

Abstract
Nuclear aggregates of polyglutamine (polyQ)-expanded proteins are associated with a number of neurodegenerative diseases including Huntington’s disease (HD) and spinocerebellar ataxias (SCAs). The nuclear deposition of polyQ proteins correlates with rearrangements of nuclear matrix, transcriptional dysregulation, and cell death. To explore the requirement for polyQ tracks in educing such cellular responses, we examined whether a non-polyQ protein can deposit as nuclear aggregates and elicit similar responses. We report that a protein chimera (GFP170*) composed of the green fluorescent protein (GFP) fused to an internal fragment of the Golgi Complex Protein (GCP-170) forms nuclear aggregates analogous to those formed by polyQ proteins. Like the polyQ nuclear aggregates, GFP170* inclusions recruit molecular chaperones and proteasomal components, alter nuclear structures containing the promyelocytic leukemia protein (PML), recruit transcriptional factors such as CREB-binding protein (CBP) and p53, repress p53 transcriptional activity, and induce cell death. Our results indicate that nuclear aggregation and transcriptional effects are not unique to polyQ-containing proteins and may represent a general response to misfolded proteins in the nucleus.
Keywords: Nuclear aggregates, Nuclear inclusions, Polyglutamine, Huntingtin, Ataxin, CBP, p53, GCP-170, Golgin 160
Introduction
At least nine neurodegenerative diseases, including Huntington’s disease (HD), spinobulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy (DRPLA), and spinocerebellar ataxias (SCA) 1, 2, 3, 6, 7, and 17, are caused by a single type of mutation, the expansion of CAG repeats encoding for a polyglutamine (polyQ) track in unrelated proteins (Zoghbi and Orr, 2000). The mutant proteins form protein aggregates or inclusions that are the hallmark of polyQ diseases (Ross, 2002). Unlike other mutant proteins that form cytoplasmic aggregates in diseases such as Alzheimer’ and Parkinson’s diseases or amyo-trophic lateral sclerosis, polyQ proteins can be deposited as cytoplasmic inclusions as well as nuclear inclusions. The nuclear deposition of polyQ proteins has been correlated with cytotoxicity. Transgenic mice expressing polyQ human huntingtin develop neuronal intranuclear inclusions prior to developing a neurological phenotype (Davies et al., 1997). Similarly, nuclear localization of polyQ proteins is essential to induce cell death in cultured cell and transgenic mouse models (Katsuno et al., 2002; Klement et al., 1998; Takeyama et al., 2002). However, the exact correlation between nuclear polyQ aggregates and pathology remains elusive. It is apparent that not all neurons containing polyQ aggregates die (Ross et al., 1998), and temporal studies of the appearance of aggregates and the onset of clinical pathology suggest that tissue damage and pathology can manifest before detection of aggregates (Saudou et al., 1998).
PolyQ pathogenesis may be linked to the sequestration and inactivation of proteins essential for cellular functions. Analyses of human post-mortem brains, animal models, and cell culture systems have shown that polyQ deposits recruit various cellular components. Invariably, proteins involved in protein folding and degradation, as well as transcriptional regulators are associated with polyQ aggregates (Li and Li, 2004). All the major classes of chaperones including members of the Hsp70 family (Hsc70 and Hsp70) and the Hsp40 family (Hdj1 and Hdj2) are recruited. Like chaperones, proteasomes have been shown to be associated with polyQ aggregates (Waelter et al., 2001). Often, polyQ inclusions are ubiquitin positive (DiFiglia et al., 1997). The sequestration of folding/degradative machinery to protein aggregates results in compromised proteasomal degradation (Bence et al., 2001).
Nuclear factors also have been shown to interact with polyQ nuclear inclusions (Okazawa, 2003). Nuclear aggregates of ataxin-1 recruit the promyelocytic leukemia protein (PML), a component of nuclear PML bodies (Skinner et al., 1997). Direct association of the CREB binding protein (CBP) with polyQ aggregates has been observed in HD cell culture models, HD transgenic mice, and human HD post-mortem brain (Nucifora et al., 2001). Sp1 and p53 also interact with polyQ huntingtin fragments (Nucifora et al., 2001; Steffan et al., 2000).
The sequestration of transcription factors by aggregates appears to alter their transcriptional activity. Specifically, polyQ expanded huntingtin and atrophin-1 (responsible for DRPLA) decrease CBP-mediated transcription in transfected primary cortical neurons (Nucifora et al., 2001). Similarly, polyQ expanded huntingtin represses transcription of a p53 reporter construct (Steffan et al., 2000). Genomic screens have shown that CBP-regulated genes, such as eukepholin and Jun, are downregulated in HD transgenic mice and in HD post-mortem brains (Luthi-Carter et al., 2000; Richfield et al., 1995). The inhibition of transcription may be a consequence of direct binding since CBP and p53 interact directly with polyQ tracks of huntingtin (Nucifora et al., 2001; Steffan et al., 2000) and with the polyQ tracks of androgen receptor that causes SBMA (McCampbell et al., 2000). Transcriptional regulators such as CBP and TATA binding protein (TBP) contain polyQ stretches, suggesting that complementary polyQ–polyQ interactions may mediate the sequestration and the inactivation.
The ability of nuclear polyQ aggregates to recruit folding/degradative cellular components, disrupt nuclear architecture, sequester transcriptionally relevant proteins, and alter transcriptional activity of the sequestered factors may be specific to the polyQ content or may represent a general cellular response to nuclear inclusions. To address this question, it is necessary to examine whether non-polyQ proteins can form nuclear inclusions and elicit similar cellular effects as polyQ aggregates. Here, we document that a non-polyQ protein (GFP170*), which contains GFP fused to an internal segment (amino acids 566 to 1375) of the Golgi Complex Protein 170 (GCP170), forms nuclear aggregates analogous to those deposited by polyQ proteins. GCP-170, also known as golgin-160, is a Golgi localized protein that associates peripherally with the cytoplasmic side of Golgi membranes (Hicks and Machamer, 2002; Misumi et al., 1997). GCP-170 was identified as a Golgi auto-antigen in sera of patients suffering from Sjorgen Syndrome (Fritzler et al., 1993). The cellular function of GCP170 is currently unknown. The internal fragment of GCP170 used here to generate GFP170* represents an artificial substrate that lacks a polyQ-expanded sequence. The ability of GFP170* to form nuclear aggregates suggests that the formation of nuclear inclusions is not a polyQ-specific process. Furthermore, like deposits of polyQ proteins, the nuclear aggregates of GFP170* recruit molecular chaperones and proteasomal components, cause a redistribution of PML bodies, and sequester transcription factors such as CBP and p53. In addition, expression of GFP170* represses p53 transcriptional activity and causes cell death. The similarity in cellular responses elicited by polyQ proteins and our non-polyQ GFP170* is consistent with the hypothesis that those responses are common to the presence of any misfolded proteins in the nucleus. Our findings raise the possibility that the etiology of diverse polyQ diseases such as HD, SBMA, DRPLA, and ataxias may share a set of common cytopathologies elicited solely by nuclear inclusions (irrespective of the polyQ content of the protein), in addition to specific responses elicited by distinct polyQ proteins.
Materials and methods
Antibodies and reagents
Polyclonal anti-GFP antibody was from Abcam Inc. (Cat. # AB-290). Anti-CBP (A-22) (Cat. # sc-369) polyclonal antibody and anti-SC35 (Y-16) (Cat. # sc-10251) monoclonal antibody were purchased from Santa Cruz Biotechnology, Inc. Anti-Hsp70 (Cat. # SPA-810) monoclonal antibody was purchased from Stressgen Biotechnologies. Anti-Hdj2 polyclonal antibody was a gift from Dr. Douglas Cyr (University of North Carolina at Chapel Hill). Anti-20S proteasome (α-subunit) polyclonal antibody was purchased from Calbiochem-Novabiochem. Mouse anti-GMP-1 monoclonal antibody (clone 21C7) recognizes the SUMO-1 protein was from Zymed Laboratories Inc. A combined monoclonal anti-p53 antibody, 1801 and 421, was kindly provided by Dr. Xinbin Chen (University of Alabama at Birmingham). Texas red-labeled goat anti-mouse IgG antibody, Texas red-labeled goat anti-rabbit IgG antibody, and Hoechst 33258 were from Molecular Probes, Inc. Restriction enzymes and molecular reagents were from Promega, New England BioLabs, Inc., or QIAGEN. All other chemicals were from Sigma-Aldrich or Fisher Scientific.
DNA constructs
To make a chimera of GFP and GCP-170, an XhoI restriction enzyme site was generated in front of the start codon of GCP-170. The 770-base pair PCR fragment containing sequences from the start codon of GCP-170 to the EcoRI site of FQSY1024 (Misumi et al., 1997) was cloned into the XhoI and EcoRI sites of pEGFP-C2 plasmid (Clontech Laboratories Inc.). A 5558-base-pair EcoRI fragment from FQSY1024 was then cloned to the EcoRI sites of the plasmid above to generate an EGFP-tagged full-length GCP-170 (GFP-GCP170FL). GFP170* construct was then generated by removing the BglII fragment and SacII fragment from the N-terminal and C-terminal end of GCP-170, respectively. The resulted construct expresses an EGFP-tagged GCP-170 fragment from amino acid 566 to 1375. The Q80-GFP construct has been described previously (Ding et al., 2002) and was kindly provided by Dr. Qunxing Ding (University of Kentucky). The construct expressing firefly luciferase under the control of the p21 promoter with two p53-responsive elements was provided by Dr. Xinbin Chen and has been described previously (Chinery et al., 1997).
Cell culture, transfections, and immunofluorescence microscopy
COS-7 cells were grown in DMEM with glucose and glutamine (Mediatech, Inc.) supplemented with 10% FBS (Life Technologies), 100 U/ml penicillin, and 100 μg/ml streptomycin (Life Technologies). Cortical neurons isolated from mouse were cultured in Neurobasal Media (Cat.#21103-049, GIBCO) supplemented with B27 (Cat.#17504-010, GIBCO). Cells were transfected with the Fugene transfection reagent (Roche) or with TransIT polyamine transfection reagents (Mirus Corporation), according to manufacturer protocols. 18–48 h after transfection, cells were fixed with 3% paraformaldehyde and processed for immunofluorescence microscopy as previously described (Alvarez et al., 1999).
Electron microscopy and immunogold labeling
Cells were transfected with the GFP170* construct or the Q80-GFP construct. 48 h after transfection, cells were washed with PBS, detached from the plate by trypsinization, and collected by centrifugation at 300 × g for 5 min at 4°C. Cells were washed twice with PBS and then fixed for 90 min with 1.5% glutaraldehyde in 0.1 M sodium cacodylate pH 7.4. Cells were then washed three times with sodium cacodylate and postfixed with 1% OsO4 in 0.1 M sodium cacodylate pH 7.4 for 60 min on ice. After washing three times with 0.1 M sodium cacodylate pH 7.4, cells were dehydrated with a series of ethanol solutions (30, 50, 70, 90, 95, and 3 × 100%) followed by 2 h incubation in 1:1 Spurr’s resin/propylene oxide. After two changes of fresh 100% resin, the cell pellets were transferred to gelatin molds and polymerized in fresh resin overnight at 60°C. Gold epoxy sections (100 nm thick) were generated with a Reichert Ultracut ultramicrotome and collected on 200 mesh copper grids. The grid specimens were stained for 20 min with saturated aqueous uranyl acetate (3.5%) diluted 1:1 with ethanol just before use followed by staining with lead citrate for 10 min. Stained samples were examined on a JEOL 100CX electron microscope.
For immunogold electron microscopy, cells expressing GFP170* were harvested by trypsinization 24 h after transfection. Cells were washed with PBS and pre-fixed with 3% formaldehyde and 0.2% glutaraldehyde for 40 min followed by dehydration with series of graded ethanol at room temperature. The cells were then infiltrated and embedded with LR white. After polymerization, sections were cut with ultramicrotome and collected onto nickel grids. The grids were incubated with anti-GFP primary antibody and goat anti-rabbit IgG conjugated to 6-nm gold particles (Jackson ImmunoResearch Laboratories, Inc.) followed by postfixation with 2% glutaraldehyde and counterstaining with uranyl acetate. Samples were then examined on a JEOL 100CX electron microscope.
Analysis of soluble and insoluble GFP170*
COS-7 cells were either mock-transfected with PBS or transfected with GFP170* construct. 48 h after transfection, cells were washed and harvested in ice-cold PBS. Cells were then lysed for 1 h on ice with RIPA buffer (50 mM Tris–HCl, pH 8.0, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, and 150 mM NaCl) supplemented with protease inhibitor cocktail and 1.0 mM PMSF. Lysates were sonicated for 5 s with microtip sonicator followed by 15 min centrifugation at 15,000 × g. Pellets were washed 2× with RIPA buffer and resuspended with equal volume of RIPA buffer. Equal volumes of samples from the total cell lysate, supernatant, and pellet fractions were boiled in SDS-PAGE sample buffer and resolved on 8% SDS-PAGE. The gel was transferred to nitro-cellulose membrane and processed for Western blotting as previously described (Gao and Sztul, 2001).
Measurement of DNA synthesis
COS-7 cells were transfected with either GFP170* or pEGFP-C2 (BD Bioscience). 32 h after transfection, the cells were incubated with 30 μM BrdU for 14 h followed by immunofluorescent staining with anti-BrdU monoclonal antibody, PRB-1 (Molecular Probes).
Measurement of cell viability by FACS analysis
COS-7 cells were mock-transfected or transfected with GFP170* or Q80-GFP. 48 h after transfection, cells were detached from the plate by trypsinization. Cells were then incubated with a red fluorescent dye L-23102 for 30 min at room temperature. Live cells exclude the dye and therefore can be separated from dead cells based on their low fluorescence intensity. Cells were then fixed with formaldehyde and washed with PBS followed by FACS analysis. Cells were first gated according to the intensity of green fluorescence. Dead cells in GFP-negative or GFP-positive groups were counted separately.
Luciferase assay
COS7 cells in 6-well plates were transfected with 300 ng luciferase expressing vector and 300 ng of pcDNA3.1 vector alone or vector expressing Q80-GFP or GFP170*. 48 h after transfection, cell lysates were made using the passive lysis buffer in the Dual Luciferase Assay system form Promega according to the manufacturer’s instructions. Luciferase activity in the lysate was measured with a Luminometer from Promega. The protein concentrations of the lysates were determined by Bradford analysis, and luciferase activity was calculated per milligram of protein and then normalized to the activity in the control sample.
Results
GFP170* forms cytoplasmic and nuclear aggregates
GCP-170 contains 1530 amino acids, arranged into an N-terminal head domain followed by a long stalk regions and a short C-terminal tail. The stalk region consists of 6 coiled-coil domains. The coiled-coil domains of GCP170 may be responsible for its dimerization (Hicks and Machamer, 2002; Misumi et al., 1997). Coiled-coil domains are known to mediate protein–protein interactions and may enhance the propensity of a protein to aggregate. GCP170 has been shown to be aggregation-prone in vitro (Misumi et al., 1997). In agreement, GFP-tagged full-length GCP-170 protein (GFP-GCP170FL) forms aggregates when over-expressed in COS-7 cells (Fig. 1A, insert, arrows).
Fig. 1.
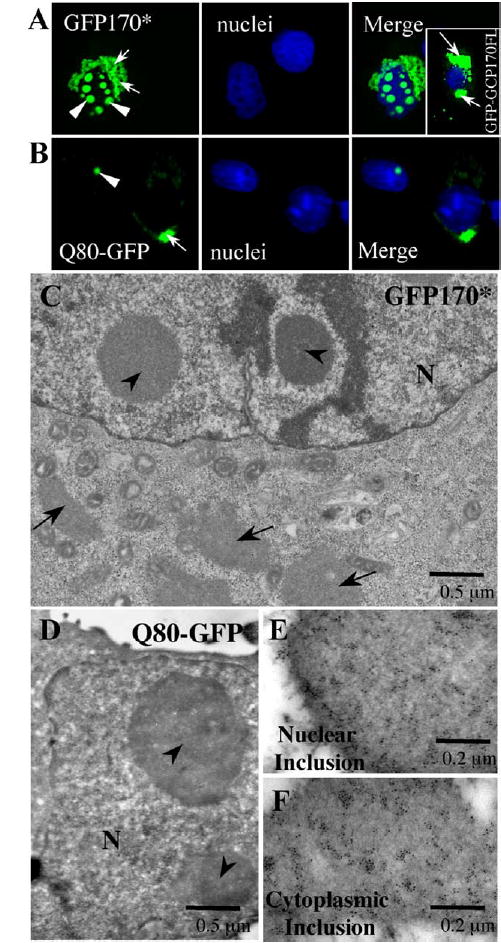
GFP170* forms cytoplasmic and nuclear aggregates. COS-7 cells were transfected with GFP170*, GFP-GCP170FL, or Q80-GFP construct. 48 h after transfection, cells were fixed and processed for fluorescence (A, B), electron microscopy (C, D), or immunogold labeling (E, F). (A and B) Light microscopic analysis of GFP170*, GFP-GCP170FL, and Q80-GFP aggregates. The nuclei are stained with Hoechst 33258. Arrows point to cytoplasmic aggregates. Arrowheads point to nuclear inclusions. (C and D) Ultrastructure of the GFP170* and the Q80-GFP aggregates. The cytoplasmic GFP170* aggregates (arrows in C) have irregular shapes and are frequently surrounded by mitochondria. Nuclear aggregates (arrowheads in panels C and D) range in size from 0.5 to 3 μm and are spherical or ovoid. (E and F) Cytoplasmic (E) and nuclear (F) inclusions of GFP170* are labeled with anti-GFP antibodies conjugated to gold particles. N, nucleus.
Here, we have generated a chimera by fusing in frame an internal segment of the coiled-coil region of GCP170 composed of amino acids 566 to 1375 to the C-terminus of the enhanced green fluorescent protein (GFP). The resulting construct is called GFP170*. GFP170* does not contain polyQ repeats (Misumi et al., 1997). When transiently expressed in COS-7 cells, GFP170* deposits as cytoplasmic aggregates in the peri-nuclear region (Fig. 1A, arrows). The aggregates appear “ribbon-like” and are significantly more dispersed than the ‘‘ball-like’’ aggregates formed by GFP-250 (Garcia-Mata et al., 1999) or CFTR (Johnston et al., 1998). The GFP170* aggregates appear concentrated around the nucleus, but in some cases extend into the periphery of the cell. In addition to the cytoplasmic inclusions, GFP170* deposits in spherical foci within the nucleus (Fig. 1A, arrowheads). The morphology of cytoplasmic and nuclear GFP170* aggregates was compared to those formed by a model polyQ protein (Q80-GFP) (Fig. 1B). Q80-GFP encodes a fusion protein containing an 80-glutamine expansion fused to the amino-terminus of GFP. Q80-GFP has been shown to deposit in characteristic cytoplasmic and nuclear aggregates (Onodera et al., 1997). The cytoplasmic inclusions formed by Q80-GFP are irregular in shape (arrow), while the nuclear aggregates are spherical (arrowheads) and resemble the GFP170* aggregates. The Q80-GFP cytoplasmic inclusions localize to the peri-centriolar region but appear more compact than those of GFP170*. Q80-GFP forms one or two aggregates per nucleus, while GFP170* forms multiple inclusions per nucleus.
We examined the ultrastructure of GFP170* aggregates by transmission electron microscopy (Fig. 1C). The cytoplasmic aggregates (arrows) can extend to more than 15 μm in length. They are often surrounded by mitochondria, similar to the close association of mitochondria with the cytoplasmic aggregates formed by the HDQ83 huntingtin mutant (Waelter et al., 2001). The nuclear aggregates of GFP170* (arrowheads) are spherical or ovoid and range from 0.5 μm to 3 μm in diameter. They are similar to the nuclear inclusions formed by the Q80-GFP (Fig. 1D). In both cases, the nuclear aggregates appear as homogenous accumulations of granular material, without apparent fibrillar content or subdomain structures. Non-transfected control cells never contain cytoplasmic or nuclear aggregates (data not shown). The deposition of GFP170* within the morphologically defined cytoplasmic and nuclear aggregates was confirmed by immunogold labeling with anti-GFP antibodies. Gold particles label the cytoplasmic and nuclear aggregates (Figs. 1E and F).
The clinically relevant deposits of polyQ proteins occur in neuronal cells (Ross, 2002). To test if GFP170* aggregates also form in neuronal cells, we expressed GFP170* in mouse primary cortical neurons. Q80-GFP was used in analogous transfections to allow direct comparisons. As shown in Fig. 2, both GFP170* and Q80-GFP form cytoplasmic and nuclear aggregates in cultured mouse primary cortical neuronal cells. Their morphologies are similar. Like in COS-7 cells, GFP170* forms multiple nuclear inclusions, while Q80-GFP deposits within a single structure. Similar results were also obtained with PC12 cells, a rat neuronal cell line (data not shown). These results indicate that GFP170* forms aggregates in neuronal cells that are morphologically indistinguishable from those formed in non-neuronal cells. This suggests that results obtained in COS-7 cells may be applicable to neuronal cells. In our studies, COS-7 cells are used because of their ease of culture and transfection.
Fig. 2.
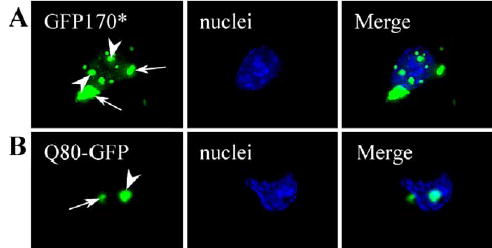
GFP170* and Q80-GFP form cytoplasmic and nuclear aggregates in neuronal cells. Mouse primary cortical neurons were transfected with GFP170* (A) or Q80-GFP (B). 48 h after transfection, cells were fixed, and the nuclei were stained with Hoechst 33258. Arrows point to cytoplasmic aggregates. Arrowheads point to nuclear inclusions.
GFP170* aggregates recruit chaperones and proteasomal components
A characteristic feature of polyQ aggregates that parallels cytopathology is the recruitment of various cellular components. Specifically, polyQ aggregates have been shown to recruit molecular chaperones and proteasomes. Inclusions of polyQ-expanded huntingtin recruit proteasomal subcomplexes 20S, 11S, and 19S and the chaperones BIP, HSP70, and HSP40 (Waelter et al., 2001). Similarly, polyQ-expanded androgen receptor aggregates recruit HSP70 (Kobayashi et al., 2000). This may facilitate the degradative clearance of the aggregates (Cummings et al., 1998). Therefore, we examined the recruitment of similar components by GFP170* aggregates. Hsp70 and Hdj2, representatives of the Hsp70 and the Hsp40 families of chaperones, respectively, are recruited to the nuclear as well as cytoplasmic GFP170* deposits (Figs. 3A and B). In addition to chaperones, proteasomal components are also recruited to the cytoplasmic and nuclear GFP170* aggregates (Fig. 3C). Another important feature of the polyQ aggregates is that they are usually detergent insoluble (Perez et al., 1998; Waelter et al., 2001). We examined the solubility of GFP170* aggregates after lysing cells in buffer containing detergent. GFP170* is largely insoluble in a RIPA buffer containing 0.1% SDS (Fig. 3D). The recovery of cytosolic β-tubulin in the soluble fraction provides an internal control for the efficacy of the solubilization.
Fig. 3.
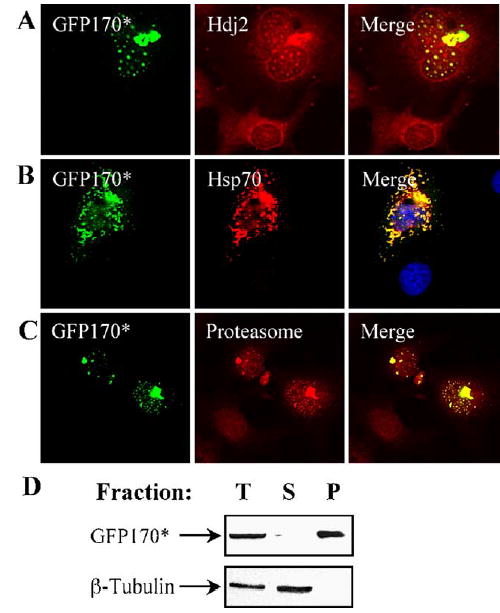
GFP170* aggregates recruit chaperones and proteasomes. COS-7 cells were transfected with GFP170*. 48 h after transfection, cells were processed for immunofluorescence using antibodies against Hdj2 (A), Hsp70 (B), or the proteasomal α-subunit (C). Recruitment to cytosolic and/or nuclear aggregates is evident. (D) COS-7 cells were transfected with GFP170* and were lysed 48 h later in RIPA buffer containing 0.1% SDS. The soluble and insoluble fractions were separated by centrifugation. Equivalent amounts of total cell lysate (T) and the soluble (S) and the insoluble pellet (P) fractions were resolved by SDS-PAGE and immunoblotted with antibodies against GFP or β-tubulin. Most of GFP170* is present in the insoluble fraction.
Nuclear aggregates of GFP170* cause redistribution of PML bodies
Nuclear inclusions of polyQ proteins have been shown to recruit nuclear structures containing the promyelocytic leukemia protein (PML bodies) (Skinner et al., 1997). PML bodies are also called nuclear domain 10 (ND10) bodies or PML oncogenic domains (PODs). The mammalian nucleus contains 10 to 30 PML bodies, which vary in size from 0.2 to 1 μm. They are thought to function in transcriptional regulation, cell cycle progression, and apoptosis, based on their content of proteins such as Sp100, PML, Daxx, pRB, CBP, and p53 that are involved in these processes (Maul et al., 2000; Yasuda et al., 1999; Zhong et al., 2000). Disruption of PML bodies caused by the (t15;17:q22;q21) translocation that results in a fusion of the PML protein and the retinoic acid receptor alpha (RARα) leads to acute promyelocytic leukemia (Weis et al., 1994).
GFP170* aggregates also recruit PML bodies (Figs. 4A and B). In normal untransfected cells, PML bodies are detected as numerous small nuclear foci (Figs. 4A and B, arrowheads). PML bodies with similar morphology are also evident in a cell expressing GFP170* at low levels (Fig. 4A, arrows). A distinct phenotype is observed in cells expressing high levels of GFP170* and displaying large nuclear aggregates (Fig. 4B). In such cells, PML bodies re-distribute to the surface of the GFP170* aggregates (Fig. 4B, arrows). Our results indicate that, like polyQ proteins, GFP170* causes changes in the nuclear architecture of PML bodies.
Fig. 4.
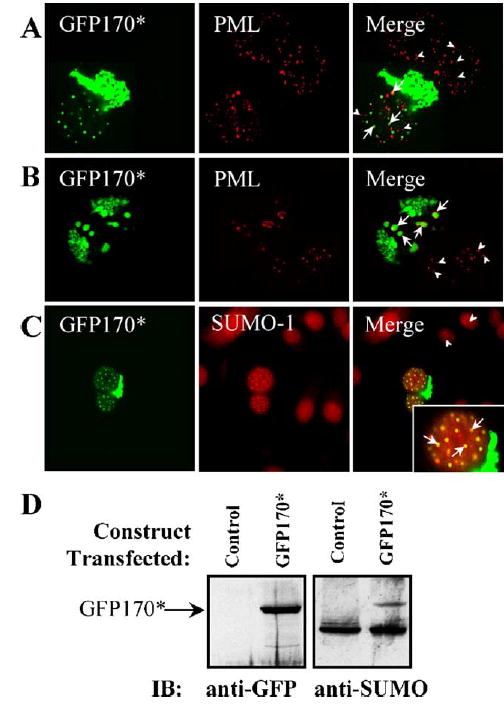
Nuclear GFP170* aggregates recruit PML protein and SUMO-1. COS-7 cells were transfected with GFP170*. 48 h after transfection, cells were processed for indirect immunofluorescence using antibodies to PML and SUMO-1. PML distribution is normal in cells expressing low levels of GFP170* (A). PML distribution is altered in cells expressing high levels of GFP170* (B). PML structures are surrounding the large GFP170* aggregates. Arrowheads point to PMLs in the control cells. Arrows point to PMLs in a nucleus containing GFP170* aggregates. (C) SUMO-1 co-localizes with nuclear GFP170* aggregates. (D) COS-7 cells were mock-transfected (control lanes) or transfected with GFP170* (GFP170* lanes) for 48 h. Cells were lysed, and the lysates were processed by SDS-PAGE and immunoblotting with anti-GFP antibody (anti-GFP panel). A GFP170* band (~124 kDa) is present only in the transfected cells. The same membrane was stripped and reprobed with anti-SUMO antibody (anti-SUMO panel). The GFP170* band is recognized. An additional band (~98 kDa) is recognized in control and in transfected cells and may represent PML.
Recently, it has been shown that the mutant huntingtin, Httex1p, is modified with the small-ubiquitin-related modifier (SUMO) (Steffan et al., 2004). Like ubiquitin, SUMO is ligated to lysine residues of a variety of proteins involved in multiple cellular pathways (Verger et al., 2003). For example, the PML protein in the PML bodies is sumoylated (Duprez et al., 1999). We therefore tested the relationship between GFP170* aggregates and SUMO-1. SUMO-1 appears diffusely distributed in the nuclei of control cells (Fig. 4C, arrows). SUMO-1 appears to be recruited to the GFP170* nuclear aggregates, but not the cytoplasmic GFP170* aggregates in cells expressing GFP170* (Fig. 4C, arrows). The insert shows extensive co-localization of GFP170* and SUMO-1 in the nuclear aggregates.
To explore whether GFP170*, like Httex1p, is sumoylated, we performed immunoblotting experiments. GFP170* is detected as a ~124-kDa band in transfected cells (Fig. 4D, anti-GFP panel), and this protein is also detected by anti-SUMO-1 antibodies (anti-SUMO panel). A major sumoylated ~98-kDa band detected in non-transfected and in transfected cells corresponds in molecular weight to sumoylated PML (Muller and Dejean, 1999).
Nuclear aggregates of GFP170* recruit transcription factors
Nuclear inclusions of polyQ proteins have been shown to recruit transcriptional regulators. Specifically, CBP, the coactivator for CREB-mediated transcription, redistributes to huntingtin polyQ (Htt-N63-148Q) protein aggregates (Nucifora et al., 2001). Similarly, the tumor supressor p53 interacts with aggregates of a pathogenic amino-terminal region of huntingtin, httex1p (Steffan et al., 2000; Suhr et al., 2001). We therefore tested if CBP and p53 also relocate in response to GFP170* aggregates that do not contain polyQ tracks. CBP is diffusely distributed in the nucleus of control cells (Fig. 5A, arrowhead) but redistributes to the GFP170* nuclear aggregates in GFP170* transfected cells (arrows). The overall level of CBP seems to be increased in transfected cells, suggesting that transcription of CBP-responsive genes might be altered. The levels of p53 are significantly increased in cells containing GFP170* aggregates since p53 is barely visible in non-transfected cells (Fig. 5B). Like CBP, p53 is recruited to nuclear inclusions of GFP170* (Fig. 5B).
Fig. 5.
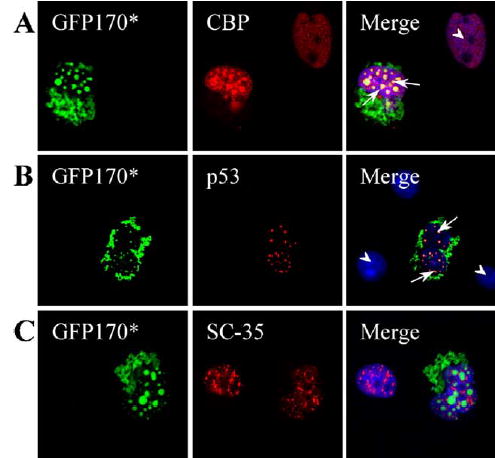
Nuclear GFP170* aggregates recruit transcriptional regulators. COS-7 cells were transfected with GFP170*. 48 h after transfection, cells were processed for indirect immunofluorescence using antibodies to CBP (A), p53 (B), and SC35 (C). Levels of CBP and p53 are increased in cells expressing GFP170*, and both proteins are recruited to nuclear inclusions of GFP170*. The levels and distribution of SC35 appear similar in non-transfected cells and in cells containing GFP170* aggregates.
In addition to transcriptional factors, the mRNA splicing factor, SC-35, which normally localizes in the nucleus as nuclear speckles (Fu and Maniatis, 1990), relocates to nuclear inclusions formed by a truncated form of ataxin-3 (HA-Q78) (Chai et al., 2001). This response appears uncommon, and SC-35 is not recruited to aggregates formed by the full-length ataxin-3 (myc-ataxin-3-Q84) (Chai et al., 2001). We examined the relationship between nuclear aggregates of GFP170* and nuclear speckles. GFP170* aggregates do not significantly influence the distribution of nuclear speckles marked by SC35 (Fig. 5C).
Expression of GFP170* alters function of transcription factors and is cytotoxic
The nuclear deposition of polyQ proteins has been linked to alterations in transcription (Okazawa, 2003). For example, a mutant form of huntingtin, httex1p, that sequesters p53 in inclusions, represses transcription of the p53-regulated proteins, p21WAF1/CIP1 (Steffan et al., 2000). Since p53 is also sequestered by nuclear GFP170* inclusions, we tested the effect of this sequestration on p53 transcriptional activity. The activity of p53 was analyzed by measuring transcription from a reporter construct composed of firefly luciferase fused to p21 promoter with two p53-responsive elements (p21-Luc) (Chinery et al., 1997). Analogous experiments were performed with Q80-GFP to allow direct comparisons. COS-7 cells were co-transfected with p21-Luc and either GFP170*, Q80-GFP, or a plasmid control. 48 h after transfection, luciferase activity in cell lysates was measured. COS-7 cells transfected with the control plasmid have wild type p53 activity, which is consistent with the results described previously (Ray et al., 1997). The luciferase activity in cells co-transfected with the GFP170* and the p21-Luc constructs is reduced to 30% of that in control cells co-transfected with control plasmid and the p21-Luc construct (Fig. 6A). This value is comparable to the luciferase activity in COS-7 cells expressing Q80-GFP, in which luciferase is reduced to 10% of control cells (Fig. 6A). These results indicate that p53 transcriptional activity can be repressed by GFP170* to a level similar to that caused by a polyQ protein.
Fig. 6.
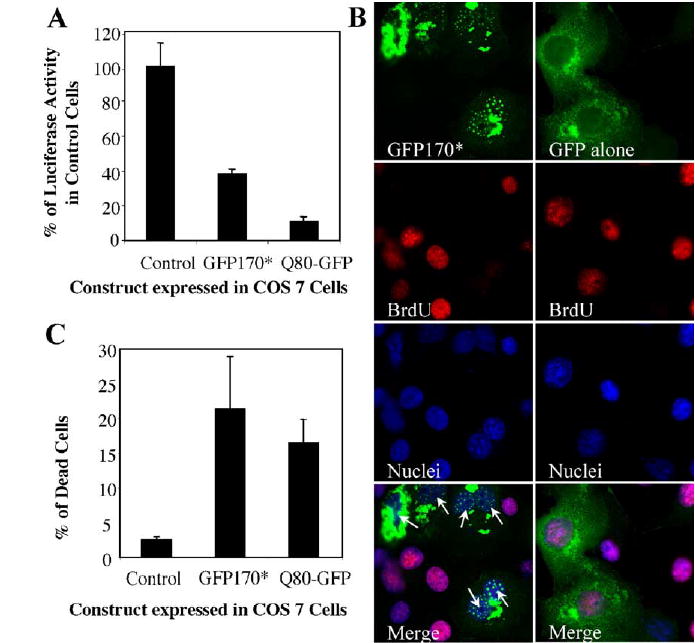
Expression of GFP170* represses transcription and is cytotoxic. (A) COS7 cells were co-transfected with the p21-Luc vector expressing firefly luciferase and either a control plasmid, GFP170*, or Q80-GFP. 48 h after transfection, cell lysates were made, and luciferase activity was measured. The activity in each sample was calculated as arbitrary units per milligram protein and normalized to that in the control sample. The p53 transcriptional activity is inhibited by GFP170* or Q80-GFP. (B) COS-7 cells were transfected with GFP170* or pEGFP-C2 (GFP alone). 32 h after transfection, cells were incubated with 30 μM BrdU followed by immunofluorescent staining with anti-BrdU monoclonal antibody. Nuclei were stained with Hoechst 33258. GFP170*-containing cells do not incorporate BrdU. (C) COS-7 cells were mock-transfected (control) or transfected with GFP170* or Q80-GFP. 48 h after transfection, cells were incubated with a viability indicator dye and sorted by FACS. The number of dead cells was counted and is plotted as % of total cells. Expression of GFP170* or Q80-GFP results in cell death.
Expression of polyQ proteins such as mutant huntingtin and atrophin-1 in cultured cells or in animal disease models leads to cellular toxicity (Nucifora et al., 2001). We therefore analyzed the effects of expressing GFP170* on cell cycle regulation and cell viability. We first examined the effects of GFP170* on DNA synthesis by measuring the incorporation of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) in COS-7 cells expressing GFP170*. BrdU incorporation during the S phase of mitosis is an indirect measure of cell proliferation. BrdU is incorporated in COS-7 cells expressing the GFP control plasmid (Fig. 6B). In contrast, BrdU is not incorporated in cells with GFP170* aggregates (Fig. 6B; arrows), suggesting a defect in DNA synthesis or a cell cycle arrest.
The effect of GFP170* expression was further analyzed by measuring the viability of cells expressing GFP170* by fluorescent-associated cell sorting (FACS) analysis. Control cells (mock-transfected) have a death rate of ~2.5%, probably due to transfection and experimental damage (Fig. 6C). In contrast, ~21% of cells containing GFP170* aggregates die 48 h after transfection. This number is similar to the ~17% of dead cells containing Q80-GFP aggregates 48 h after transfection. The percentage of dead cells expressing GFP170* or Q80-GFP is similar to that expressing the HD83Q huntingtin mutant (Waelter et al., 2001).
Discussion
Neurodegenerative disorders, including HD, SBMA, DRPLA, and SCAs 1, 2, 3, 6, 7, and 17, are characterized by the formation of cytoplasmic and/or nuclear inclusions of polyQ proteins. The nuclear aggregates recruit molecular chaperones, ubiquitin, and proteasome proteins (Cummings et al., 1998; Davies et al., 1997; Paulson et al., 1997) and cause significant alterations in the nuclear matrix-associated structures containing PML (Skinner et al., 1997). In addition, transcription factors such as TAF (TATA-binding protein-associated factor), CREB (cAMP-responsive element-binding protein), and CBP (CREB-binding protein) are recruited to inclusions of polyQ proteins in vitro and in vivo (McCampbell et al., 2000; Nucifora et al., 2001; Shimohata et al., 2000). This recruitment influences transcriptional regulation (Dunah et al., 2002; Nucifora et al., 2001; Obrietan and Hoyt, 2004; Shimohata et al., 2000; Steffan et al., 2000; Suhr et al., 2001) and has been correlated with cytopathology. The cellular responses to the polyQ proteins appear directly linked to the polyQ tracks because the wild type proteins without extended polyQ tracks do not form nuclear inclusions and do not elicit cellular defects (Michalik and Van Broeckhoven, 2003; Orr, 2001; Ross, 2002). Some transcriptional factors, exemplified by CBP, contain poly-Q stretches, leading to the proposal that the recruitment and direct interactions with transcriptional regulators may be mediated through the polyQ tracks. It has not been addressed whether proteins lacking polyQ tracks can form nuclear inclusions, alter nuclear structure, and induce transcriptional responses analogous to those caused by polyQ proteins.
We report that nuclear aggregates are formed by GFP170*, a GFP-tagged fragment of the Golgi protein GCP170 that lacks a polyQ tract. Like aggregates of polyQ proteins, the nuclear aggregates of GFP170* recruit molecular chaperones and proteasomal components and cause redistribution of PML bodies (Davies et al., 1997; Ross, 2002; Schilling et al., 1999; Skinner et al., 1997; Waelter et al., 2001). It is therefore unlikely that the recruitment of these proteins is directly mediated through the polyQ tracks. Rather, it may involve mechanisms that recognize any misfolded protein as part of the cellular responses to either re-fold or clear aggregated proteins. PML bodies have been proposed to be depots (Maul et al., 2000; Negorev and Maul, 2001), and our findings confirm that they associate with aggregated proteins in the nucleus.
Like aggregates of polyQ proteins, the nuclear aggregates of GFP170* recruit CBP and p53 (McCampbell et al., 2000; Steffan et al., 2000; Suhr et al., 2001). The interaction between polyQ proteins and CBP requires polyQ repeats in both proteins since altering the polyQ domain in either protein prevents association (Nucifora et al., 2001). Whether polyQ proteins bind directly to CBP is unknown. Although polyQ-expanded huntingtin fragment can be co-immunoprecipitated with CBP from cell lysates, suggesting a direct interaction, the co-immunoprecipitation may reflect the recovery of a larger complex of proteins. The nature of GFP170* association with CBP remains to be defined. The interaction is unlikely to be direct since the Golgi-localized GCP170 has not been proposed to be involved in transcriptional regulation. It is more likely that binding of chaperones and proteasomes or sumoylation provides the link with CBP recruitment. The interaction of polyQ proteins with p53 may be direct or mediated through other cofactors (Steffan et al., 2000). The nature of GFP170* association with p53 remains to be characterized and may or may not involve mechanisms analogous to those recruiting CBP. Irrespective of the actual mechanisms, our results suggest that the cellular models used to examine the role of polyQ inclusions in pathogenesis reveal general (rather than polyQ-specific) cellular responses to the accumulation of misfolded protein in the nucleus.
Significantly, like the expression of polyQ proteins, the expression of GFP170* represses p53 transcriptional activity and causes cell death. The cell death pathways induced by GFP170* remain to be analyzed. Cell death may be due to the alteration of p53 activity since it has been documented that p53 may induce apoptosis through transcriptional repression (Oren, 2003). Some polyQ proteins appear to induce apoptosis (Ellerby et al., 1999; de Cristofaro et al., 2000; Saudou et al., 1998; Wanker, 2002). However, there are reports of non-apoptotic death caused by polyQ proteins (Moulder et al., 1999), indicating that the pathways leading to cell death are complex (Sawa et al., 2003).
Aggregation of non-polyQ proteins other than GFP170* has been linked to human diseases. For example, expression of mutant forms of aggregation-prone proteins such as superoxide dismutase (Bruijn et al., 1998; Durham et al., 1997), α-synuclein (Masliah et al., 2000), glial fibrillary acidic protein (GFAP) (Messing et al., 1998), or β-amyloid (Harper and Lansbury, 1997; Selkoe, 2003) in transgenic mice models of human disease results in the formation of large aggregates in selected neurons and neurodegeneration of the same neurons that mimic the pathology of FALS, Parkinson’s disease, Alexander’s disease or Alzheimer’s disease, respectively. However, in all known cases on non-polyQ proteins, the aggregates are either cytoplasmic or extracellular. To our knowledge, GFP170* represents the only non-polyQ protein that deposits in cytoplasmic as well as nuclear aggregates. Our findings indicate that nuclear aggregation may be caused by general features of misfolded proteins rather than the presence of polyQ tracks. Aggregate-induced nuclear alterations and transcriptional dysregulation may represent a general cellular response to the accumulation of any (polyQ or non-polyQ) aggregated protein in the nucleus. This suggests that the mechanisms for the neuropathology of polyQ neurodegenerative diseases may include common (polyQ-independent) events in addition to polyQ-specific responses.
Acknowledgments
We thank Dr. Susan Lyons for her help on preparing the primary neuronal cells from mice. We thank Dr. Xinbin Chen for the anti-p53 antibody and Ms. Kelly Harms in his laboratory for help on the luciferase assay. We thank Ms. Leigh Millican for technical support during the electron microscopy study. We thank Drs. Gail Johnson and Peter Detloff for helpful discussions. This work was partially supported by a NIH grant DK68074 (to L.F.).
References
- Alvarez C, Fujita H, Hubbard A, Sztul E. ER to Golgi transport: requirement for p115 at a pre-Golgi VTC stage. J Cell Biol. 1999;147:1205– 1222. doi: 10.1083/jcb.147.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin– proteasome system by protein aggregation. Science. 2001;292:1552– 1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851– 1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Chai Y, Wu L, Griffin JD, Paulson HL. The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease. J Biol Chem. 2001;276:44889– 44897. doi: 10.1074/jbc.M106575200. [DOI] [PubMed] [Google Scholar]
- Chinery R, Brockman JA, Peeler MO, Shyr Y, Beauchamp RD, Coffey RJ. Antioxidants enhance the cytotoxicity of chemotherapeutic agents in colorectal cancer: a p53-independent induction of p21WAF1/CIP1 via C/EBPbeta. Nat Med. 1997;3:1233– 1241. doi: 10.1038/nm1197-1233. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- de Cristofaro T, Affaitati A, Feliciello A, Avvedimento EV, Varrone S. Polyglutamine-mediated aggregation and cell death. Biochem Biophys Res Commun. 2000;272:816– 821. doi: 10.1006/bbrc.2000.2843. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990– 1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Ding Q, Lewis JJ, Strum KM, Dimayuga E, Bruce-Keller AJ, Dunn JC, Keller JN. Polyglutamine expansion, protein aggregation, proteasome activity, and neural survival. J Biol Chem. 2002;277:13935– 13942. doi: 10.1074/jbc.M107706200. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Duprez E, Saurin AJ, Desterro JM, Lallemand-Breitenbach V, Howe K, Boddy MN, Solomon E, de The H, Hay RT, Freemont PS. SUMO-1 modification of the acute promyelocytic leukaemia protein PML: implications for nuclear localisation. J Cell Sci. 1999;112 (Pt 3):381– 393. doi: 10.1242/jcs.112.3.381. [DOI] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- Ellerby LM, Hackam AS, Propp SS, Ellerby HM, Rabizadeh S, Cashman NR, Trifiro MA, Pinsky L, Wellington CL, Salvesen GS, Hayden MR, Bredesen DE. Kennedy’s disease: caspase cleavage of the androgen receptor is a crucial event in cytotoxicity. J Neurochem. 1999;72:185– 195. doi: 10.1046/j.1471-4159.1999.0720185.x. [DOI] [PubMed] [Google Scholar]
- Fritzler MJ, Hamel JC, Ochs RL, Chan EK. Molecular characterization of two human autoantigens: unique cDNAs encoding 95- and 160-kD proteins of a putative family in the Golgi complex. J Exp Med. 1993;178:49– 62. doi: 10.1084/jem.178.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Maniatis T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature. 1990;343:437– 441. doi: 10.1038/343437a0. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sztul E. A novel interaction of the Golgi complex with the vimentin intermediate filament cytoskeleton. J Cell Biol. 2001;152:877– 894. doi: 10.1083/jcb.152.5.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic GFP-chimera. J Cell Biol. 1999;146:1239– 1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385– 407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- Hicks SW, Machamer CE. The NH2-terminal domain of Golgin-160 contains both Golgi and nuclear targeting information. J Biol Chem. 2002;277:35833–35839. doi: 10.1074/jbc.M206280200. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883– 1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843– 854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41– 53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772– 8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Huntingtin– protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146– 154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Maul GG, Negorev D, Bell P, Ishov AM. Review: properties and assembly mechanisms of ND10, PML bodies, or PODs. J Struct Biol. 2000;129:278– 287. doi: 10.1006/jsbi.2000.4239. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- Messing A, Head MW, Galles K, Galbreath EJ, Goldman JE, Brenner M. Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am J Pathol. 1998;152:391– 398. [PMC free article] [PubMed] [Google Scholar]
- Michalik A, Van Broeckhoven C. Pathogenesis of polyglutamine disorders: aggregation revisited. Hum Mol Genet. 2003;12 (Spec 2):R173–R186. doi: 10.1093/hmg/ddg295. [DOI] [PubMed] [Google Scholar]
- Misumi Y, Sohda M, Yano A, Fujiwara T, Ikehara Y. Molecular characterization of GCP170, a 170-kDa protein associated with the cytoplasmic face of the Golgi membrane. J Biol Chem. 1997;272:23851– 23858. doi: 10.1074/jbc.272.38.23851. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Onodera O, Burke JR, Strittmatter WJ, Johnson EM., Jr Generation of neuronal intranuclear inclusions by polyglutamine-GFP: analysis of inclusion clearance and toxicity as a function of polyglutamine length. J Neurosci. 1999;19:705– 715. doi: 10.1523/JNEUROSCI.19-02-00705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Dejean A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J Virol. 1999;73:5137– 5143. doi: 10.1128/jvi.73.6.5137-5143.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negorev D, Maul GG. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene. 2001;20:7234–7242. doi: 10.1038/sj.onc.1204764. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington’s disease transgenic mice. J Neurosci. 2004;24:791– 796. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazawa H. Polyglutamine diseases: a transcription disorder? Cell Mol Life Sci. 2003;60:1427– 1439. doi: 10.1007/s00018-003-3013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera O, Burke JR, Miller SE, Hester S, Tsuji S, Roses AD, Strittmatter WJ. Oligomerization of expanded-polyglutamine domain fluorescent fusion proteins in cultured mammalian cells. Biochem Biophys Res Commun. 1997;238:599– 605. doi: 10.1006/bbrc.1997.7337. [DOI] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925– 932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457– 1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RB, Steele R, Meyer K, Ray R. Transcriptional repression of p53 promoter by hepatitis C virus core protein. J Biol Chem. 1997;272:10983– 10986. doi: 10.1074/jbc.272.17.10983. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Cox C, Gilmore J, Voorn P. Reduced expression of preproenkephalin in striatal neurons from Huntington’s disease patients. Ann Neurol. 1995;37:335–343. doi: 10.1002/ana.410370309. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819– 822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Becher MW, Wood JD, Engelender S, Cooper JK, Sharp AH. Pathogenesis of neurodegenerative diseases associated with expanded glutamine repeats: new answers, new questions. Prog Brain Res. 1998;117:397–419. doi: 10.1016/s0079-6123(08)64029-7. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Sawa A, Tomoda T, Bae BI. Mechanisms of neuronal cell death in Huntington’s disease. Cytogenet Genome Res. 2003;100:287– 295. doi: 10.1159/000072864. [DOI] [PubMed] [Google Scholar]
- Schilling G, Wood JD, Duan K, Slunt HH, Gonzales V, Yamada M, Cooper JK, Margolis RL, Jenkins NA, Copeland NG, Takahashi H, Tsuji S, Price DL, Borchelt DR, Ross CA. Nuclear accumulation of truncated atrophin-1 fragments in a transgenic mouse model of DRPLA. Neuron. 1999;24:275– 286. doi: 10.1016/s0896-6273(00)80839-9. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900– 904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T, Takiyama Y, Nishizawa M, Goto J, Kanazawa I, Davidson I, Tanese N, Takahashi H, Tsuji S. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000;26:29– 36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- Skinner PJ, Koshy BT, Cummings CJ, Klement IA, Helin K, Servadio A, Zoghbi HY, Orr HT. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature. 1997;389:971–974. doi: 10.1038/40153. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- Verger A, Perdomo J, Crossley M. Modification with SUMO. A role in transcriptional regulation. EMBO Rep. 2003;4:137–142. doi: 10.1038/sj.embor.embor738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393– 1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE. Hip1 and Hippi participate in a novel cell death-signaling pathway. Dev Cell. 2002;2:126–128. doi: 10.1016/s1534-5807(02)00121-1. [DOI] [PubMed] [Google Scholar]
- Weis K, Rambaud S, Lavau C, Jansen J, Carvalho T, Carmo-Fonseca M, Lamond A, Dejean A. Retinoic acid regulates aberrant nuclear localization of PML-RAR alpha in acute promyelocytic leukemia cells. Cell. 1994;76:345– 356. doi: 10.1016/0092-8674(94)90341-7. [DOI] [PubMed] [Google Scholar]
- Yasuda S, Inoue K, Hirabayashi M, Higashiyama H, Yamamoto Y, Fuyuhiro H, Komure O, Tanaka F, Sobue G, Tsuchiya K, Hamada K, Sasaki H, Takeda K, Ichijo H, Kakizuka A. Triggering of neuronal cell death by accumulation of activated SEK1 on nuclear polyglutamine aggregations in PML bodies. Genes Cells. 1999;4:743– 756. doi: 10.1046/j.1365-2443.1999.00294.x. [DOI] [PubMed] [Google Scholar]
- Zhong S, Salomoni P, Pandolfi PP. The transcriptional role of PML and the nuclear body. Nat Cell Biol. 2000;2:E85– E90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]