

Abstract
Measurement of fluorescence resonance energy transfer (FRET) efficiency and the relative concentration of donor and acceptor fluorophores in living cells using the three-filter cube approach requires the determination of two constants: 1), the ratio of sensitized acceptor emission to donor fluorescence quenching (G factor) and 2), the ratio of donor/acceptor fluorescence intensity for equimolar concentrations in the absence of FRET (k factor). We have developed a method to determine G and k that utilizes two donor-acceptor fusion proteins with differing FRET efficiencies—the value of which need not be known. We validated the method by measuring the FRET efficiency and concentration ratio of the fluorescent proteins Cerulean and Venus in mammalian cells expressing a series of fusion proteins with varying stoichiometries. The method greatly simplifies quantitative FRET measurement in living cells as it does not require cell fixation, acceptor photobleaching, protein purification, or specialized equipment for determining fluorescence spectra or lifetime.
Fluorescence resonance energy transfer (FRET) occurs when a donor fluorophore in the excited state transfers energy nonradiatively to an acceptor fluorophore in the ground state (1). FRET efficiency, defined as the proportion of the donor molecules that have transferred excitation state energy to the acceptor molecules, increases with decreasing intermolecluar distance (typically over the range 1–10 nm for fluorescent proteins). Thus FRET-based imaging can be used to assess fluorophore proximity, and by inference, protein-protein interaction, in living cells.
FRET measurements in living cells using “three-cube FRET” fluorescence microscopy (2–5) has become increasingly popular as the method is fast, simple, nondestructive, and requires only a standard fluorescence imaging microscope. With this method, images are acquired using three different fluorescence filter cubes: 1), the donor channel (IDD, donor excitation and emission), 2), the FRET channel (IDA, donor excitation, acceptor emission), and 3), the acceptor channel (IAA, acceptor excitation and emission). Because of spectral overlap between donor and acceptor fluorescent proteins (FP), procedures ((3–6), see Supplementary Materials) are used to isolate the donor (Idd), sensitized acceptor (Fc, i.e., fraction of IDA resulting from FRET), and direct acceptor (Iaa) fluorescence intensities from the uncorrected intensity images (IDD, IDA, and IAA).
FRET indices based on normalized Fc are often used to report experimental results. Unfortunately, such indices are instrument-dependent and thus results generated from different imaging setups are not directly comparable. However, Fc can be converted to FRET efficiency, an instrument-independent parameter, using a proportionality constant termed G factor (3,5) or α (6,7). G factor represents the ratio of sensitized acceptor emission, Fc, to quenched (i.e., lost) donor emission due to FRET and is constant for a particular fluorophore pair and imaging setup.
Three methods have been reported for determining the G factor of an FP pair. First, Hoppe et al. (8) determined the G factor (termed γ/ξ in their article) by using a donor-acceptor fusion protein with predetermined FRET efficiency (from fluorescence lifetime measurements) as a reference point. Fluorescence lifetime measurements require sophisticated and expensive instruments not available in most laboratories. Second, Zal and Gascoigne (5) determined the G factor for a CFP-YFP pair by gradually photobleaching the acceptor while monitoring the ratio of the decrease in Fc to increase in Idd. This approach requires the donor to be photostable and the acceptor photolabile. If the donor is not completely photostable, as has been reported for CFP (9), the G factor will be overestimated, resulting in an underestimation of FRET efficiency. In addition, photobleaching is often performed on formaldehyde treated cells to eliminate cell movement and diffusion of FP from unbleached areas. Whether a G factor determined from fixed cells is valid for living cells is unclear. It is noteworthy that GFP fluorescence is quenched by formaldehyde fixation (10). We also found that fixation differentially quenched the FP variants Venus and Cerulean ((11,12), Supplementary Material Fig. 1). Finally, Nagy et al. (7) determined G factor (termed α in their article) using three CFP-YFP fusion constructs differing in linker length. The mean-squared difference in calculated FRET efficiency using two formulae was determined for a range of hypothetical G factors and the minimum of this relationship used to estimate the actual G factor. Although the minimum was well-defined for small G factor values, the topography of the function was shallow for larger values, making determination of the minimum under these circumstances difficult.
FIGURE 1 .

Validation of G and k factor. (A) Representative pseudocolor images illustrating FRET efficiency and Cerulean and Venus concentration ratio [C]/[V], in HeLa cells transfected with the fusion constructs as indicated. (B) Bar graphs summarizing the mean FRET efficiency (left) and the [C]/[V] ratio (right) for the indicated construct. Data are presented as mean ± SEM. The number of cells for each group is indicated in parentheses. Scale bar, 30 μm.
To avoid issues inherent to these methods, we developed an alternative approach for determining the G factor. The method requires preparation of cDNA constructs encoding donor-acceptor fusion FPs differing, as widely as possible, in FRET efficiency. This was accomplished by varying the length and composition of the linker residues connecting the CFP variant Cerulean and the YFP variant Venus. We reasoned that if the two constructs were expressed at the same level in two different cells, then the G factor would equal ΔFc/ΔIdd (see Supplementary Material for derivation). Because protein expression varies among cells, implementation of this idea is not practical. However, as FRET does not alter Iaa, sensitized acceptor emission and donor fluorescence intensity can be normalized to Iaa. Thus
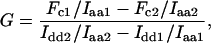 |
(1) |
where the parameters derived from each FP fusion construct (i.e., 1 and 2) are denoted in the subscript. Fc, Iaa, and Idd from 24 HeLa cells transiently expressing either C5V or CTV were determined. C5V and CTV were fusion constructs in which Cerulean was connected to Venus by a 5- and 236-residue linker, respectively (13). Using Eq. 1, the G factor for Cerulean and Venus on our imaging microscope was calculated to be 1.815 ± 0.067. Once the G factor is determined, sensitized acceptor emission intensity can be converted to FRET efficiency (E) using the formalism of Zal and Gascoigne (5):
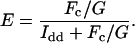 |
(2) |
Note that Fc/G represents the quenched donor fluorescence and thus Idd + Fc/G represents the total donor fluorescence that would be present in the absence of FRET.
We have also developed a method to determine the [donor]/[acceptor] ratio from data obtained in three-cube FRET experiments based on the k factor, the ratio of donor/acceptor fluorescence intensity for equimolar concentrations in the absence of FRET. Although Iaa is proportional to acceptor concentration regardless of FRET, Idd is not proportional to donor concentration in the presence of FRET due to quenching of donor fluorescence. Once the G factor is determined, the total donor fluorescence can be numerically restored. We can thus determine the k factor using a 1:1 donor-acceptor fusion construct from
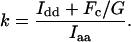 |
(3) |
For C5V, the mean k factor determined using Eq. 3 was 0.2168 ± 0.0014 (n = 24).
Once the k and G factor are determined for a particular donor and acceptor FP pair, one can measure the relative abundance of the donor and acceptor FP or FP-tagged proteins regardless of stoichiometry from
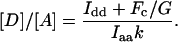 |
(4) |
Hoppe et al. (8) also derived a formula to convert donor and acceptor fluorescence intensities to a concentration ratio in the presence of FRET. However, their method required a donor-acceptor fusion protein with FRET efficiency previously determined from fluorescence lifetime measurements.
We examined the validity of our formulae by measuring the FRET efficiency and [donor]/[acceptor] in HeLa cells expressing fusion proteins with varying linker lengths and stoichiometries. In Fig. 1 A, representative pseudocolored images illustrate the FRET efficiency and [C]/[V] ratio for fields of cells transfected with cDNA constructs encoding C32V, C40V, C50V, CVC, or VCV as indicated. CnV constructs were Cerulean-Venus fusion constructs in which n is the number of residues in the linker separating the fluorophores. CVC and VCV were fusion constructs with 2:1 and 1:2 donor/acceptor stoichiometries, respectively. Details of the constructs are documented in the Supplementary Material. The calculation of FRET efficiency and [C]/[V] ratio for each pixel was based on the G and k factors determined using CTV and C5V. As summarized in Fig. 1 B, the mean FRET efficiency measured from cells expressing C32V, C40V, and C50V was 31.2 ± 0.2, 21.4 ± 0.4 and 12.9 ± 0.2%, respectively. Thus, increasing the linker length by 8 or 10 residues significantly reduced FRET efficiency consistent with an increased distance between donor and acceptor. The mean [C]/[V] ratios measured from cells expressing C32V, C40V, and C50V were 0.98 ± 0.01, 0.99 ± 0.02 and 0.97 ± 0.01, respectively, values nearly identical to the expected value of 1. The mean FRET efficiency measured from cells expressing CVC and VCV was 40.0 ± 0.7 and 69.3 ± 1.0%, respectively, which is comparable to the FRET efficiency determined using spectral unmixing methodology and FLIM (13). Similarly, the mean [C]/[V] ratios measured from cells expressing CVC and VCV were 2.1 ± 0.04 and 0.47 ± 0.01, respectively, again comparable with the [C]/[V] ratios measured using spectral unmixing (13), although slightly different from the predicted values of 2.0 and 0.5. Taken together, our measurements of FRET efficiency and [C]/[V] ratio were quite accurate, thus validating our methods for determining the G and k factor.
In summary, we have developed and validated a simple calibration method that does not require cell fixation, acceptor photobleaching, purification of proteins, or specialized equipment for determining fluorescence spectra or lifetime. The method greatly simplifies the determination of FRET efficiency and the relative concentration of donor to acceptor molecules in living cells.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Dr. Shui-lin Niu for many helpful discussions.
This work was supported by the intramural program of the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD 20892.
References
- 1.Wallrabe, H., and A. Periasamy. 2005. Imaging protein molecules with FRET and FLIM microscopy. Curr. Opin. Biotechnol. 16:19–27. [DOI] [PubMed] [Google Scholar]
- 2.Mitra, R. D., C. M. Silva, and D. C. Youvan. 1996. Fluorescence resonance energy transfer between blue-emitting and red-shifted excitation derivatives of the green fluorescent protein. Gene. 173:13–17. [DOI] [PubMed] [Google Scholar]
- 3.Gordon, G. W., G. Berry, X. H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson, M. G., B. A. Alseikhan, B. Z. Peterson, and D. T. Yue. 2001. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 31:973–985. [DOI] [PubMed] [Google Scholar]
- 5.Zal, T., and N. R. Gascoigne. 2004. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys. J. 86:3923–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tron, L., J. Szollosi, S. Damjanovich, S. H. Helliwell, D. J. Arndt-Jovin, and T. M. Jovin. 1984. Flow cytometric measurement of fluorescence resonance energy transfer on cell surfaces. Quantitative evaluation of the transfer efficiency on a cell-by-cell basis. Biophys. J. 45:939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy, P., L. Bene, W. C. Hyun, G. Vereb, M. Braun, C. Antz, J. Paysan, S. Damjanovich, J. W. Park, and J. Szollsi. 2005. Novel calibration method for flow cytometric fluorescence resonance energy transfer measurements between visible fluorescent proteins. Cytometry A. 67:86–96. [DOI] [PubMed] [Google Scholar]
- 8.Hoppe, A., K. Christensen, and J. A. Swanson. 2002. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83:3652–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Munster, E. B., G. J. Kremers, M. J. Adjobo-Hermans, and T. W. Gadella Jr. 2005. Fluorescence resonance energy transfer (FRET) measurement by gradual acceptor photobleaching. J. Microsc. 218:253–262. [DOI] [PubMed] [Google Scholar]
- 10.Brewis, N., A. Phelan, J. Webb, J. Drew, G. Elliott, and P. O'Hare. 2000. Evaluation of VP22 spread in tissue culture. J. Virol. 74:1051–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagai, T., K. Ibata, E. S. Park, M. Kubota, K. Mikoshiba, and A. Miyawaki. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20:87–90. [DOI] [PubMed] [Google Scholar]
- 12.Rizzo, M. A., G. H. Springer, B. Granada, and D. W. Piston. 2004. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 22:445–449. [DOI] [PubMed] [Google Scholar]
- 13.Thaler, C., S. V. Koushik, P. S. Blank, and S. S. Vogel. 2005. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys. J. 89:2736–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]