

Abstract
Structural characterization of biomolecules by NMR methods frequently requires the enrichment of magnetically active isotopes at particular molecular sites. Introduction is usually achieved biosynthetically through the use of bacterial cultures grown on isotopically enriched media, but for certain types of molecules—cell-surface carbohydrates of mammalian origin, for example—this is not practical. Here we explore a means of introducing 13C-enriched sites, postisolation in natural carbohydrate products, and illustrate an ability to acquire sufficient information to select appropriate conformational models from among energetically allowed sets. The application presented involves replacement of native N-acetyl groups with 13C-labeled acetyl groups in a simple disaccharide derivative, (GlcNAc)2-OBu, or O-butyl-chitobiose. The assignment of the two acetyl groups introduced is based on a novel combination of NMR and mass spectrometry data. Structural information is obtained from chemical shift anisotropy offsets of 13C carbonyl resonances and 13C-13C dipolar couplings between the labeled methyl and carbonyl carbons of the acetyl groups. Although the application is to a relatively simple system, it lays the groundwork for application to biologically important complex carbohydrate systems.
INTRODUCTION
Carbohydrates that occur naturally as parts of glycoproteins, glycolipids, and glycoaminoglycans play crucial roles in biological processes that range from cell differentiation to inflammation to fertilization to protein turnover to pathogen infection (1–4). Very often specific recognition of structural features of carbohydrates by proteins is a key step in these processes, and structural characterization of the carbohydrate becomes important to the understanding and control of the processes. However, structural characterization has not been easy (5,6). Many carbohydrates do not crystallize easily, and NMR spectra are complex because of the functional similarity of the sugars that combine to make complex carbohydrates. 13C-NMR spectra and multi-dimensional versions of these spectra offer higher resolution than 1H spectra, but without enrichment in 13C, sensitivity is often inadequate. Enrichment in 13C has been accomplished in a few cases (7,8), but for many carbohydrates, complexity and species specificity prohibits synthetic or biosynthetic introduction of 13C.
Here we illustrate a procedure for introducing 13C-labeled sites in complex carbohydrates through the process of exchange of acetyl groups in isolated natural products. We also include a novel assignment strategy for cases with multiple acetyl groups; this strategy is based on a combination of NMR and mass spectrometry methods. We demonstrate that adequate structural information can be obtained from 13C labels in a few enriched sites to distinguish energetically reasonable from unreasonable structural models. Application is to a simple disaccharide of N-acetyl glucosamine (chitobiose) anchored to a model membrane through the addition of a butyl chain at the reducing end.
Chitin, a β(1–4) polymer of N-acetyl glucosamine, is a common component of the cell walls of fungi and the exoskeletons of insects. There has been a substantial interest in the conformational properties of derived oligomers as they relate to the binding of plant defense proteins such as hevein. A substantial amount of prior work using primarily homonuclear nuclear Overhauser effect (NOE)-based NMR methods to deduce conformational properties of chitin oligomers has, in fact, been undertaken (9,10). Here we use the simple disaccharide unit from chitin as a representative of more complex N-acetyl containing oligosaccharides that may be less amenable to study by homonuclear NOE methods, particularly when interacting with proteins or other cellular elements. Acetyl groups are, for example, widely distributed in glycosaminoglycans such as heparin and chondroitin sulfate where every second sugar could carry an acetyl group (11,12).
The structural characterization that we demonstrate exploits orientationally dependent parameters in a manner similar to our recent work on a galactosyl-mannosyl disaccharide (13). This work relied on residual dipolar couplings (RDCs) that could be retrieved from a number of sites in each sugar ring; it benefited from a substantial amount of prior work using RDCs to investigate conformations of oligosaccharides (7,14–17). This work differs in its focus on data that can be retrieved from spectra of 13C-labeled acetyl groups in acetylated saccharides. One of the problems that must be overcome is removing native acetyls and replacing them with a 13C-enriched version. Here we use a carefully controlled base-catalyzed hydrolysis reaction to remove acetyls, and we replace them with acetyls from 13C-enriched acetic anhydride. It is however likely that suitable enzyme-catalyzed steps for removal or addition could replace these steps in applications to more sensitive targets.
13C-labeled acetyl groups are actually rich in structural information. A group labeled at the carbonyl carbon has a large chemical shift anisotropy (CSA) that can provide orientational information (18). Likewise, a group labeled at both methyl and carbonyl carbons can exhibit 13C-13C RDC that provides complementary information (19,20). In both cases, preferential orientation of the molecule in media such as aqueous dispersions of lipid or detergent bicelles is required to make these interactions measurable. For illustration purposes, we also found it advantageous to enhance orientation of our disaccharide by promoting association with bicelles through the addition of a butyl chain.
The structural information, although rich, is not so abundant that introduction of a single group is adequate; so multiple acetyl groups (two in our case) are introduced. This immediately presents an assignment problem. Usually NMR assignments are accomplished by through-bond magnetization transfer (total correlation spectroscopy or coupling correlated spectroscopy, for example) to make sequential connections between acetyl atoms and a series of atoms in the backbones of the carbohydrate (6,21). Without uniform 13C enrichment, these assignment strategies are often impractical especially for larger homooligomers and oligomers with resonances broadened by association with proteins or lipid membranes. To avoid this problem, a novel approach based on distinguishing the different isotope enrichment level of each acetyl group was introduced. Both MS and NMR are capable of detecting the presence of 13C isotopes. NMR does this through resonance intensity; MS does this through mass shifts. NMR provides, in addition, structural information, but MS can distinguish residues, even in homopolymers, through well-characterized fragmentation patterns (22).
The data presented here prove adequate to distinguish reasonable from unreasonable models for the structure of our (GlcNAc)2-OBu disaccharide. Although this characterization falls short of a de novo structural determination method, it sets a precedent for a new structural strategy that may be of particular utility in the investigation of protein interactions with complex carbohydrate systems.
MATERIALS AND METHODS
Synthesis
Acetic anhydride-13C4 was purchased from Cambridge Isotope Laboratories (Andover, MA), and iatrobeads were purchased from Iatron Laboratories (Tokyo, Japan). All the other reagents used in preparation of the NMR samples and used in the synthesis of the glycosides were purchased from Sigma-Aldrich (St. Louis, MO).
O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl)-(1→4)-2-acetamido-1,3,6-tri-O-acetyl-2-deoxy-α, β-D-glucopyranose (2)
The synthesis of the butyl analog of chitobiose 1 followed approximately the procedure outlined in Hare et al. (23) (Fig. 1). To a solution of compound 1 (50 mg, 0.12 mmol) in pyridine (1 ml) was added Ac2O (0.4 mL, 4.2 mmol), and the solution was stirred at room temperature overnight. The residue was put on a silica gel column and eluted (EtOAc/methanol = 10:1) to yield 2 (74 mg, 0.11 mmol, 90%).
FIGURE 1.

Synthesis of 13C (GlcNAc)2-OBu from chitobiose. Asterisk denotes 13C label.
O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl)-(1→4)-2-methyl-(3,6-di-O-acetyl-1,2-dideoxy-α-D-glucopyrano)-[2,1-d]-2-oxazoline (3)
To a solution of compound 2 (74 mg, 0.11 mmol) in dry dichloroethane was added TMS triflate (19 μL, 0.11 mmol, 1 equiv). The reaction flask was flushed with dry argon gas, and the reaction was stirred for 5 h at 90°. Triethylamine (43 μL, 0.33 mmol, 3 equiv) was added at 0° to halt the reaction, and the solvent was evaporated with additions of toluene. It must be noted that compound 3 is not very stable in the presence of water and can be lost if eluted on a silica gel column. To improve yield, the crude product was moved to the next step without purification.
Butyl-O-(2-acetamido-3,4,6-tri-O-acetyl-2-deoxy-β-D-glucopyranosyl)-(1→4)-2-acetamido-3,6-di-O-acetyl-2-deoxy-β-D-glucopyranoside (4)
To a solution of 3 in dry n-butanol (1 mL) trifluoromethanesulfonic acid (2.65 μL, 0.03 mmol) was added. The reaction was protected by overlaying dry argon gas and was stirred at 60° for 20 min. Pyridine (1.1 mL) was added to halt the reaction, and the solution was evaporated with additions of toluene. To improve yield, the crude product was again moved to the next step without purification.
Butyl-O-(2-amine-2-deoxy-β-D-glucopyranosyl)-(1→4)-2-amine-2-deoxy-β-D-glucopyranoside (5)
To a solution of 4 in H2O (1 mL) was added anhydrous BaO (77 mg), and the solution was stirred at 90° overnight to promote hydrolysis of acetyl groups (24). H2SO4/H2O (0.5 N) was added at 0° until pH = 7 to quench the reaction. Removal of N-acetyls at this point is not complete, leading to a percentage of 12C-N-acetyls in the final product. Precipitated BaSO2 was removed by filtration, and the supernatant was freeze-dried to give 5 as an amorphous solid (16 mg).
Butyl-O-(2-[1,2-13C2]acetamido-2-deoxy-β-D-glucopyranosyl)-(1→4)-2-[1,2-13C2]acetamido-2-deoxy-β-D-glucopyranoside (6)
To a solution of 5 in dry methanol (2 mL) was added acetic anhydride-13C4 (7.5 μL, 0.09 mmol, 1.5 equil). The reaction was stirred for 3 h and halted when complete as judged by TLC. The product was purified by iatrobeads column chromatography (CHCl3/methanol = 2:1) to give 6 as colorless solid (19 mg, 95%).
Preparation of NMR samples
To introduce alignment in the sample and mimic a lipid bilayer environment that would promote association of the alkylated sugar, a liquid crystal solvent was prepared from pentaethylene glycol monododecyl ether (C12E5) (25). An 8% (w/w) alignment medium was made by adding 47 uL C12E5 and 17 uL hexanol in 500 uL D2O. A total of 2.4 mg (GlcNAc)-OBu was added to make a 10-mM field alignable sample. We also made another 10-mM sample in pure D2O for isotropic measurements. A total of 5 mg tetramethylammonium bromide was added to both samples as a chemical shift reference.
NMR methods
The NMR spectra were recorded on a Varian (Palo Alto, CA) Inova spectrometer operating at 125.67 MHz for 13C. Proton homonuclear decoupling was achieved using WALTZ-16 at a decoupling power of 37 W. For the sample in D2O, spectra were acquired under near fully relaxed conditions since the signal intensity of each N-acetyl group was used to determine the isotopic ratio 13C/12C in acetyl groups of each sugar residue,. To achieve this, the repetition delay was set to 4 s. For the sample in the aligned liquid crystal environment, the acquisition time was kept short (100 ms) and the relaxation delay relatively long (2 s) to avoid heating of the sample by the decoupling field.
MS methods
Matrix-assisted laser desorption ionization (MALDI-MS) spectra were obtained on an Applied Biosystems (Foster City, CA) 4700 time of flight/time of flight (TOF/TOF) Proteomics Analyzer, equipped with a nitrogen laser (337 nm). A total of 0.1 M 2,5-dihydroxybenzoic acid in acetonitrile was used as the ionization matrix. Collision-induced dissociation was applied to the molecular ions generated by the first TOF stage to fragment the ions for residue-specific analysis (16). The mass selection window was set relatively wide (10 Da) to allow isotope analysis of the fragmented ions in the second stage.
Molecular simulations
Molecular structures to be used in analysis of NMR data were first generated with a web-based carbohydrate-building tool (26). Structures were then energy minimized using AMBER 7 (27) using the GLYCAM_04 force field (26). This program was also used to generate an energy map as a function of φ and ψ glycosidic torsion angles so that energetically unreasonable structures could be eliminated from consideration. To produce structures with variations in glycosidic torsion angles, the bond rotation tools in the program CHIMERA were used (28).
RESULTS
Synthesis
The first step in our strategy for the structural characterization of the (GlcNAc)2-OBu disaccharide is the introduction of 13C-labeled acetyl groups. Both enzymatic and chemical methods are available for the removal of the N-acetyl groups (29,30). The methodology we illustrated in Fig. 1 was based on strong base deacetylation. The primary side reaction involves degredation from the reducing end. Hence, we found it both necessary and convenient to protect the reducing end by preparing an O-alkyl derivative before deacetylation. In our case the introduction of an O-butyl group also provides a means of increasing the alignment of the sample (13) (Fig. 2). The methodology based on the oxazoline chemistry allowed synthesis in a reasonable yield and gave almost exclusively the β-anomer as product.
FIGURE 2.

Association of the (GlcNAc)2-OBu with oriented bicelles.
To facilitate assignment, it was desirable to have the 13C/12C isotopic ratio for each N-acetyl group different. Thus, partial deacetylation relying on inherent differences in reactivity was required. Here, the temperature for the deacetylation reaction was kept lower than 100° and the reaction time was limited to <12 h. Reacetylation with doubly 13C-labeled acetic anhydride was accomplished with preferential acetylation of the amino group as opposed to hydroxyl groups. This selectivity is based on the higher reactivity of the amino group and the higher stability of the amide bond.
Mass spectrometry data
Assaying the extent of 13C-label introduction was done via mass spectrometry. Expansions of mass regions corresponding to B1 and Y1 ions of the natriated form of the glycoside are shown in Fig. 3, a and b, respectively (22). These ions contain single N-acetylglucosamine rings from the nonreducing and alkylated parts of the molecule, respectively. The monoisotopic peaks at lower mass represent 12C-containing molecules; the peaks two mass units higher represent those containing a 13C2-acetyl group. Considering the small number of the carbon atoms in the molecule, the contribution from natural abundance 13C to the higher mass peak can be ignored. Therefore, the intensity of the later peak comes primarily from the doubly 13C-labeled N-acetyl group. The N-acetylglucosamine at the nonreducing end is also highly enriched. Fitting the data suggests the isotopic ratios for two N-acetyl groups are 68% and 47% for the nonreducing (II) and alkylated (I) end sugars, respectively.
FIGURE 3.

MASS spectra of B1 (a) and Y1 (b) ions of the natriated form of the oligosaccharide. These show different isotopic ratios for each sugar residue.
NMR spectra of N-acetyl groups
The carbonyl region of 13C-NMR spectra for the sample in an isotropic D2O solution is presented in Fig. 4 A. Doublets with 50.2-Hz splittings from the N-acetyl carbonyls of each residue were observed at 174.70 and 174.81 ppm, respectively. These arise from coupling to a second 13C in the directly bonded acetyl methyl group. The intensities of these doublets are different, with the lower field doublet being ∼1.5 times as intense. Given that in well-relaxed spectra intensities reflect isotopic abundance and given that the mass spectrometry data show the N-acetylglucosamine at the nonreducing end of the disaccharide to be more highly enriched, the downfield doublet can be assigned to the nonreducing or terminal site (II).
FIGURE 4.

1H-decoupled 13C-NMR spectrum (125 MHz) of (GlcNAc)2-OBu 13C-labeled in the acetyl carbons. The carbonyl regions of the spectrum are shown. (A) Sample in D2O. (B) Sample in oriented medium. The total acquisition time for b was ∼8 h.
The same region of the spectrum from a sample in an aligned C12E5 liquid crystal medium is presented in Fig. 4 B. Both doublets are shifted downfield but by different amounts (0.29 and 0.39 ppm, respectively). Also, close inspection shows that the splittings of the doublets have changed (by −6.8 and −10.5 Hz, respectively). These changes are typical of the effects of incomplete averaging of anisotropic parameters in aligned media. The change in splitting arises from an RDC, and the change in chemical shift is suggested to arise from a CSA offset. Changes in splittings can also be seen in the acetyl methyl region of the spectrum (not show). The changes in splittings, as expected, are identical to those measured from the carbonyl resonances. The changes in chemical shift for the methyl resonances are negligible because of the small CSA of the methyl carbons and the apparent lack of direct medium effects on chemical shift. To confirm the lack of medium effects on chemical shifts, we have also examined shifts of carbonyls in a nearly identical medium that does not align (altered by the addition of small extra amounts of hexanol). Changes in shifts are <0.01 ppm, confirming the origin of chemical shift changes as CSA offsets.
DISCUSSION
Theory
For a doubly 13C-labeled N-acetyl group, the RDCs and CSA offsets measured above potentially contain a great deal of structural information. In principle, both are functions of the molecular structure and the nature of motional averaging. The latter can, in general, be anisotropic and requires specification of both a principal order parameter and an asymmetry parameter. It can also reflect averaging by internal motions that make order different for different rings of an oligosaccharide. Because of the limited amount of data in our study, and our primary objective of illustrating principles of analysis, we will make the simplifying assumption of axially symmetric motion of a single rigid entity for our bicelle-associated disaccharide. Under this assumption, the 13C-13C RDCs measured between the 13C-labeled carbonyl and methyl carbons may be related to structural and order variables as follows (20):
 |
(2) |
Here γi and γj are the gyromagnetic ratios of the two interacting nuclei; rij is the distance between the two bonded nuclei; SDD is an order parameter describing the degree of motional order; and θ is the angle between a given internuclear vector ij and the axial averaging axis. The order parameter, SDD, and the angles θij are the only unknowns.
Similar arguments pertain to CSA offsets of the N-acetyl group (20). The deviation of the observed chemical shift of a nucleus from its isotropic value, δCSA, in a molecule undergoing axially symmetric motion is given by
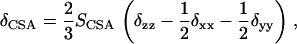 |
(3) |
where SCSA is an order parameter analogous to that described above, and δzz, δxx, and δyy are the diagonal elements of the chemical shift tensor for the carbonyl carbon of an N-acetyl group written in a frame with the z axis coincident with the axial averaging axis. For any orientation of the N-acetyl group, δzz, δxx, and δyy can be calculated by transforming the chemical shift tensor from its principal frame for a given acetyl group to the molecular averaging frame. Principal values for the shift tensor can be taken from model compounds in the literature. In our case these were taken from the experimentally determined 13C carbonyl chemical shift tensor for glycinylglycine hydrogen chloride monohydrate (31). The principal values for δ′xx, δ′yy, and δ′zz are 244, 171, and 95 ppm, respectively. Two of the principal axes, corresponding to the principal values δ′xx and δ′yy, lie in the peptide plane with that for δ′yy aligned approximately along the C=O bond. The orientation of the principal axis corresponding to δ′zz is nearly perpendicular to the peptide plane. Once transformed to the molecular averaging frame, using molecular geometry information, a value for SCSA can be calculated from measured CSA offsets.
For our molecule we assume that each sugar residue is rigid and that the alkyl chain at the reducing end anchors the disaccharide to the lipid bilayer with the alkyl chain extending along the bilayer normal (Fig. 2). Moreover, we assume that the orientation of the acetyl group relative to the sugar ring is fixed in a geometry consistent with three bond scalar couplings between the amide proton and the sugar ring 2-proton. Thus, the orientation of residue I and its attached acetyl group are determined by the terminal torsion angles ϕ and ψ, defined as O5(I)-C1(I)-O1(I)-C1′ and C1(I)-O1(I)-C1′-C2′, respectively. Here C1′ and C2′ refer to the two carbons in the butyl chain and the numbers in parentheses refer to residue I. The orientation of residue II and its attached acetyl group are determined by the combination of the terminal torsion angles and the glycosidic angles ϕ′ and ψ′, which are defined as O5(II)-C1(II)-O1(II)-C4(I) and C1(II)-O1(II)-C4(I)-C3(I), respectively.
If particular geometries for the sugars in our disaccharide (sets of φ and ψ angles) are chosen, the only unknowns left in Eqs. 2 and 3 are the order parameters SDD and SCSA. So, once the dipolar coupling and the CSA offsets are measured, for a given N-acetyl group we can calculate the order parameters SDD and SCSA for each sugar residue in a particular geometry. It is important to note that for any proper orientation of an approximately rigid disaccharide, the numeric values of the two order parameters must be identical. We can define a structural deviation factor which measures the variation in these parameters, as given below and use this factor as an indicator of acceptable geometries (f close to 1):
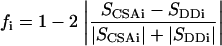 |
(4) |
Conformational analysis
To generate structures and calculate structural deviation factors, we need a molecular structure for our (GlcNAc)2-OBu disaccharide. The initial structure was obtained from the online carbohydrate builder on the CCRC website (http://glycam.ccrc.uga.edu/AMBER/index.html) and this structure was energy minimized using AMBER (27). The H-N-C2-H torsion angle connecting the N-acetyl group to the glucose backbone, 142°, is consistent with the crystal structural torsion angle of 140°. However the glycosidic torsion angles −60/60 deviate significantly from x-ray crystal data (32). Here we initially chose a structure consistent with the x-ray data (−80/110) for the intersaccharide glycosidic torsion angles and generated structures by incrementing the alkylation torsion angles, φ and ψ, in 10° increments from 0° to 360°. Fig. 5 a shows the contour level diagram of the structural deviation factor, f, for residue I as a function of the alkylation torsion angle. From this figure we see that there are four possible torsion angle sets that are consistent with RDC and CSA offset data for the first sugar ring. One of these (80/20) proves unreasonable based on energy calculations (see Fig. 6) and another (−100/320) is moderately high in energy and would direct the saccharide portion into the hydrophobic portion of the bicelle. After excluding these, two possible torsion angle regions are left. They are around −110/160 and 70/190. Both fall in low energy regions of an energy calculation diagram (23) (Fig. 6).
FIGURE 5.

Contour diagrams for the structural deviation factors, f, as the function of the torsion angles. (a) Shows f for residue I as the function of sugar-chain torsion angles. (b) Shows the superimposed diagram of f2 using data from residue II (solid) as a function of sugar-chain torsion angles superimposed on f1 using data from residue I (shaded). (c) Shows the f for residue II as the function of the glycosidic torsion angles with the terminal ones fixed at −100/160. (d) Shows the superimposed diagram of f from residue I and II after the optimization of the gycosidic torsion angles.
FIGURE 6.

Experimental solutions of the molecular conformations (dots) superimposed on a potential energy map as the function of terminal torsion angles generated by AMBER (21).
We also calculated the structural deviation factor from residue II, f2, using the intersaccharide glycosidic torsion angles as found in the crystal structure and varying the sugar-chain torsion angles. The contour diagram for f2 is shown in black overlaid on f1, shaded (Fig. 5 b). The agreement is not ideal. Although this may reflect imperfections in the model, such as the assumption of axially symmetric motion or rigidity of the disaccharide unit, we decided to first explore the possibility of slight alterations in the intersaccharide glycosidic torsion angle. To do this, we fixed the terminal torsion angles at one of the two possibilities (−110/160) and calculated f values for ring II as a function of the glycosidic torsion angles ϕ′ and ψ′ (Fig. 5 c). Again there are four possible torsion angle sets based on the calculation. None of these agree with the crystal structure set. Three of the four torsion angle sets lead to energetically unreasonable glycosidic conformations. But the fourth represents a torsion angle set (−70/70) that is very close to the −60/60 set of the original energy-minimized structure. The same process for optimization of the glycosidic torsion angles was performed with the terminal torsion angles fixed at 70/190. This also gave an allowed set of intersaccharide glycosidic torsion angles near −60/60. Given the two possible terminal torsion angle sets and one possible intersaccharide glycosidic torsion angle set, two possible conformations of the (GlcNAc)2-OBu disaccharide, as anchored to a bilayer analog, exist. These conformations are shown in Fig. 7.
FIGURE 7.

Two possible conformations of (GlcNAc)2-OBu.
The order parameter values calculated for each residue, for each of the above geometries, are given in Table 1. For conformation II, the order parameters are nearly identical for both rings. This is consistent with modeling based on a nearly rigid intersaccharide glycosidic bond. For conformation I, the order parameters are slightly different for each ring and in fact slightly larger for the more remote ring. To the extent that we would expect the more remote ring to be less ordered, we would exclude conformation I. The differences are however minor and may well be within experimental/computational errors, given our assumptions of axial symmetry.
TABLE 1.
Order parameters for two possible conformations with the intersaccharide glycosidic torsion angles optimized at −70/70
| Order parameters | Conformation I(ϕ = −100, ψ = 160) | Conformation II(ϕ = 70, ψ = 190) |
|---|---|---|
| SDD(I) | 0.0041 ± 0.0002 | 0.0063 ± 0.0010 |
| SCSA(I) | 0.0040 ± 0.0002 | 0.0061 ± 0.0010 |
| SDD(II) | 0.0055 ± 0.0002 | 0.0061 ± 0.0001 |
| SCSA(II) | 0.0056 ± 0.0002 | 0.0060 ± 0.0004 |
CONCLUSION
Hence, we are able, with a small amount of NMR orientational information, to select reasonable conformational representations of an alkylated disaccharide bound to a lipid bilayer-like surface. This is in itself not surprising; there have been several other similar accomplishments reported in the literature (14,16,17,33). However, in the case presented here, the information comes from just two 13C2-N-acetyl groups that replaced native N-acetyl groups in an alkylated oligosaccharide. We have also provided a novel way of assigning resonances from these groups based on measuring differential isotope incorporation by mass spectrometry. The application is to a relatively small molecule with just two acetyl groups. However, the methodology should be easily extendable to systems with three to four acetyls. The limitation is likely to be in resolution of resonances from chemically similar acetyl groups rather than accuracy of isotope ratio measurements, but even here, variation in CSA offsets due to orientational differences will improve resolution.
The introduction and assignment of isotopically labeled acetyl groups is important because of the widespread distribution of N-acetyl groups in naturally occurring oligosaccharides and the difficulties normally encountered in synthesizing analogs of these oligosaccharides in isotopically labeled forms. Developing methodology applicable to acetyl replacement in gangliosides, N-glycosides, and glycosaminoglycans may provide routes to structural characterization of these molecules as they exist in disease-relevant complexes with proteins that recognize these molecules.
Acknowledgments
The authors thank Xinghai Ning for valuable suggestions on organic syntheses.
This work was supported by a grant from the National Institute of General Medical Sciences, GM33225, and used computational facilities supported by a grant from the National Centers for Research Resources, RR05351.
References
- 1.Haltiwanger, R. S., and J. B. Lowe. 2004. Role of glycosylation in development. Annu. Rev. Biochem. 73:491–537. [DOI] [PubMed] [Google Scholar]
- 2.Helenius, A., and M. Aebi. 2004. Roles of n-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73:1019–1049. [DOI] [PubMed] [Google Scholar]
- 3.Dube, D. H., and C. R. Bertozzi. 2005. Glycans in cancer and inflammation. Potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 4:477–488. [DOI] [PubMed] [Google Scholar]
- 4.Rudd, P. M., M. R. Wormald, and R. A. Dwek. 2004. Sugar-mediated ligand-receptor interactions in the immune system. Trends Biotechnol. 22:524–530. [DOI] [PubMed] [Google Scholar]
- 5.Wormald, M. R., A. J. Petrescu, Y. L. Pao, A. Glithero, T. Elliott, and R. A. Dwek. 2002. Conformational studies of oligosaccharides and glycopeptides: complementarity of NMR, x-ray crystallography, and molecular modelling. Chem. Rev. 102:371–386. [DOI] [PubMed] [Google Scholar]
- 6.Duus, J. O., C. H. Gotfredsen, and K. Bock. 2000. Carbohydrate structural determination by NMR spectroscopy: modern methods and limitations. Chem. Rev. 100:4589–4614. [DOI] [PubMed] [Google Scholar]
- 7.Martin-Pastor, M., and C. A. Bush. 2001. Refined structure of a flexible heptasaccharide using h-1-c-13 and h-1-h-1 NMR residual dipolar couplings in concert with NOE and long range scalar coupling constants. J. Biomol. NMR. 19:125–139. [DOI] [PubMed] [Google Scholar]
- 8.Colebrooke, S. A., C. D. Blundell, P. L. DeAngelis, I. D. Campbell, and A. Almond. 2005. Exploiting the carboxylate chemical shift to resolve degenerate resonances in spectra of c-13-labelled glycosaminoglycans. Magn. Reson. Chem. 43:805–815. [DOI] [PubMed] [Google Scholar]
- 9.Aboitiz, N., M. Vila-Perello, P. Groves, J. L. Asensio, D. Andreu, F. J. Canada, and J. Jimenez-Barbero. 2004. NMR and modeling studies of protein-carbohydrate interactions: synthesis, three-dimensional structure, and recognition properties of a minimum hevein domain with binding affinity for chitooligosaccharides. Chembiochem. 5:1245–1255. [DOI] [PubMed] [Google Scholar]
- 10.Chavez, M. I., C. Andreu, P. Vidal, N. Aboitiz, F. Freire, P. Groves, J. L. Asensio, G. Asensio, M. Muraki, F. J. Canada, and J. Jimenez-Barbero. 2005. On the importance of carbohydrate-aromatic interactions for the molecular recognition of oligosaccharides by proteins: NMR studies of the structure and binding affinity of acamp2-like peptides with non-natural naphthyl and fluoroaromatic residues. Chem-Eur. J. 11:7060–7074. [DOI] [PubMed] [Google Scholar]
- 11.Raman, R., V. Sasisekharan, and R. Sasisekharan. 2005. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 12:267–277. [DOI] [PubMed] [Google Scholar]
- 12.Linhardt, R. J., and T. Toida. 2004. Role of glycosaminoglycans in cellular communication. Acc. Chem. Res. 37:431–438. [DOI] [PubMed] [Google Scholar]
- 13.Yi, X. B., A. Venot, J. Glushka, and J. H. Prestegard. 2004. Glycosidic torsional motions in a bicelle-associated disaccharide from residual dipolar couplings. J. Am. Chem. Soc. 126:13636–13638. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Pastor, M., A. Canales, F. Corzana, J. L. Asensio, and J. Jimenez-Barbero. 2005. Limited flexibility of lactose detected from residual dipolar couplings using molecular dynamics simulations and steric alignment methods. J. Am. Chem. Soc. 127:3589–3595. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Pastor, M., A. Canales-Mayordomo, and J. Jimenez-Barbero. 2003. NMR experiments for the measurement of proton-proton and carbon-carbon residual dipolar couplings in uniformly labelled oligosaccharides. J. Biomol. NMR. 26:345–353. [DOI] [PubMed] [Google Scholar]
- 16.Tian, F., H. M. Al-Hashimi, J. L. Craighead, and J. H. Prestegard. 2001. Conformational analysis of a flexible oligosaccharide using residual dipolar couplings. J. Am. Chem. Soc. 123:485–492. [DOI] [PubMed] [Google Scholar]
- 17.Lycknert, K., A. Maliniak, and G. Widmalm. 2001. Analysis of oligosaccharide conformation by NMR spectroscopy utilizing h-1,h-1 and h-1,c-13 residual dipolar couplings in a dilute liquid crystalline phase. J. Phys. Chem. A. 105:5119–5122. [Google Scholar]
- 18.Lipsitz, R. S., and N. Tjandra. 2001. Carbonyl CSA restraints from solution NMR for protein structure refinement. J. Am. Chem. Soc. 123:11065–11066. [DOI] [PubMed] [Google Scholar]
- 19.Lipsitz, R. S., and N. Tjandra. 2004. Residual dipolar couplings in NMR structure analysis. Annu. Rev. Biophys. Biomol. Struct. 33:387–413. [DOI] [PubMed] [Google Scholar]
- 20.Prestegard, J. H., C. M. Bougault, and A. I. Kishore. 2004. Residual dipolar couplings in structure determination of biomolecules. Chem. Rev. 104:3519–3540. [DOI] [PubMed] [Google Scholar]
- 21.Bendiak, B., T. T. Fang, and D. N. M. Jones. 2002. An effective strategy for structural elucidation of oligosaccharides through NMR spectroscopy combined with peracetylation using doubly c-13-labeled acetyl groups. Can. J. Chem.. 80:1032–1050. [Google Scholar]
- 22.Zaia, J. 2004. Mass spectrometry of oligosaccharides. Mass Spectrom. Rev. 23:161–227. [DOI] [PubMed] [Google Scholar]
- 23.Hare, B. J., F. Rise, Y. Aubin, and J. H. Prestegard. 1994. C-13 NMR-studies of wheat-germ-agglutinin interactions with n-acetylglucosamine at a magnetically oriented bilayer surface. Biochemistry. 33:10137–10148. [DOI] [PubMed] [Google Scholar]
- 24.Horton, D., and W. Weckerle. 1976. Synthesis of 3-amino-2,3,6-trideoxy-d-ribo-hexose hydrochloride. Carbohydr. Res. 46:227–235. [DOI] [PubMed] [Google Scholar]
- 25.Ruckert, M., and G. Otting. 2000. Alignment of biological macromolecules in novel nonionic liquid crystalline media for NMR experiments. J. Am. Chem. Soc. 122:7793–7797. [Google Scholar]
- 26.Woods, R. J. 2006. Glycam_04. http://www.glycam.ccrc.uga.edu.
- 27.Case, D. A., T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang, and R. J. Woods. 2005. The amber biomolecular simulation programs. J. Comput. Chem. 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen, E. F., T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, and T. E. Ferrin. 2004. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 29.Kafetzopoulos, D., A. Martinou, and V. Bouriotis. 1993. Bioconversion of chitin to chitosan—purification and characterization of chitin deacetylase from mucor-rouxii. Proc. Natl. Acad. Sci. USA. 90:2564–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Methacanon, P., M. Prasitsilp, T. Pothsree, and J. Pattaraarchachai. 2003. Heterogeneous n-deacetylation of squid chitin in alkaline solution. Carbohyd. Polym. 52:119–123. [Google Scholar]
- 31.Oas, T. G., C. J. Hartzell, F. W. Dahlquist, and G. P. Drobny. 1987. The amide n-15 chemical-shift tensors of 4 peptides determined from c-13 dipole-coupled chemical-shift powder patterns. J. Am. Chem. Soc. 109:5962–5966. [Google Scholar]
- 32.Muraki, M., K. Harata, N. Sugita, and K. Sato. 2000. Protein-carbohydrate interactions in human lysozyme probed by combining site-directed mutagenesis and affinity labeling. Biochemistry. 39:292–299. [DOI] [PubMed] [Google Scholar]
- 33.Martin-Pastor, M., and C. A. Bush. 2000. The use of NMR residual dipolar couplings in aqueous dilute liquid crystalline medium for conformational studies of complex oligosaccharides. Carbohydr. Res. 323:147–155. [DOI] [PubMed] [Google Scholar]