropic regulator PlcR. Although a reverse Spo0A box has been identified downstream of the inhA2 promoter (13) (Fig. 2A), there is no evidence that Spo0A directly represses inhA2 expression, and inhA2 repression might be due to the inhibitory effect of Spo0A on plcR. Indeed, the transcription of plcR is repressed by Spo0A, resulting in the complete loss of plcR expression when the cells are grown in a sporulation-specific medium (26).
PlcR activates the expression of several potential virulence genes in B. cereus and B. thuringiensis at the end of the exponential growth phase. These genes encode secreted proteins, including phospholipases C, hemolysins, enterotoxins, and proteases (1, 25, 30). PlcR acts by binding to a highly conserved palindromic sequence (TATGNAN4TNCATA) located in the promoter region of PlcR-regulated genes (1, 36). Our footprinting experiments show that PlcR activates inhA2 transcription directly by binding to the DNA sequence (5′-CATGNAN4TNCATA-3′) located just upstream of the −35 consensus promoter box of inhA2. This sequence differs from the previously defined PlcR box by one residue (C1 versus T1) (1), but this substitution does not result in a loss of function. Thus, this DNA sequence is a PlcR recognition target for inhA2 activation. This is the first example of a functional PlcR box that differs from that previously published. A similar DNA motif is present upstream of the inhA2 coding sequence in B. cereus strain ATCC 14579. The observation that InhA2 production was significantly diminished in this B. cereus strain carrying a disrupted plcR gene (15) is consistent with the expression of inhA2 being dependent on PlcR in B. cereus.
B. thuringiensis spores significantly synergize the insecticidal activity of Cry proteins when coingested by susceptible larvae, and this synergy is abolished by the disruption of the plcR and inhA2 genes (13, 33). PlcR-regulated toxins and degradative enzymes may facilitate the spread of the bacterium through host tissues, thus allowing bacterial cells to gain access to alternative sources of nutrients and to cause septicemia. Since phospholipases C, enterotoxins, or hemolysins were not found to be essential on their own in providing synergism (D. Lereclus, unpublished data), inhA2 is the first example of PlcR-regulated gene shown to be essential for virulence. This raised the question as to whether the absence of InhA2 alone could explain the avirulent phenotype of the ΔplcR mutant. To answer this question, we tested the virulence of a ΔplcR strain complemented with the inhA2 gene. The results showed that spores from this strain were as inefficient as the ΔplcR spores in potentiating the toxic effect of crystals. InhA2 was, however, able to complement the ΔinhA2 virulence defect. There are two hypotheses to explain why InhA2 was unable to compensate for the absence of PlcR. First, the correct maturation of InhA2 requires the product of a PlcR-regulated gene, and thus InhA2 would be not functional in B. thuringiensis strain 407 Cry− [ΔplcR (pinhA2)]. Alternatively, InhA2 might have to cooperate with one or several unidentified PlcR-regulated factors. The possible cooperative properties of members of the PlcR regulon highlighted the multifactorial characteristic of B. thuringiensis virulence against orally infected insects. The identification of these factors may improve our understanding of the genetic and biochemical bases of the interaction between B. thuringiensis and its host after oral infection.
Acknowledgments
We are grateful to Alexei Sorokine for kindly providing us with the inhA2 nucleotide sequence from the finished B. cereus genome database. We thank Alex Edelman and Associates for correcting the English.
This work was supported by a grant from INRA (Département Santé des Plantes et Environnement). S.F. was supported by grants from the Tunisian government and INRA.
REFERENCES
- 1.Agaisse, H., M. Gominet, O. A. Økstad, A. B. Kolstø, and D. Lereclus. 1999. PlcR is a pleiotropic regulator of extracellular virulence factor gene expression in Bacillus thuringiensis. Mol. Microbiol. 32:1043-1053. [DOI] [PubMed] [Google Scholar]
- 2.Agaisse, H., and D. Lereclus. 1994. Expression in Bacillus subtilis of the Bacillus thuringiensis cryIIIA toxin gene is not dependent on a sporulation-specific sigma factor and is increased in a spo0A mutant. J. Bacteriol. 176:4734-4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arantes, O., and D. Lereclus. 1991. Construction of cloning vectors for Bacillus thuringiensis. Gene 108:115-119. [DOI] [PubMed] [Google Scholar]
- 4.Beecher, D. J., J. S. Pulido, N. P. Barney, and A. C. Wong. 1995. Extracellular virulence factors in Bacillus cereus endophthalmitis: methods and implication of involvement of hemolysin BL. Infect. Immun. 63:632-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bravo, A., H. Agaisse, S. Salamitou, and D. Lereclus. 1996. Analysis of cryIAa expression in sigE and sigK mutants of Bacillus thuringiensis. Mol. Gen. Genet. 250:734-741. [DOI] [PubMed] [Google Scholar]
- 6.Callegan, M. C., D. C. Cochran, S. T. Kane, M. S. Gilmore, M. Gominet, and D. Lereclus. 2002. Contribution of membrane-damaging toxins to Bacillus endophthalmitis pathogenesis. Infect. Immun. 70:5381-5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlson, C. R., T. Johansen, and A. B. Kolsto. 1996. The chromosome map of Bacillus thuringiensis subsp. canadensis HD224 is highly similar to that of the Bacillus cereus type strain ATCC 14579. FEMS Microbiol. Lett. 141:163-167. [DOI] [PubMed] [Google Scholar]
- 8.Daffonchio, D., A. Cherif, and S. Borin. 2000. Homoduplex and heteroduplex polymorphisms of the amplified ribosomal 16S-23S internal transcribed spacers describe genetic relationships in the “Bacillus cereus group”. Appl. Environ. Microbiol. 66:5460-5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalhammar, G., and H. Steiner. 1984. Characterization of inhibitor A, a protease from Bacillus thuringiensis which degrades attacins and cecropins, two classes of antibacterial proteins in insects. Eur. J. Biochem. 139:247-252. [DOI] [PubMed] [Google Scholar]
- 10.Donovan, W. P., J. C. Donovan, and J. T. Engleman. 2001. Gene knockout demonstrates that vip3A contributes to the pathogenesis of Bacillus thuringiensis toward Agrotis ipsilon and Spodoptera exigua. J. Invertebr. Pathol. 78:45-51. [DOI] [PubMed] [Google Scholar]
- 11.Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high-voltage electroporation. Nucleic Acids Res. 16:6127-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edlund, T., I. Siden, and H. G. Boman. 1976. Evidence for two immune inhibitors from Bacillus thuringiensis interfering with the humoral defense system of saturniid pupae. Infect. Immun. 14:934-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fedhila, S., P. Nel, and D. Lereclus. 2002. The InhA2 metalloprotease of Bacillus thuringiensis strain 407 is required for pathogenicity in insects infected via the oral route. J. Bacteriol. 184:3296-3304. [DOI] [PMC free article] [PubMed]
- 14.Gibson, T. J. 1984. Ph.D thesis. University of Cambridge, Cambridge, United Kingdom.
- 15.Gohar, M., O. A. Okstad, N. Gilois, V. Sanchis, A.-B. Kolsto, and D. Lereclus. 2002. Two-dimensional electrphoresis analysis of the extracellular proteome of Bacillus cereus reveals the importance of the PlcR regulon. Proteomics 2:784-791. [DOI] [PubMed] [Google Scholar]
- 16.Gominet, M., L. Slamti, N. Gilois, M. Rose, and D. Lereclus. 2001. Oligopeptide permease is required for expression of the Bacillus thuringiensis plcR regulon and for virulence. Mol. Microbiol. 40:963-975. [DOI] [PubMed] [Google Scholar]
- 17.Grandvalet, C., M. Gominet, and D. Lereclus. 2001. Identification of genes involved in the activation of the Bacillus thuringiensis inhA metalloprotease gene at the onset of sporulation. Microbiology 147:1805-1813. [DOI] [PubMed] [Google Scholar]
- 18.Granum, P. E., and T. Lund. 1997. Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Lett. 157:223-228. [DOI] [PubMed] [Google Scholar]
- 19.Hansen, B. M., and S. Salamitou. 2000. Virulence of Bacillus thuringiensis, p. 41-63. In J. F. Charles, A. Delécluse, and C. Nielson-Leroux (ed.), Entomopathogenic bacteria: from laboratory to field application.Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 20.Heierson, A., I. Sidén, A. Kivaisi, and H. G. Boman. 1986. Bacteriophage-resistant mutants of Bacillus thuringiensis with decreased virulence in pupae of Hyalophora cecropia. J. Bacteriol. 167:18-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helgason, E., O. A. Okstad, D. A. Caugant, H. A. Johansen, A. Fouet, M. Mock, I. Hegna, and Kolsto. 2000. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis—one species on the basis of genetic evidence. Appl. Environ. Microbiol. 66:2627-2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernandez, E., F. Ramisse, T. Cruel, R. le Vagueresse, and J. D. Cavallo. 1999. Bacillus thuringiensis serotype H34 isolated from human and insecticidal strains serotypes 3a3b and H14 can lead to death of immunocompetent mice after pulmonary infection. FEMS Immunol. Med. Microbiol. 24:43-47. [DOI] [PubMed] [Google Scholar]
- 23.Kotiranta, A., K. Lounatmaa, and M. Haapasalo. 2000. Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2:189-198. [DOI] [PubMed] [Google Scholar]
- 24.Lecadet, M. M., M. O. Blondel, and J. Ribier. 1980. Generalized transduction in Bacillus thuringiensis var. berliner 1715, using bacteriophage CP54 Ber. J. Gen. Microbiol. 121:203-212. [DOI] [PubMed] [Google Scholar]
- 25.Lereclus, D., H. Agaisse, M. Gominet, S. Salamitou, and V. Sanchis. 1996. Identification of a Bacillus thuringiensis gene that positively regulates transcription of the phosphatidylinositol-specific phospholipase C gene at the onset of the stationary phase. J. Bacteriol. 178:2749-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lereclus, D., H. Agaisse, C. Grandvalet, S. Salamitou, and M. Gominet. 2000. Regulation of toxin and virulence gene transcription in Bacillus thuringiensis. Int. J. Med. Microbiol. 290:295-299. [DOI] [PubMed] [Google Scholar]
- 27.Lereclus, D., O. Arantes, J. Chaufaux, and M.-M. Lecadet. 1989. Transformation and expression of a cloned δ-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol. Lett. 60:211-218. [DOI] [PubMed] [Google Scholar]
- 28.Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 65:499-560. [DOI] [PubMed] [Google Scholar]
- 29.Msadek, T., F. Kunst, D. Henner, A. Klier, G. Rapoport, and R. Dedonder. 1990. Signal transduction pathway controlling synthesis of a class of degradative enzymes in Bacillus subtilis: expression of the regulatory genes and analysis of mutations in degS and degU. J. Bacteriol. 172:824-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Økstad, O. A., M. Gominet, B. Purnelle, M. Rose, D. Lereclus, and A.-B. Kolstø. 1999. Sequence analysis of three Bacillus cereus loci under PlcR virulence gene regulator control. Microbiology 145:3129-3138. [DOI] [PubMed] [Google Scholar]
- 31.Peterson, G. L. 1983. Determination of total protein. Methods Enzymol. 91:95-119. [DOI] [PubMed] [Google Scholar]
- 32.Rahmet-Alla, M., and A. F. Rowley. 1989. Studies on the cellular defense reactions of the Madeira cockroach, Leucophaea maderae: nodule formation in response to injected bacteria. J. Invertebr. Pathol. 54:200-207. [DOI] [PubMed] [Google Scholar]
- 33.Salamitou, S., F. Ramisse, M. Brehelin, D. Bourguet, N. Gilois, M. Gominet, E. Hernandez, and D. Lereclus. 2000. The PlcR regulon is involved in the opportunistic properties of Bacillus thuringiensis and Bacillus cereus in mice and insects. Microbiology 146:2825-2832. [DOI] [PubMed] [Google Scholar]
- 34.Sanchis, V., H. Agaisse, J. Chaufaux, and D. Lereclus. 1996. Construction of new insecticidal Bacillus thuringiensis recombinant strains by using the sporulation non-dependent expression system of cryIIIA and a site specific recombination vector. J. Biotechnol. 48:81-96. [DOI] [PubMed] [Google Scholar]
- 35.Schnepf, E., N. Crickmore, J. Van Rie, D. Lereclus, J. Baum, J. Feitelson, D. R. Zeigler, and D. H. Dean. 1998. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol. Mol. Biol. Rev. 62:775-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Slamti, L., and D. Lereclus. 2002. A cell-cell signaling peptide activates the PlcR virulence regulon in bacteria of the Bacillus cereus group. EMBO J. 21:4550-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephens, J. M. 1952. Disease in codling moth larvae produced by several strains of Bacillus cereus. Can. J. Zool. 30:30-40. [Google Scholar]
- 38.Trieu-Cuot, P., and P. Courvalin. 1983. Nucleotide sequence of the Streptococcus faecalis plasmid gene encoding the 3′5"-aminoglycoside phosphotransferase type III. Gene 23:331-341. [DOI] [PubMed] [Google Scholar]
- 39.Zhang, M.-Y., A. Lövgren, M. G. Low, and R. Landén. 1993. Characterization of an avirulent pleitropic mutant of the insect pathogen Bacillus thuringiensis: reduced expression of flagellin and phospholipases. Infect. Immun. 61:4947-4954. [DOI] [PMC free article] [PubMed] [Google Scholar]