

Abstract
PSII catalyzes the oxidation of water and reduction of plastoquinone in oxygenic photosynthesis. PSII contains an oxygen-evolving complex, which is located on the lumenal side of the PSII reaction center and which contains manganese, calcium, and chloride. Four sequential photooxidation reactions are required to generate oxygen. This process produces five Sn-states, where n refers to the number of oxidizing equivalents stored. Calcium is required for oxygen production. Strontium is the only divalent cation that replaces calcium and maintains activity. In our previous FT-IR work, we assessed the effect of strontium substitution on substrate-limited PSII preparations, which were inhibited at the S3 to S0 transition. In this work, we report reaction-induced FT-IR studies of hydrated PSII preparations, which undergo the full S-state cycle. The observed difference FT-IR spectra reflect long-lived photoinduced conformational changes in the oxygen-evolving complex; strontium exchange identifies vibrational bands sensitive to substitutions at the calcium site. During the S1′ to S2′ transition, the data are consistent with an electrostatic or structural perturbation of the calcium site. During the S3′ to S0′ and S0′ to S1′ transitions, the data are consistent with a perturbation of a hydrogen bonding network, which contains calcium, water, and peptide carbonyl groups. To explain our data, persistent shifts in divalent cation coordination must occur when strontium is substituted for calcium. A modified S-state model is proposed to explain these results and results in the literature.
INTRODUCTION
Photosystem II (PSII) catalyzes the light-driven oxidation of water and the reduction of plastoquinone. The oxidation of water to oxygen occurs within the PSII OEC. Four sequential oxidation reactions are required to oxidize water and to generate oxygen. The sequentially oxidized forms of the OEC are called the S-states. From the dark adapted S1-state, three flashes produce S4, which releases oxygen and converts to S0 in the dark (1).
The oxidation state of Mn as a function of S-state has been studied by EPR and XANES. The S1-state contains a MnIII2MnIV2 cluster and the S2-state contains a MnIII1MnIV3 cluster (see Britt et al. (2) and references therein). The oxidation state of the Mn ions in the S3-state has been controversial, with some XANES studies suggesting that a Mn oxidation occurs during the S2 to S3 transition (3,4) and some studies suggesting that a ligand or μ-oxo bridge to the Mn is oxidized instead (5,6). EXAFS and kinetic studies suggest that a structural change in the OEC occurs during the S2 to S3 transition (7–9).
Calcium is an essential cofactor in PSII (reviewed in (10)). EXAFS and 87Sr+2 ESEEM measurements report Ca-Mn distances of 3–5 Å (11–13). Four x-ray diffraction studies of PSII crystals from thermophilic cyanobacteria have been presented at 3.8–3.0 Å (14–17). In the 3.5 and 3.0 Å structures, electron density has been assigned to calcium and manganese, and calcium is an integral component of the manganese cluster (16,17). However, many of the calcium and manganese ligands are not yet assigned in these structures (18,19). A 113Cd NMR study suggests that Ca+2 has symmetric array of ligands, containing oxygen, nitrogen, and/or chlorine (20).
Calcium removal blocks the S-state cycle (reviewed in Yocum (10)). Strontium is the only divalent ion that can restore oxygen evolution (21). However, preparations containing strontium exhibit a slower turnover of the S-state cycle (21–24), and substitution of strontium for calcium alters the S2-state EPR signals (for an example, see Kim et al. (13) and references therein). To explain these results, three possible roles for calcium in PSII have been suggested; these roles are not mutually exclusive. First, it has been suggested that calcium plays an electrostatic role in maintaining the correct reduction potential of the Mn cluster (25). Second, it has been suggested that calcium maintains a hydrogen-bonded network, which is essential for activity (26,27). Third, it has also been suggested that calcium binds a water or hydroxide ion, which acts as a nucleophile during formation of the oxygen-oxygen bond (28–30).
In this report, we employ reaction-induced or difference FT-IR spectroscopy to identify structural changes, which occur on each S-state transition and involve the calcium site. Recently, microsecond infrared measurements have been reported, which are on the timescale of the water oxidation chemistry (31). Alternatively, as is done here, FT-IR spectra can be recorded on long timescales (s) before and after a saturating flash, and persistent changes in electrostatics, conformation, protonation, and ligation changes can be identified (for examples, see (27,32–34)). These long-lived structural changes have been reported to be modulated in amplitude and frequency as a function of flash number (32,33). Some of the bands in these spectra have been assigned by global 15N and 13C labeling and by specific isotopic labeling of a PSII extrinsic subunit (34–37). However, the origin of these spectra and the assignment of the many of the FT-IR bands are still under investigation.
In our previous strontium exchange and FT-IR experiments (27), we studied PSII samples that were substrate water limited. Under these conditions, the OEC can undergo the S1 to S2 and the S2 to S3 transitions and cannot complete the oxygen evolution cycle. Our data showed that both the S1 to S2 and the S2 to S3 transitions produce vibrational bands, which are sensitive to strontium substitution on the seconds timescale. In addition, the vibrational spectra on the first and second flash were distinct.
To understand the origin of these spectral changes in more detail, we have conducted reaction-induced FT-IR studies under hydrated conditions in which the complete S-state cycle can occur. Divalent cation editing identifies vibrational bands that are sensitive to strontium substitution. To explain our results, we propose a new model for the photosynthetic water oxidizing cycle.
MATERIALS AND METHODS
PSII membranes were prepared from market spinach, suspended in SMN buffer (0.40 M sucrose, 50 mM MES-NaOH, 15 mM NaCl, pH 6.0) at 3–5 mg chlorophyll per ml, and then stored at −70°C (38,39). To deplete the 18 and 24 kDa subunits, PSII membranes were prepared by dark incubation of PSII samples in high-salt (2.0 M NaCl) SMN buffer for 30 min at 4°C (40). These PSII membranes were then washed with SMN buffer. To exchange strontium for calcium, this salt-washed PSII was resuspended in and pelleted from a SMN buffer that contained either 20 mM CaCl2, as a control, or 20 mM SrCl2 (27). Aliquots of these samples were stored at −70°C (27).
Oxygen evolution activity was monitored using a Clark electrode, as previously described (41). Average oxygen rates for calcium-substituted samples (in calcium-containing buffers) were 390 ± 60 μmol O2 (mg chl-h)−1; average oxygen rates for strontium-substituted samples (in strontium-containing buffers) were 120 ± 60 μmol O2 (mg chl-h)−1. The lower steady state rate observed in the presence of strontium is consistent with substitution of strontium for calcium at the active site for water oxidation (27,30,42).
EPR spectroscopy was used to verify the replacement of calcium with strontium in SMN buffer (13). Data acquisition was conducted at 10 K using a Bruker (Billerica, MA) EMX EPR spectrometer, Oxford (Concord, MA) cryostat, and standard Bruker TE cavity. Samples contained 500 μM potassium ferricyanide and 250 μM recrystallized DCBQ as electron acceptors. An EPR spectrum was recorded before illumination to generate the dark background. To generate signals from the S2-state, samples were illuminated outside the EPR cavity at 200 K with red-filtered and heat-filtered illumination from a Dolan-Jenner (Lawrence, MA) fiber optic illuminator. EPR parameters were: 9.44 GHz frequency, 3.2 mW microwave power, 16 G field modulation, 5.2 s time constant, and 336 s sweep time. Four scans were averaged, and light-minus-dark spectra were constructed.
For FT-IR spectroscopy, samples were defrosted on ice immediately before use, and the electron acceptors, potassium ferricyanide, and recrystallized DCBQ, were added to the suspension to give 760 and 67 mol equivalents per mole reaction center, respectively. The concentrated DCBQ stock was made up in ethanol; the final ethanol concentration in the FT-IR samples was ≤0.3%. After the addition of the electron acceptors, samples were pelleted (48,000 × G for 6 min at 4°C). The FT-IR sample was created as previously described (27), except that a 1 mg chl sample was concentrated under nitrogen at 4°C for 2–3 min, instead of ∼20 min. The amplitude ratios of the OH stretching (3400 cm−1) and amide II (1545 cm−1) bands were indistinguishable in the calcium-containing and strontium-exchanged samples (3.0 ± 0.4 and 2.7 ± 0.3, respectively). The sample was sealed with silicone high-vacuum grease, applied in a continuous bead that did not contact the sample, and a layer of parafilm was used to seal the circumference of the windows. Using this procedure, dehydration of the sample over the course of data acquisition was minimal (<2.5%), as previously described (27).
FT-IR measurements were performed with a Bruker 66v spectrometer (27,43,44). Samples were maintained at a constant temperature of 4°C through the use of a Harrick (Ossining, NY) sample cell and a recirculating water bath. Samples were transferred to the cell at 14°C and immediately cooled. The sample compartment was purged with dry nitrogen for 60 min before data acquisition began and during data acquisition. Samples were irradiated with a depolarized Nd-YAG laser spot (Continuum, Santa Clara, CA, 7 ns, 532 nm), with a laser intensity between 79 and 35 mJ/cm2. In this range, the laser energy density was saturating, because there was no significant change in spectral frequencies and amplitudes in this laser intensity range.
The measurement cycle began with dark adaptation of the samples for 60 min. This mixture was then given a single flash and allowed to equilibrate for 20 min to achieve a maximal concentration of the S1-state (27). Single-channel spectra (100 scans, 15 s total) were collected before S-state advancement flashes and after each of six advancement flashes. There was a 20-min equilibration between each set of six flashes. All single channel spectra were collected at 4 mm aperture, 8 cm−1 resolution, with a Happ-Genzel apodization function, four levels of zero-filling, and a Mertz phase correction. Difference spectra were calculated from the single channel spectra collected before and after each photolysis flash by ratioing the single beam data directly and then calculating the absorbance. A FT-IR absorption spectrum of each sample was also measured before and after data collection by ratioing the single beam spectrum to an open beam background. Difference spectra were normalized to the amide II absorbance of 0.5, correcting for pathlength and concentration. The strontium data were scaled to account for any small difference in the intensity of the ferricyanide/ferrocyanide bands (2114/2037 cm−1), compared to the corresponding calcium spectra (27,45). The calcium-exchanged data is an average of 78 spectra, and the strontium-exchanged data is an average of 59 spectra.
RESULTS
Fig. 1 shows light-minus-dark EPR spectra derived from calcium-containing (A) and strontium-exchanged (B) PSII in SMN buffer. To facilitate divalent ion exchange, strontium-exchanged samples were incubated four hours in the presence of the divalent ion. The control (Fig. 1 A) shows the multi-line signal at g = 2 and the g = 4.1 signal. Both of these EPR signals derive from the S2 state (46). The increase in the g = 4.1 signal and the change in the multi-line hyperfine splittings observed in Fig. 1 B demonstrate that strontium has been exchanged into the OEC calcium site (see Kim et al. (13) and references therein).
FIGURE 1.

Light-minus-dark EPR spectra of calcium-containing (A) and strontium-exchanged (B) PSII in SMN buffer. The frequency was 9.44 GHz. Data points in the g = 2.0 region, which contains a large signal from oxidized tyrosine residues, have been deleted for presentation purposes.
Fig. 2 shows the 1800–1200 cm−1 region of difference FT-IR spectra acquired from calcium-containing PSII samples with one (Fig. 2 A, black line), two (Fig. 2 B, black line) three (Fig. 2 C, black line), and four (Fig. 2 D, black line) saturating, 532-nm flashes. The data have previously been assigned to S2-minus-S1, S3-minus-S2, S0-minus-S3, and S1-minus-S0 spectra, respectively (32,33). A dark-dark spectrum in Fig. 2 E exhibits the level of noise in the calcium-containing sample (black line). The corresponding difference FT-IR spectra acquired from strontium-exchanged samples are shown superimposed as the red lines (Fig. 2, A–E).
FIGURE 2.

Difference FT-IR spectra of calcium-containing (black traces) and strontium-exchanged (red traces) in the 1800–1200 cm−1 region. The spectra are associated with the S1′ to S2′ (A), S2′ to S3′ (B), S3′ to S0′ (C), and S0′ to S1′ (D) transitions. In E, a dark-minus-dark control is presented. The tick marks on the y axis are 2 × 10−4 absorbance units. The red traces are labeled.
Fig. 3 shows a comparison of the 2200–1900 cm−1 region of the difference FT-IR spectra obtained from calcium-containing (black trace) and strontium-exchanged (red trace) PSII, as a function of flash number. As observed, the amplitudes of the ferricyanide/ferrocyanide CN vibrational bands (2114/2037 cm−1) are similar on every flash in the calcium-exchanged samples. This can be taken as a measure that the amount of charge separation is similar (45) on each photo-induced reaction in calcium-containing PSII. As shown in Fig. 3, the spectra acquired from the strontium-exchanged sample have been normalized to the calcium-containing spectra to give the same ferricyanide/ferricyanide intensity on each flash.
FIGURE 3.

Difference FT-IR spectra of calcium-containing (black traces) and strontium-exchanged (red traces) in the 2200–1900 cm−1 region. The spectra are associated with the S1′ to S2′ (A), S2′ to S3′ (B), S3′ to S0′ (C), and S0′ to S1′ (D) transitions. In E, a dark-minus-dark control is presented. The tick marks on the y axis are 2 × 10−4 absorbance units.
The spectral differences in the 1800−1200 cm−1 region are more readily observable in Fig. 4, which presents the strontium-edited double difference spectra. To derive these data, difference spectra acquired from strontium-exchanged PSII (Fig. 2, A–D, red lines) were subtracted from difference spectra acquired from calcium-containing PSII (Fig. 2, A–D, black lines). The spectrum in Fig. 4 A was obtained with one flash, the spectrum in Fig. 4 B was obtained with two flashes, the spectrum in Fig. 4 C was obtained with three flashes, and the spectrum in Fig. 4 D was obtained with four flashes. Fig. 4 E is a representative control double difference spectrum, in which no vibrational bands are expected (see Fig. 4, legend). Fig. 4 E is presented as an estimate of the noise in the measurements. Spectral features, with signals above the noise in the measurements, are observed in all the double difference spectra. Vibrational bands are only present in these double difference spectra if substitution of strontium for calcium and S-state advancement perturb the vibrational spectrum.
FIGURE 4.

Strontium-edited double difference spectra constructed from the data in Fig. 2 (calcium-minus-strontium) in the 1850–1250 cm−1 region. The spectra are associated with the S1′ to S2′ (A), S2′ to S3′ (B), S3′ to S0′ (C), and S0′ to S1′ (D) transitions. In E, a representative double difference control is presented in which no vibrational bands are expected. This control was constructed from the S2′ to S3′ data set by subtracting one half of the calcium data set from the other half, by subtracting one half of the strontium data set from the other half, and by averaging. The tick marks on the y axis are 1 × 10−4 absorbance units.
As shown in Fig. 4 A, one photolysis flash and substitution of strontium for calcium perturb the vibrational frequency of bands throughout the entire 1850–1250 cm−1 region of the infrared spectrum. In contrast, two photolysis flashes and strontium substitution perturb only vibrational bands between 1661 and 1638 cm−1 (Fig. 4 B). Experiments were conducted in 18OH2 buffer to assess whether these bands are assignable to water. Although background interference in the 1650 cm−1 region was high under these hydrated conditions, no downshifted bands were observed in this region after 18OH2 exchange (data not shown). Therefore, we favor the assignment of these bands to amide I frequencies, arising from the C=O vibration of the peptide bond regions (47), although amino acid side chain absorption is also possible in this region (for example, see Braiman et al. (48)). The strontium edited spectrum acquired with three flashes (Fig. 4 C) spectrum is also perturbed by calcium/strontium exchange, and the strontium-edited spectrum reflects perturbations of amide I and II bands in the 1650 and 1545 cm−1 regions (47). Finally, amide I and II bands may also be observed in the strontium-edited spectrum acquired with four flashes (Fig. 4 D), but with a lower signal/noise ratio compared to the other transitions.
DISCUSSION
In this work, we report the effects of strontium exchange on the S-state transitions in hydrated PSII. To appear in the strontium edited spectrum, vibrational bands must be perturbed by the photooxidation reaction and by strontium substitution. A possible origin of these strontium-sensitive bands, given the timescale of the measurements, is described below.
Timescale of the rapid scan FT-IR measurements
In our rapid scan FT-IR measurements, data are recorded for 15 s after each photolysis flash, with a ∼100-ms average gap between the photolysis pulse and the beginning of data acquisition. A 10–100 ms delay is typical of published FT-IR studies using rapid scan techniques (for examples, see (32,33,49,50)). In some cases the gap between photolysis and data acquisition is longer (see for example, Debus et al. (50)). In contrast, the Mn redox reactions associated with the S-state transitions are complete on the microsecond to millisecond timescale, even in these highly concentrated samples. For example, a recent transient IR study, conducted in 2H2O containing buffers, reported microsecond and millisecond time constants for OEC-related processes (31). A recent XANES study reports half times of 70, 190, 1100, and 30 μs for Mn redox reactions in the S1 to S2, S2 to S3, S3 to S0, and S0 to S1 transitions, respectively (4). Results from UV transient spectroscopy are in qualitative agreement with the XANES results (51). Therefore, one must conclude that electron transfer events involving the OEC are largely completed before the beginning of data acquisition in this type of rapid scan FT-IR experiment.
Recent transient IR measurements give insight into the origin of the rapid scan spectra. In these microsecond timescale measurements, a transient bleach at 1483 cm−1 was detected (31). This transient bleach occurred with the 1-ms response time of the instrument. This initial bleach was followed by an increase in absorbance over baseline, and this increase in absorption then decayed on the timescale of hundreds of milliseconds. Rate constants derived from fits to multiple flashes showed four flash oscillations (31), demonstrating that these transients derive from structural changes in the OEC. These spectral alterations, which are on the microsecond-millisecond timescale of the water splitting reactions, are not expected to be detectable on the timescale employed here or in other published rapid scan studies.
Therefore, these rapid scan FT-IR measurements must be detecting persistent structural alterations in PSII, which have been reported to be modulated in frequency and amplitude by flash number and S-state changes (32,33). These spectra are eliminated by removal of Mn (43) and also affected by calcium substitution (as shown in (27,37) and this work), showing that these long-lived structural changes are tightly coupled to water oxidation.
Speculative model to explain persistent structural changes during the S state cycle
A model to explain the persistent structural changes observed with rapid scan FT-IR spectroscopy is proposed in Fig. 5. In the model, each photo-induced transition produces the appropriate Sn state. Each Sn state then relaxes spontaneously in the dark to generate a Sn′ state, which is at the same oxidation state but distinct from the corresponding Sn state, as assessed by FT-IR spectroscopy. FT-IR spectroscopy detects proton transfer events, changes in hydrogen bonding, electrostatic alterations, and changes in metal ligation, so it reasonable to propose that these structural alterations distinguish each Sn′- and Sn-state.
FIGURE 5.

Proposed model for the S state transitions, which includes thermally accessible, conformationally relaxed forms of the S1, S2, S3, and S0 states, denoted as Sn′. The S4′ state was proposed in Haumann et al. (4) and is consistent with the results of Barry et al. (31).
Fig. 6 is an energy diagram, which illustrates this proposed process for the S1 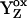 to S2 transition (first flash). In Fig. 6, the S2-state is a kinetic product, which will predominate at low temperature and in measurements conducted on rapid timescales. At higher temperature or with longer measuring times, the transition from S2 to S2′, the thermodynamic product, will be observed. Note that deactivation of S2 back to the S1YZ state (Fig. 6) will compete with S2′ formation. The effect of this S2 recombination reaction is to reduce the overall amplitude of the difference FT-IR signal, which only detects photolysis-induced changes in vibrational bands.
to S2 transition (first flash). In Fig. 6, the S2-state is a kinetic product, which will predominate at low temperature and in measurements conducted on rapid timescales. At higher temperature or with longer measuring times, the transition from S2 to S2′, the thermodynamic product, will be observed. Note that deactivation of S2 back to the S1YZ state (Fig. 6) will compete with S2′ formation. The effect of this S2 recombination reaction is to reduce the overall amplitude of the difference FT-IR signal, which only detects photolysis-induced changes in vibrational bands.
FIGURE 6.

Speculative energy diagram showing the proposed production of kinetic (S2) and thermodynamic (S2′) states from the 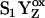 state. For a more general discussion of kinetic versus thermodynamics processes, see Carey and Sundberg (75).
state. For a more general discussion of kinetic versus thermodynamics processes, see Carey and Sundberg (75).
In Fig. 5, the proposed relaxation events that populate local thermodynamic minima are proposed to be reversible. For example, S2′ is linked to recombination of the S2-state with electrons on the acceptor side, which generates the S1YZ state, and with the next photolysis-induced reaction, which generates the S3-state. Therefore, the structural changes induced by dark relaxation will fluctuate with flash number. If the S2 and S2′ do not differ in oxidation state or in magnetic coupling, the two states will not be distinguished by magnetic resonance techniques.
Experimental evidence supporting this model
Previously published data are consistent with the model shown in Fig. 5. For example, previous FT-IR work showed that prolonged dark adaptation in the S1-state causes spontaneous changes in the S2-minus-S1 FT-IR difference spectrum (43). Work from other groups has also suggested that extended dark adaptation results in structural changes in PSII (see Beck et al. (52) and references therein).
The model in Fig. 5 can also explain the observation of different infrared signatures on different timescales and with different repetition rates. On the microsecond timescale, with a rapid repetition rates, as in Barry et al. (31), the model would propose that population of the Sn′ states is prevented kinetically because thermal equilibration between Sn and Sn′ is slow (Fig. 6) relative to the rate of S-state oxidation. The exception would be the population of the S1′ and S0′ states, which can occur during dark adaptation before the beginning of data acquisition.
With longer scan times and longer repetition rates, the model in Fig. 5 predicts that difference FT-IR spectra will actually reflect differences between the Sn′ states and in the concentrations of the Sn′ states. For example, the 15-s difference spectrum obtained with the second flash reflects S3′-minus-S2′ structural changes. These structural changes will include those associated with relaxation events and with the photoinduced transition from the S2- to S3-state. Because the structurally relaxed states are linked to the S-state cycle, these difference spectra would be modulated by flash number, as previously reported (32,33). The observed, overall decrease in amplitude with increasing flash number is attributed to Sn recombination in some centers.
The model in Fig. 5 can also explain differences in FT-IR spectra acquired at different temperatures. Low temperatures would be expected to favor kinetically accessible product states (Fig. 6, S2); higher temperatures and longer relaxation times would favor thermodynamically accessible product states (Fig. 6, S2′). In previous work, S2-minus-S1 spectra were recorded at 200 K in an optical glass, which required high concentrations of osmotic agents (53–55). At this temperature, all S-state transitions but the S1 to S2 transition are blocked, and 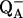 to QB electron transfer cannot occur (see Steenhuis et al. (53–55) and references therein). Therefore, the difference spectrum associated with
to QB electron transfer cannot occur (see Steenhuis et al. (53–55) and references therein). Therefore, the difference spectrum associated with 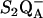 -minus-S1QA can be obtained under continuous illumination at 200 K. Control experiments showed that there was no illumination-induced heating of the sample and that spectra acquired immediately after illumination were identical to spectra acquired under illumination (56). These 200 K spectra exhibited broad lines and significant frequency shifts relative to spectra acquired with a single flash at higher temperature (56). Effects of osmotic agents could explain part of the observed spectral changes, but could not account for all frequency shifts and for the observation of broad vibrational bands (44). The model in Fig. 5 can explain increased heterogeneity and broad line widths because the spontaneous equilibration between S2 and S2′ is predicted to be temperature dependent. To explain these results in the literature, the model would propose that the 200 K conditions favor a more mixed population of the S2′- and S2-states, compared to the experimental conditions at higher temperature, which may reflect mainly the S2′-state. The mixed population of the S2′- and S2-states is proposed to have the effect of broadening vibrational bands and shifting vibrational frequencies.
-minus-S1QA can be obtained under continuous illumination at 200 K. Control experiments showed that there was no illumination-induced heating of the sample and that spectra acquired immediately after illumination were identical to spectra acquired under illumination (56). These 200 K spectra exhibited broad lines and significant frequency shifts relative to spectra acquired with a single flash at higher temperature (56). Effects of osmotic agents could explain part of the observed spectral changes, but could not account for all frequency shifts and for the observation of broad vibrational bands (44). The model in Fig. 5 can explain increased heterogeneity and broad line widths because the spontaneous equilibration between S2 and S2′ is predicted to be temperature dependent. To explain these results in the literature, the model would propose that the 200 K conditions favor a more mixed population of the S2′- and S2-states, compared to the experimental conditions at higher temperature, which may reflect mainly the S2′-state. The mixed population of the S2′- and S2-states is proposed to have the effect of broadening vibrational bands and shifting vibrational frequencies.
The model in Fig. 5 can potentially explain differing results concerning PSII mutations at aspartate 170 in the D1 subunit (57). The phenotype of the DE170D1 mutant suggested that this aspartate is close to or ligating Mn (57). X-ray diffraction studies of PSII also suggest that aspartate 170 is close to or ligating the manganese cluster ((16,17), but see (18,19)). In 200 K cryogenic FT-IR experiments conducted of the DE170D1 mutant, this mutation changed the light-induced difference spectrum dramatically (58). In experiments conducted with a single flash at higher temperature and scan times on the seconds timescale, no apparent effects of a DH170D1 mutation on the light-induced difference spectra were observed in the 1800–1200 cm−1 region (50). These discrepancies can be rationalized if photoinduced structural changes at D170 decay in the 330 ms before the beginning of data acquisition in Debus et al. (50).
Interpretation of strontium editing and the S1 to S2 transition
Given the model in Fig. 5, the difference FT-IR spectrum, acquired with 15-s scans and one flash, will correspond to a S2′-minus-S1′ spectrum (Fig. 5). In our experiments, dark adaptation times were held constant at 20 min to prevent variability due to evolution of the dark S1′ state. The rapid scan FT-IR spectra will reflect both photooxidation induced changes that occur when S1′ converts to S2 and also any protein structural changes that occur when S2 converts to S2′ and S1 converts to S1′. The structural change must persist on the seconds timescale to be detectable.
The strontium-edited S1′ to S2′ transition shows complex bands throughout the 1800 and 1200 cm−1 region (Fig. 4 A). The S1 to S2 transition is known to correspond to a MnIII to MnIV oxidation reaction (2). This oxidation reaction should slightly alter bond lengths in the manganese-calcium cluster (59) and could lead to perturbations of the calcium-binding site through bridging ligands. These changes would persist upon formation of the putative S2′ state, because no change in oxidation is proposed to occur (27). Available PSII structures may show bridging carboxylate amino acid ligation between calcium and manganese (16,17).
Alternatively, vibrational bands may undergo a Stark shift, due to the generation of additional charge near the calcium site, on this transition. Stark shifts in proteins can induce substantial frequency shifts in vibrational bands; the direction of the shift can be either positive or negative, depending on the relative orientations of the electric field and transition dipole (60,61). It has been proposed that the S1 to S2 transition builds up charge on the OEC, because the transition does not involve compensatory proton transfer (62). This idea may explain the unique spectral changes observed on this transition, when compared to the other S-states (see discussion below), on which compensatory proton transfer reactions are proposed to occur (62).
Strontium substitution is also expected to perturb the OEC, because PSII x-ray structures are consistent with calcium as an intrinsic component of the manganese cluster. Although all metal ligands have not been assigned in the PSII structures, peptide carbonyl oxygens, as well as water, aspartate side chains, and glutamate side chains, are likely protein ligands for calcium ions (63–66). Previous x-ray diffraction studies of the calcium binding protein, blood coagulation factor XIII, showed that strontium substitution alters the ligating environment of the divalent cation (67). Other model studies support the conclusion that coordination changes, including an increase in the number of divalent ion ligands, occur when strontium is exchanged into a calcium-binding site (68,69). Therefore, it seems possible that a strontium-induced change in the number and type of ligands may occur in the OEC and that these alterations may persist in the S2′ state. Changes in the number and type of ligands will result in a strontium-induced perturbation in the FT-IR spectrum. Note that some of the spectral perturbations are most likely due to the putative S2 to S2′ thermal transition (Fig. 5) and not due to the photo-induced oxidation reaction itself. This idea can be tested by experiments on faster timescales.
Interpretation of strontium editing and the other S-state transitions
Given the model in Fig. 5, the difference FT-IR spectrum, acquired with 15-s scans and two flashes, will correspond to an S3′-minus-S2′ spectrum (Fig. 5). Therefore, the rapid scan FT-IR spectra will reflect both photooxidation-induced changes that occur when S2 converts to S3 and also any protein conformational changes, protonation changes, or structural relaxation events that occur when S2′ converts to S2 and S3 converts to S3′. Difference spectra constructed with three and four flashes will correspond to S0′-minus-S3′ and S1′-minus-S0′ data, respectively. The structural change must persist on the seconds timescale to be detectable.
The strontium-edited data acquired with two (Fig. 4 B) and three (Fig. 4 C) flashes exhibit bands in the amide I region (1660–1640 cm−1) and bands in the amide I and II regions (1690–1620; 1589–1545 cm−1), respectively (47). Although the signal/noise ratio is lower, strontium-edited spectra acquired with four flashes also shows bands in the amide I and II regions. In each case, the observed frequencies are distinct. These data suggest two conclusions. First, because these strontium-edited spectra do not exhibit the complex spectral features observed in the S2′-minus-S1′, we propose that electrostatic Stark shifts (or perturbations of bridging ligands, see above) do not occur on these transitions. Although Mn redox reactions have been reported to occur on all the transitions at room temperature (4), proton transfer reactions may compensate for the electrostatic changes on the second, third, or fourth flash. Second, because amide bands are observed, we propose that these transitions perturb the vibrational frequencies of peptide carbonyl groups. This alteration could occur through a change in hydrogen bonding network that involves calcium and C=O ligands to calcium. Note that some of the spectral perturbations may be due to relaxation to generate the Sn′ states during the putative thermal transitions (Fig. 5) and not due to the photoinduced oxidation reactions themselves.
Comparison to data acquired under water limiting conditions and other previous results
When the data in this work are compared to strontium-edited spectra acquired under water limiting conditions in SM buffers (27), the spectra associated with the S1 to S2 transition are similar, except for an increase in overall amplitude in the hydrated double difference spectra. However, the spectra associated with the S2 to S3 transition are distinct from the previously reported data. In those previous FT-IR experiments, the water content of the PSII sample was limiting for the reaction mechanism (27). The comparison of the results suggests that water removal and strontium substitution have synergistic effects on the S2′ to S3′ transition. One possible explanation for this synergy is that this Sn′ state transition perturbs a hydrogen-bonded network that contains calcium, water, and peptide carboxyl groups (see discussion in Barry et al. (27)). Such a hydrogen-bonded network has been observed in structures of a calcium and strontium exchanged blood coagulation factor XIII (67).
Previous FT-IR experiments have disagreed about the effect of strontium substitution on the S2′-minus-S1′ spectrum (reviewed in Barry et al. (27)). In Strickler et al. (37), FT-IR data were acquired from cyanobacterial PSII; the cyanobacteria were cultured on strontium instead of calcium to generate the strontium-exchanged samples. In the cyanobacterial samples as in the plant samples employed here, complex changes are observed throughout the 1800–1200 cm−1 region. Although the resulting double difference spectra is similar to the data reported here (Fig. 4 A), spectral differences are observed. One possible explanation for these small differences is the content of PSII extrinsic subunits. In our plant samples, the 24 and 18 kDa proteins are removed to facilitate exchange. Cyanobacteria have a different complement of extrinsic subunits when compared to plants (70). Alternatively, differences in the temperature or length of data acquisition may cause the small spectral shifts observed. These factors could slightly alter the relative contributions of the S2 and S2′ states to the data.
Interestingly, our data are also distinct from the results of Kimura et al. (71), which were obtained from plant PSII membranes. In Kimura et al. (71), little effect of strontium substitution was observed in plant PSII membranes during the S1 to S2 transition. In Kimura et al. (71), plant PSII was calcium depleted, and then strontium was reconstituted. The work reported here shows that the difference between the two results is not the result of different hydration levels, because we observe significant strontium-induced changes in both hydrated (this work) and dehydrated (27) preparations on the first flash. The origin of this difference is under investigation, but may be due to differences in calcium substitution protocols. Our method causes the expected strontium-induced changes in the EPR signals from the S2-state (Fig. 1), showing that our method effectively substitutes strontium into the OEC.
SUMMARY
In this work, we propose a new model for the photosynthetic water oxidizing cycle, which proposes that relaxed conformation states are thermodynamically accessible from each photoinduced intermediate. These relaxed conformational states are proposed to remain at the same oxidation state as the photoinduced S-state. This model can explain many apparent contradictions in the literature concerning PSII function. Also, there is precedent in the literature showing that membrane proteins, such as bacteriorhodopsin (72), cytochrome bo (73), and cytochrome c oxidase (74), can undergo conformational relaxation to generate kinetically distinct states. This model implies that substantial protein conformational changes are linked to the photoinduced transitions in the water oxidizing cycle.
Our work also shows that the calcium binding site in the OEC is perturbed on each photooxidation induced transition, although the perturbative mechanism differs on the S1′ to S2′ transition compared to later transitions. During the S1′ to S2′ transition, the data are consistent with an electrostatic effect of Mn oxidation on the calcium site or with a perturbation of bridging ligands between Mn and Ca+2. During the S3′ to S0′ and S0′ to S1′ transitions, the data are consistent with perturbations of a hydrogen-bonded network, which involves water, peptide C=O groups, and calcium. To explain our results, persistent changes in divalent ion coordination must occur when strontium is substituted for calcium.
Acknowledgments
This work was supported by National Science Foundation grant No. MCB 03-55421.
Abbreviations used: PSII, photosystem II; chl, chlorophyll; DCBQ, 2,6-dichloro-1,4-benzoquinone; EPR, electron paramagnetic resonance; ESEEM, electron spin echo envelope modulation; EXAFS, extended x-ray absorption fine structure; FT-IR, Fourier transform infrared; OEC, oxygen-evolving complex of photosystem II; NMR, nuclear magnetic resonance; XANES, x-ray absorption near edge structure.
References
- 1.Joliot, P., and B. Kok. 1975. Oxygen evolution in photosynthesis. In Bioenergetics of Photosynthesis. Govindjee, editor. Academic Press, New York. 388–412.
- 2.Britt, R. D., K. A. Campbell, J. M. Peloquin, M. L. Gilchrist, C. P. Aznar, M. M. Dicus, J. Robblee, and J. Messinger. 2004. Recent pulsed EPR studies of the photosystem II oxygen-evolving complex: implications as to water oxidation mechanisms. Biochim. Biophys. Acta. 1655:158–171. [DOI] [PubMed] [Google Scholar]
- 3.Ono, T., T. Noguchi, Y. Inoue, M. Kusunoki, T. Matsushita, and H. Oyanagi. 1992. X-ray detection of the period-four cycling of the manganese cluster in photosynthetic water oxidizing enzyme. Science. 258:1335–1337. [DOI] [PubMed] [Google Scholar]
- 4.Haumann, M., P. Liebisch, C. Muller, M. Barra, M. Grabolle, and H. Dau. 2005. Photosynthetic O2 formation tracked by time-resolved x-ray experiments. Science. 310:1019–1021. [DOI] [PubMed] [Google Scholar]
- 5.Roelofs, T. A., W. Liang, M. L. Latimer, R. M. Cinco, A. Rompel, J. C. Andrews, K. Sauer, V. K. Yachandra, and M. P. Klein. 1996. Oxidation states of the manganese cluster during the flash-induced S-state cycle of the photosynthetic oxygen-evolving complex. Proc. Natl. Acad. Sci. USA. 93:3335–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Messinger, J., J. H. Robblee, U. Bergmann, C. Fernandez, P. Glatzel, H. Visser, R. Cinco, K. L. McFarlane, E. Bellacchio, S. A. Pizarro, S. P. Cramer, K. Sauer, M. Klein, and V. K. Yachandra. 2001. Absence of Mn-center oxidation in the S2–S3 transition: implications for the mechanism of photosynthetic water oxidation. J. Am. Chem. Soc. 123:7804–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang, W., M. J. Latimer, H. Dau, T. A. Roelofs, V. K. Yachandra, K. Sauer, and M. P. Klein. 1994. Correlation between structure and magnetic spin state of the manganese cluster in the oxygen-evolving complex of photosystem II in the S2 state: determination by x-ray absorption spectroscopy. Biochemistry. 33:4923–4932. [DOI] [PubMed] [Google Scholar]
- 8.Karge, M., K.-D. Irrgang, and G. Renger. 1997. Analysis of the reaction coordinate of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O. Biochemistry. 36:8904–8913. [DOI] [PubMed] [Google Scholar]
- 9.Liang, W., T. A. Roelofs, R. M. Cinco, A. Rompel, M. J. Latimer, W. O. Yu, K. Sauer, M. P. Klein, and V. K. Yachandra. 2000. Structural change of the Mn cluster during the S2 to S3 state transition of the oxygen-evolving complex of photosystem II. Does it reflect the onset of water/substrate oxidation? Determination by Mn x-ray absorption spectroscopy. J. Am. Chem. Soc. 122:3399–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yocum, C. F. 1992. The calcium and chloride requirements for photosynthetic water oxidation. In Manganese Redox Enzymes. V. L. Pecoraro, editor. VCH Publishers, New York. 71–83.
- 11.Penner-Hahn, J. E., R. M. Fronko, V. L. Pecoraro, C. F. Yocum, S. D. Betts, and N. R. Bowlby. 1990. Structural characterization of the manganese sites in the photosynthetic oxygen-evolving complex using x-ray absorption spectroscopy. J. Am. Chem. Soc. 112:2549–2557. [Google Scholar]
- 12.Latimer, M. J., V. J. DeRose, V. K. Yachandra, K. Sauer, and M. P. Klein. 1998. Structural effects of calcium depletion on the manganese cluster of photosystem II: determination by x-ray absorption spectroscopy. J. Phys. Chem. B. 102:8257–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim, S. H., W. Gregor, J. M. Peloquin, M. Brynda, and R. D. Britt. 2004. Investigation of the calcium-binding site of the oxygen evolving complex of photosystem II using 87Sr ESEEM spectroscopy. J. Am. Chem. Soc. 126:7228–7237. [DOI] [PubMed] [Google Scholar]
- 14.Zouni, A., H.-T. Witt, J. Kern, P. Fromme, N. Krauß, W. Saenger, and P. Orth. 2001. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 409:739–743. [DOI] [PubMed] [Google Scholar]
- 15.Kamiya, N., and J.-R. Shen. 2003. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 Å resolution. Proc. Natl. Acad. Sci. USA. 100:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferreira, K. N., T. M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata. 2004. Architecture of the photosynthetic oxygen-evolving center. Science. 303:1831–1837. [DOI] [PubMed] [Google Scholar]
- 17.Loll, B., J. Kern, W. Saenger, A. Zouni, and J. Biesiadka. 2005. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 438:1040–1044. [DOI] [PubMed] [Google Scholar]
- 18.Yano, J., J. Kern, K.-D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni, and V. K. Yachandra. 2005. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc. Natl. Acad. Sci. USA. 102:12047–12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grabolle, M., M. Haumann, C. Muller, P. Liebisch, and H. Dau. 2006. Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by X-ray irradiation at 10–300 K. J. Biol. Chem. 281:4580–4588. [DOI] [PubMed] [Google Scholar]
- 20.Matysik, J., A. Alia, G. Nachtegaal, H. J. van Gorkom, A. J. Hoff, and H. J. M. de Groot. 2000. Exploring the calcium-binding site in photosystem II membranes by solid-state 113Cd NMR. Biochemistry. 39:6751–6755. [DOI] [PubMed] [Google Scholar]
- 21.Ghanotakis, D. F., G. T. Babcock, and C. F. Yocum. 1984. Calcium reconstitutes high rates of oxygen evolution in polypeptide depleted photosystem II preparations. FEBS Lett. 167:127–130. [Google Scholar]
- 22.Boussac, A., J.-L. Zimmermann, and A. W. Rutherford. 1989. EPR signals from modified charge accumulation states of the oxygen evolving enzyme in Ca+2-deficient photosystem II. Biochemistry. 28:8984–8989. [DOI] [PubMed] [Google Scholar]
- 23.Westphal, K. L., N. Lydakis-Simantris, R. I. Cukier, and G. T. Babcock. 2000. Effects of Sr+2 substitution on the reduction rates of Yz∑ in PSII membranes—evidence for concerted hydrogen-atom transfer in oxygen evolution. Biochemistry. 39:16220–16229. [DOI] [PubMed] [Google Scholar]
- 24.Boussac, A., F. Rappaport, P. Carrier, J.-M. Verbavatz, R. Gobin, D. Kirilovsky, A. W. Rutherford, and M. Sugiura. 2004. Biosynthetic Ca+2/Sr+2 exchange in photosystem II oxygen-evolving enzyme of Thermosynechococcus elongatus. J. Biol. Chem. 279:22809–22819. [DOI] [PubMed] [Google Scholar]
- 25.Riggs-Gelasco, P. J., R. Mei, C. F. Yocum, and J. E. Penner-Hahn. 1996. Reduced derivatives of the Mn cluster in the oxygen-evolving complex of photosystem II: an EXAFS study. J. Am. Chem. Soc. 118:2387–2399. [Google Scholar]
- 26.Haumann, M., and W. Junge. 1999. Photosynthetic water oxidation: a simplex-scheme of its partial reactions. Biochim. Biophys. Acta. 1411:86–91. [DOI] [PubMed] [Google Scholar]
- 27.Barry, B. A., C. Hicks, A. De Riso, and D. L. Jenson. 2005. Calcium ligation in photosystem II under inhibiting conditions. Biophys. J. 89:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pecoraro, V. L., M. J. Baldwin, M. T. Caudle, W. Y. Hsieh, and N. A. Law. 1998. A proposal for water oxidation in photosystem II. Pure Appl. Chem. 70:925–929. [Google Scholar]
- 29.Hendry, G., and T. Wydrzynski. 2003. 18O isotope exchange measurements reveal that calcium is involved in the binding of one substrate-water molecule to the oxygen-evolving complex in photosystem II. Biochemistry. 42:6209–6217. [DOI] [PubMed] [Google Scholar]
- 30.Vrettos, J. S., D. A. Stone, and G. W. Brudvig. 2001. Quantifying the ion selectivity of the calcium site in photosystem II: evidence for direct involvement of Ca+2 in O2 formation. Biochemistry. 40:7937–7945. [DOI] [PubMed] [Google Scholar]
- 31.Barry, B. A., I. B. Cooper, A. De Riso, S. H. Brewer, D. Vu, and R. B. Dyer. 2006. Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle. Proc. Natl. Acad. Sci. USA. 103:7288–7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hillier, W., and G. Babcock. 2001. S-state dependent Fourier transform infrared difference spectra for the photosystem II oxygen evolving complex. Biochemistry. 40:1503–1509. [DOI] [PubMed] [Google Scholar]
- 33.Noguchi, T., T. Tomo, and C. Kato. 2001. Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry. 40:1497–1502. [DOI] [PubMed] [Google Scholar]
- 34.Yamanari, T., Y. Kimura, N. Mizusawa, A. Ishii, and T. Ono. 2004. Mid- to low-frequency Fourier transform infrared spectra of S-state cycle for photosynthetic water oxidation in Synechocystis sp. PCC 6803. Biochemistry. 43:7479–7490. [DOI] [PubMed] [Google Scholar]
- 35.Noguchi, T., and M. Sugiura. 2003. Analysis of flash-induced FTIR difference spectra of the S-state cycle in the photosynthetic water-oxidizing complex by uniform 15N and 13C isotope labeling. Biochemistry. 42:6035–6042. [DOI] [PubMed] [Google Scholar]
- 36.Sachs, R., K. M. Halverson, and B. A. Barry. 2003. Specific isotope labeling and photooxidation-linked structural changes in the manganese stabilizing subunit of photosystem II. J. Biol. Chem. 278:44222–44229. [DOI] [PubMed] [Google Scholar]
- 37.Strickler, M. A., L. M. Walker, W. Hillier, and R. J. Debus. 2005. Evidence from biosynthetically incorporatted strontium and FTIR difference spectroscopy that the C-terminus of the D1 polypeptide of photosystem II does not ligate calcium. Biochemistry. 44:8571–8577. [DOI] [PubMed] [Google Scholar]
- 38.Berthold, D. A., G. T. Babcock, and C. F. Yocum. 1981. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 134:231–234. [Google Scholar]
- 39.Anderson, L. B., A. J. A. Ouellette, and B. A. Barry. 2000. Probing the primary structure of photosystem II with amines and phenylhydrazine. J. Biol. Chem. 275:4920–4927. [DOI] [PubMed] [Google Scholar]
- 40.Ghanotakis, D. F., J. N. Topper, G. T. Babcock, and C. F. Yocum. 1984. Water-soluble 17 and 23 kDa polypeptides restore oxygen evolution activity by creating a high-affinity binding site for Ca+2 on the oxidizing side of photosystem II. FEBS Lett. 170:169–173. [Google Scholar]
- 41.Barry, B. A. 1995. Tyrosyl radicals in photosystem II. Methods Enzymol. 258:303–319. [DOI] [PubMed] [Google Scholar]
- 42.Yocum, C. F. 1991. Calcium activation of photosynthetic water oxidation. Biochim. Biophys. Acta. 1059:1–15. [Google Scholar]
- 43.Halverson, K. M., and B. A. Barry. 2003. Evidence for spontaneous structural changes in a dark-adapted state of photosystem II. Biophys. J. 85:2581–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halverson, K. M., and B. A. Barry. 2003. Sucrose and glycerol effects on photosystem II. Biophys. J. 85:1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim, S., and B. A. Barry. 1998. The protein environment surrounding tyrosyl radicals D∑ and Z∑ in photosystem II: a difference FT-IR study. Biophys. J. 74:2588–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller, A.-F., and G. W. Brudvig. 1991. A guide to electron paramagnetic resonance spectroscopy of photosystem II membranes. Biochim. Biophys. Acta. 1056:1–18. [DOI] [PubMed] [Google Scholar]
- 47.Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. In Advances in Protein Chemistry. Vol. 38. C. B. Anfinsen, J. T. Edsall, and F. M. Richards, editors. Academic Press, New York. 181–364. [DOI] [PubMed]
- 48.Braiman, M. S., D. M. Briercheck, and K. M. Kriger. 1999. Modeling vibrational spectra of amino acid side chains in proteins: effects of protonation state, counterion, and solvent on arginine C-N stretch frequencies. J. Phys. Chem. B. 103:4744–4750. [Google Scholar]
- 49.Kimura, Y., N. Mizusawa, A. Ishii, and T.-a. Ono. 2005. FTIR detection of structural changes in a histidine ligand during S-state cycling of photosynthetic oxygen-evolving complex. Biochemistry. 44:16072–16078. [DOI] [PubMed] [Google Scholar]
- 50.Debus, R. J., M. A. Strickler, L. M. Walker, and W. Hillier. 2005. No evidence from FTIR difference spectroscopy that aspartate-170 of the D1 polypeptide ligates a manganese ion that undergoes oxidation during the S0 to S1, S1 to S2, or S2 to S3 transitions in photosystem II. Biochemistry. 44:1367–1374. [DOI] [PubMed] [Google Scholar]
- 51.Dekker, J. P., J. J. Plijter, L. Ouwehand, and H. J. van Gorkom. 1984. Kinetics of manganese redox transitions in the oxygen-evolving apparatus of photosynthesis. Biochim. Biophys. Acta. 767:176–179. [Google Scholar]
- 52.Beck, W. F., J. C. de Paula, and G. W. Brudvig. 1985. Active and resting states of the O2-evolving complex of photosystem II. Biochemistry. 24:3035–3043. [DOI] [PubMed] [Google Scholar]
- 53.Steenhuis, J. J., and B. A. Barry. 1996. A difference infrared study of protein structural changes in the photosynthetic water-oxidizing complex. J. Am. Chem. Soc. 118:11927–11932. [Google Scholar]
- 54.Steenhuis, J. J., and B. A. Barry. 1997. The protein and ligand environment of the S2 state in photosynthetic oxygen evolution: a difference FT-IR study. J. Phys. Chem. 101:6652–6660. [Google Scholar]
- 55.Steenhuis, J. J., and B. A. Barry. 1998. An FT-IR study of photoassembly in the manganese catalytic site of photosystem II. J. Phys. Chem. 102:4–8 [Letter]. [Google Scholar]
- 56.Hutchison, R. S., J. J. Steenhuis, C. F. Yocum, R. M. Razeghifard, and B. A. Barry. 1999. Deprotonation of the 33 kDa, extrinsic, manganese stabilizing protein accompanies photooxidation of manganese in photosystem II. J. Biol. Chem. 274:31987–31995. [DOI] [PubMed] [Google Scholar]
- 57.Boerner, R. J., A. P. Nguyen, B. A. Barry, and R. J. Debus. 1992. Evidence from directed mutagenesis that aspartate 170 of the D1 polypeptide influences the assembly and/or stability of the manganese cluster in the photosynthetic water-splitting complex. Biochemistry. 31:6660–6672. [DOI] [PubMed] [Google Scholar]
- 58.Steenhuis, J. J., R. S. Hutchison, and B. A. Barry. 1999. Alternations in carboxylate ligation at the active site of photosystem II. J. Biol. Chem. 274:14609–14616. [DOI] [PubMed] [Google Scholar]
- 59.Smith, J. C., E. Gonzalez-Vergara, and J. B. Vincent. 1997. Detection of structural changes upon oxidation in multinuclear Mn-oxo-carboxylate assemblies by Fourier transform infrared spectroscopy: relationship to photosystem II. Inorg. Chim. Acta. 255:99–103. [Google Scholar]
- 60.Phillips, G. N., Jr., M. L. Teodoro, T. Li, B. Smith, and J. S. Olson. 1999. Bound CO is a molecular probe of electrostatic potential in the distal pocket of myoglobin. J. Phys. Chem. B. 103:8817–8829. [Google Scholar]
- 61.Suydam, I. T., and S. G. Boxer. 2003. Vibrational Stark effects calibrate the sensitivity of vibrational probes for electric fields in proteins. Biochemistry. 42:12050–12055. [DOI] [PubMed] [Google Scholar]
- 62.McEvoy, J. P., and G. W. Brudvig. 2004. Structure-based mechanism of photosynthetic water oxidation. Phys. Chem. Chem. Phys. 6:4754–4763. [Google Scholar]
- 63.Bjornson, M. E., D. C. Corson, and B. D. Sykes. 1985. 13C and 113Cd NMR studies of the chelation of metal ions by the calcium binding protein parvalbumin. J. Inorg. Biochem. 25:141–149. [DOI] [PubMed] [Google Scholar]
- 64.Szebenyi, D. M., and K. Moffat. 1986. The refined structure of vitamin D-dependent calcium-binding protein from bovine intestine. Molecular details, ion binding, and implications for the structure of other calcium-binding proteins. J. Biol. Chem. 261:8761–8777. [PubMed] [Google Scholar]
- 65.Ilag, L. L., R. McKenna, M. P. Yadav, J. N. BeMiller, N. L. Incardona, and M. G. Rossmann. 1994. Calcium ion-induced structural changes in bacteriophage phi X174. J. Mol. Biol. 244:291–300. [DOI] [PubMed] [Google Scholar]
- 66.Dudev, T., Y. Lin, M. Dudev, and C. Lim. 2003. First-second shell interactions in metal binding sites in proteins: a PDB survey and DFT/CDM calculations. J. Am. Chem. Soc. 125:3168–3180. [DOI] [PubMed] [Google Scholar]
- 67.Fox, B. A., V. C. Yee, L. C. Pedersen, I. L. Trong, P. D. Bishop, R. E. Stenkamp, and D. C. Teller. 1999. Identification of the calcium binding site and a novel yterrbium site in blood coagulation factor XIII by x-ray crystallography. J. Biol. Chem. 274:4917–4923. [DOI] [PubMed] [Google Scholar]
- 68.Schauer, C. K., and O. P. Anderson. 1987. Calcium-selective ligands. 2. Structural and spectroscopic studies on calcium and cadmium complexes of EGTA. J. Am. Chem. Soc. 109:3646–3656. [Google Scholar]
- 69.Joris, S. J., and C. H. Amberg. 1971. The nature of deficiency in nonstoichiometric hydroxyapatites. II. Spectroscopic studies of calcium and strontium hydroxyapatites. J. Phys. Chem. 75:3172–3178. [Google Scholar]
- 70.Kashino, Y., W. M. Lauber, J. A. Carroll, Q. Wang, J. Whitmarsh, K. Satoh, and H. B. Pakrasi. 2002. Proteomic analysis of a highly active photosystem II preparation from the cyanobacterium Synechocystis sp. PCC 6803 reveals the presence of novel polypeptides. Biochemistry. 41:8004–8012. [DOI] [PubMed] [Google Scholar]
- 71.Kimura, Y., K. Hasegawa, and T.-a. Ono. 2002. Characteristic changes of the S2/S1 difference FTIR spectrum induced by Ca+2 depletion and metal cation substitution in the photosyntheticc oxygen-evolving complex. Biochemistry. 41:5844–5853. [DOI] [PubMed] [Google Scholar]
- 72.Nishikawa T., M. Murakami, and T. Kouyama. 2005. Crystal structure of the 13-cis isomer of bacteriorhodopsin in the dark-adapted state. J. Mol. Biol. 352:319–328. [DOI] [PubMed] [Google Scholar]
- 73.Moody, A. J., C. E. Cooper, R. B. Gennis, J. N. Rumbley, and P. R. Rich. 1995. Interconversion of fast and slow forms of cytochrome bo from Escherichia coli. Biochemistry. 34:6838–6846. [DOI] [PubMed]
- 74.Moody, A. J., C. E. Cooper, and P. R. Rich. 1991. Characterisation of ‘fast’ and ‘slow’ forms of bovine heart cytochrome-c oxidase. Biochim. Biophys. Acta. 1059:189–207. [DOI] [PubMed] [Google Scholar]
- 75.Carey, F. A., and R. J. Sundberg. 2000. Advanced Organic Chemistry. Kluwer Academic/Plenum Publishers, New York.