

Abstract
Crystallohydrodynamics describes the domain orientation in solution of antibodies and other multidomain protein assemblies where the crystal structures may be known for the domains but not the intact structure. The approach removes the necessity for an ad hoc assumed value for protein hydration. Previous studies have involved only the sedimentation coefficient leading to considerable degeneracy or multiplicity of possible models for the conformation of a given protein assembly, all agreeing with the experimental data. This degeneracy can be considerably reduced by using additional solution parameters. Conformation charts are generated for the three universal (i.e., size-independent) shape parameters P (obtained from the sedimentation coefficient or translational diffusion coefficient), ν (from the intrinsic viscosity), and G (from the radius of gyration), and calculated for a wide range of plausible orientations of the domains (represented as bead-shell ellipsoidal models derived from their crystal structures) and after allowance for any linker or hinge regions. Matches are then sought with the set of functions P, ν, and G calculated from experimental data (allowing for experimental error). The number of solutions can be further reduced by the employment of the Dmax parameter (maximum particle dimension) from x-ray scattering data. Using this approach we are able to reduce the degeneracy of possible solution models for IgG3 to a possible representative structure in which the Fab domains are directed away from the plane of the Fc domain, a structure in accord with the recognition that IgG3 is the most efficient complement activator among human IgG subclasses.
INTRODUCTION
Assessing the conformation of flexibly linked, multidomain proteins has been an ongoing area of investigation in biochemistry, implying that conformation and domain orientation can be important for biochemical activity. The term “domain” can refer to a single independently folded protein subunit or entities. For example, the Fab and Fc parts of an antibody may be considered as domains themselves, as they consist of arrangements of a distinct and recognizable motif in immunological molecules, the “immunoglobulin fold”. High-resolution structural analysis of multidomain complexes is problematic in that multidomain proteins are high-molecular-weight molecules that preclude detailed investigations by NMR, and by crystallographic studies where the domains/subunits with even a modest degree of flexibility relative to each other can be refractory in producing suitable crystals. Further, even if crystals can be produced, the complex may retain sufficient conformational freedom even within the crystal lattice to prevent interpretation of the electron density map. Recently, cryoelectron tomography has been shown to have potential for providing an insight into the dynamics of individual antibody by three-dimensional reconstruction of individual objects from a tilt series of electron microscope images of a sample quenched to the temperature of liquid nitrogen (1,2). However, this will require a large number of snapshots of individual antibodies to be investigated to reveal the full population of possible structures in solution. Consequently, information on domain orientation in flexibly linked multidomain proteins is often obtained by combining macroscopic solution studies with various modeling strategies to identify possible conformations.
Hydrodynamic methods can in principle give conformational information in terms of orientation of domains in solution, particularly if the structure or overall shape of the domains is already known, from either x-ray crystallography or high-resolution nuclear magnetic resonance spectroscopy. The term “crystallohydrodynamics” was coined to describe the combination of this structural information from individual domains with hydrodynamic data for the domains and for the intact multidomain structure to estimate the orientation of the domains relative to each other in dilute solution (3–5), and without the requirement for an assumed ad hoc value to represent the time-averaged effects of water association to the protein referred to as “hydration” that have had to be adopted in other approaches (6–9). Hydration effects are dynamic processes (10) that cannot be ignored and that alter the effective volume and hence hydrodynamic properties of the protein. Indeed, as was repeatedly shown long ago (11–13), hydrodynamic parameters are often more sensitive to hydration than to shape.
Complications still arise, however, from uniqueness or degeneracy (the existence of more than one model for domain orientation agreeing with experimental parameters) and flexibility (predicted orientations are of necessity time-averaged). The earlier articles in this series (3–5) dealt with the complications of hydration and flexibility but focused on one particular type of hydrodynamic measurement, namely, the sedimentation coefficient from sedimentation velocity analysis in the analytical ultracentrifuge. Here we try to tackle the degeneracy problem by incorporating additional types of solution measurement. We also move from the former ad hoc approach for the generation of possible models for consideration to a much more systematic one by taking advantage of the recent Monte Carlo-type algorithm MONTESUB (14).
THEORY
An early attempt—with some success—on a multidomain structure was the first demonstration, using hydrodynamic bead modeling and the algorithm TRV (15) that the immunoglobulin IgE was cusp-shaped (16). The classical hydration problem (namely that hydrodynamic parameters like the frictional ratio depend on the time-averaged hydration as well as the conformation) was dealt with by comparison of the hydrodynamic properties of the IgE molecule with those for the hingeless mutant IgG Mcg molecule, whose crystal structure was known.
An improved method of dealing with the hydration problem for the modeling of IgG subclasses was given in the so-called crystallohydrodynamics approach by Carrasco et al. (3), which took into account known crystal structures for the Fab and Fc domains. The approach employed the latest bead-shell approach (HYDRO/SOLPRO) for modeling the domains as surface ellipsoids and calculating the appropriate hydrodynamic properties such as the sedimentation coefficient 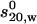 , and its corresponding universal (i.e., size-independent) shape parameter P {P = 1 for a sphere, regardless of size, and its value (>1) can be computed for any arbitrary shape or a crystal structure}. The procedure was as follows:
, and its corresponding universal (i.e., size-independent) shape parameter P {P = 1 for a sphere, regardless of size, and its value (>1) can be computed for any arbitrary shape or a crystal structure}. The procedure was as follows:
- P is calculated from the shape of each of the Fab and Fc domains from their known crystal structures. These values, when combined with their measured sedimentation coefficients allowed an estimate of the apparent time-averaged hydration δapp for the domains (and hence, from a weighted average, for the intact antibody) to be made. The relevant relations are as follows:
and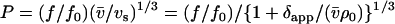
(1)
(f/fo) is the translational frictional ratio or ratio of the frictional coefficient of the macromolecule to the theoretical frictional coefficient of a spherical macromolecule of identical mass and anhydrous volume,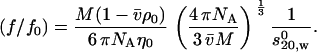
(2) 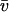 is the partial specific volume of the protein (ml/g), vs is its swollen specific volume in solution (ml/g), δapp is the “time-averaged apparent hydration”,
is the partial specific volume of the protein (ml/g), vs is its swollen specific volume in solution (ml/g), δapp is the “time-averaged apparent hydration”, 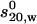 the sedimentation coefficient (s) corrected to standard solvent conditions (density ρo = 0.99823 g/ml and viscosity ηo = 0.010 Poise of water at 20°C) and extrapolated to infinite dilution, M is the molecular weight (g/mol), NA is Avogadro's number (6.02205 × 1023mol−1). In the case of human IgG antibodies, this procedure yielded values for δapp of ∼0.53 for IgG Fab′ and ∼0.70 for IgG Fc and hence a weighted average of ∼0.59 for an intact IgG antibody (3). It is worth reiterating what we mean by δapp. It is referred to as “time-averaged” in the sense that so-called “hydration” is a dynamic rather than a static process (17). Nonetheless it does influence the hydrodynamic properties of macromolecules and cannot, as has erroneously been suggested by some, be ignored. It is referred to as an “apparent” hydration because besides volume/water association, its estimation by Eqs.1 and 2 is affected by the fact that 1), the domains are not true ellipsoid structures; 2), the domains have considerable surface rugosity; and 3), small imperfections in the bead model approximation exist: the hydrodynamic parameters for a bead and bead-shell model cannot be calculated exactly, as they can for ellipsoids, but only to a very good approximation.
the sedimentation coefficient (s) corrected to standard solvent conditions (density ρo = 0.99823 g/ml and viscosity ηo = 0.010 Poise of water at 20°C) and extrapolated to infinite dilution, M is the molecular weight (g/mol), NA is Avogadro's number (6.02205 × 1023mol−1). In the case of human IgG antibodies, this procedure yielded values for δapp of ∼0.53 for IgG Fab′ and ∼0.70 for IgG Fc and hence a weighted average of ∼0.59 for an intact IgG antibody (3). It is worth reiterating what we mean by δapp. It is referred to as “time-averaged” in the sense that so-called “hydration” is a dynamic rather than a static process (17). Nonetheless it does influence the hydrodynamic properties of macromolecules and cannot, as has erroneously been suggested by some, be ignored. It is referred to as an “apparent” hydration because besides volume/water association, its estimation by Eqs.1 and 2 is affected by the fact that 1), the domains are not true ellipsoid structures; 2), the domains have considerable surface rugosity; and 3), small imperfections in the bead model approximation exist: the hydrodynamic parameters for a bead and bead-shell model cannot be calculated exactly, as they can for ellipsoids, but only to a very good approximation. Using this value of δapp combined with the experimentally measured value of s20,w for the intact antibody structure yields an experimental value for P for a particular intact IgG antibody molecule. These were evaluated for the set of human IgG subclasses IgG1, IgG2, IgG3, and IgG4 and a hingeless mutant IgG and presented by Carrasco et al. (3).
Bead models were then constructed in an ad hoc fashion for the intact antibody molecules by arranging the two bead shell Fabs and Fc in three-dimensional space, allowing for a significant hinge in IgG1 and IgG3. A variety of possible orientations were explored and the corresponding P values evaluated using the HYDRO/SOLPRO program.
Matches were then sought between the P values of the models and the experimental P values evaluated in step 2 above. Because of the large degeneracy (large number of models giving values of P agreeing with the experimental P values), only limited conclusions could be made in the Carrasco et al. (3) study, namely that “open conformations” seemed favored over “compact conformations”, although good agreement was achieved for the predicted solution conformation of the one IgG antibody studied whose crystal structure was known: the hingeless mutant IgG Mcg.
Modification of Longman and colleagues
An improvement to the crystallohydrodynamics approach was made by Longman et al. (4) to take into account that the (time-averaged) apparent hydration not only increased the volume of an antibody domain but also altered the hydrodynamic shape. This modification resulted in changes in domain dimensions to better reflect the effect of apparent hydration. Two IgG4 point mutants were studied using this modification: one with the hinge region reinforced by the point mutation of a serine to proline at position 241 in the hinge, the other with the cysteines removed to prevent disulfide bridge formation. Models found for these two antibodies occupied overlapping regions of conformational space with considerable degeneracy persisting, as a number of different models were capable of reproducing P-function values consistent with the experimental value obtained from the sedimentation coefficient (within experimental error).
We now attempt to tackle the degeneracy problem in domain orientation analysis of antibodies by using additional hydrodynamic parameters, and we also move away from the former ad hoc approach for creating plausible models by a more systematic approach of creating plausible models covering a representative range of possible domain orientations and hinge lengths, taking advantage of the new algorithms HYDROSUB (18) and MONTESUB (14), and we will illustrate the effectiveness of this new approach by application to human IgG3.
Incorporation of three universal shape functions
Besides the sedimentation coefficient and its corresponding universal shape function P, the additional solution properties we wish to use are the intrinsic viscosity [η] and its corresponding universal shape function ν (the so-called viscosity increment or Simha-Saito shape function) and the root mean-square radius of gyration Rg (or 〈s2〉1/2) and its corresponding universal shape function G (the so-called reduced radius of gyration).
The viscosity increment is evaluated from the intrinsic viscosity (ml/g) from:
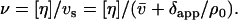 |
(3) |
The traditional method of evaluating [η] for proteins is by capillary viscometry with automatic timing facility: such a procedure has the problem of requiring high concentrations (>5 mg/ml) to give a significant flow-time difference between solution and pure solvent. The appearance of a relatively new type of viscometer based on the measurement of pressure difference between solution and solvent flow now renders it possible to measure at slightly lower concentrations (see, e.g., Harding (19)).
The reduced radius of gyration function G (20) is related to the radius of gyration Rg (cm) by:
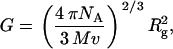 |
(4) |
where v is the scattering volume (mL/g). Two important issues reside with regard to the use of the G function. The first is the meaning of v and whether it includes associated water or not, i.e., is v ∼ 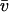 or v ∼ vs or perhaps something in between. This issue was considered by Carrasco and co-workers in relation to the modeling of seed globulins (21). For neutron scattering v ∼
or v ∼ vs or perhaps something in between. This issue was considered by Carrasco and co-workers in relation to the modeling of seed globulins (21). For neutron scattering v ∼  appears the best approximation; for solution x-ray scattering, v ∼ vs is probably better, based on the premise that associated solvent has a different density than bulk solvent (22). The associated solvent is unlikely to possess the same scattering density as bulk solvent, and we treat it as an integral part of the scattering particle, although the actual scattering density of this solvent is unlikely to be exactly the same as the macromolecule. The second issue with G is that it is derived from the whole macromolecule and not just the surface: in the calculation of G a bead-shell surface model has to be filled with interatomic scattering elements (23).
appears the best approximation; for solution x-ray scattering, v ∼ vs is probably better, based on the premise that associated solvent has a different density than bulk solvent (22). The associated solvent is unlikely to possess the same scattering density as bulk solvent, and we treat it as an integral part of the scattering particle, although the actual scattering density of this solvent is unlikely to be exactly the same as the macromolecule. The second issue with G is that it is derived from the whole macromolecule and not just the surface: in the calculation of G a bead-shell surface model has to be filled with interatomic scattering elements (23).
Use of the maximum dimension Dmax as an additional conformation filter
Besides Rg, another very useful parameter that can be obtained from solution x-ray scattering is an estimate for the maximum dimension Dmax of the scattering particle. The potential usefulness of combining Rg and Dmax together for describing the conformation of antibodies was clearly demonstrated by Svergun and collaborators (24), and Perkins and co-workers have shown how good use can be made of the full angular intensity profile (e.g., 6–9). The Dmax parameter is obtained by transformation of the scattering profile into the distance distribution function p(r) of intraparticle scattering vectors, which is obtained from the scattering profile using the algorithm GNOM (25). The distance distribution function p(r) of a macromolecule is directly related to the angular dependence of the scattering density, reflecting the shape and the mass distribution of the molecule. The longest “interatomic” scattering vector, Dmax, is taken as the distance at which the distance distribution function becomes equal to zero at a nonzero distance. For a bead model composed of rigid arrays of spheres, the distance distribution function, the distribution of interatomic scattering vectors, can be calculated as can also be done for a bead-shell model after it has been “filled” with interatomic scattering elements (23). Both the distance distribution function and Dmax will change as the spatial arrangement and relative orientation of the antibody domains are altered. The Dmax of a model is easily extracted from the p(r) profiles, provided there is no significant contribution from aggregates: this needs to be established by a sedimentation velocity experiment.
Enhanced crystallohydrodynamic approach
A summary of the new enhanced crystallohydrodynamic approach is shown in Fig. 1. The first part of the approach is the same as in our previous work, namely, the estimation of δapp from use of the known shape of the Fab and Fc fragments from crystallography combined with sedimentation coefficient data for the fragments. Then
The experimental shape functions are measured for the antibody whose domain orientation is being sought.
Bead models are constructed for plausible models using the MONTESUB program, followed by calculation of the solution properties and scattering properties of these models via HYDROSUB and SOLPRO algorithms.
The experimental shape functions are compared with those of the candidate models, rejecting unlikely conformations and selecting those models which give matches for all of P, ν, and G, allowing for reasonable experimental error.
If there is still degeneracy, i.e., more than one model that gives a match for all of P, ν, and G, use the experimental Dmax and compare this with the Dmax values calculated for the models using GNOM.
FIGURE 1.

Scheme for the enhanced crystallohydrodynamic approach for characterization of multidomain structure in solution to address the degeneracy problem. This approach includes four different experimental parameters (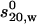 , Rg, [η], and Dmax) as opposed to just one (
, Rg, [η], and Dmax) as opposed to just one (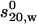 ) used earlier. The routine MONTESUB is now used to generate the range of models for consideration.
) used earlier. The routine MONTESUB is now used to generate the range of models for consideration.
If there is still degeneracy, we can suggest one last ancillary procedure: compare the features of the experimental distance distribution function with the selected models: there may exist special features facilitating further matching.
Hinge inclusion
Two approaches to hinge inclusion have been adopted, the introduction of an inflexible cylindrical bead-shell body between the Fc and the Fabs (3), and inclusion of “virtual frictionless” hinges which, having no hydrodynamic properties, effectively just maintaining the spatial separation of the domains in a preset, relative orientation (4). It has been shown that for calculating the macroscopic solution properties of flexibly linked biopolymer complexes, retaining structural detail is not as important as capturing the overall size and shape of the domains in the model (14). In the investigation reported here, the domains were linked together with a “semiflexible” linker. Although also “frictionless”, the semiflexible linker we used in this approach does allow changes in relative domain orientation.
Modeling domain orientation: HYDROSUB and MONTESUB
For a typical globular protein, fine structural details (crevices, pockets, protrusions, etc.) can make a relatively large contribution to the hydrodynamics. However, for multisubunit structures (antibodies are a paradigmatic example), it is the arrangement of the subunits or domains that dominates the hydrodynamic properties, whether or not there is a hinge, or whether the conformation is more open or closed. Given the additional complication of hydration, it is quite reasonable to reduce the complexity of the problem by making structural approximations for the subunits, thus allowing the analysis to concentrate on their spatial arrangement. This approach also facilitates the modeling of the flexibility between domains (14).
Arrangement of domains for IgG antibodies
The two Fab domains are represented as (bead-shell) prolate ellipsoids and the Fc domain is represented by an oblate ellipsoid, whose shapes and hydrated axial ratios and dimensions have been obtained as outlined above. The values estimated by Longman et al. (4) are given in Table 1.
TABLE 1.
Dimensions of the ellipsoids for representing the Fab and Fc domains
| Longest semiaxis (Å) | Shortest semiaxis (Å) | |
|---|---|---|
| Fab | 39.30 | 24.35 |
| Fc | 36.60 | 21.63 |
The hinge region, if present, is represented by three linear chains of minibeads formed into a “Y” shape. In this representation of the hinge, the Fc is linked to the “body” of a Y, and each Fab is connected with the “arm” of the Y. One bead is placed at the origin of a three-dimensional coordinate system as the connecting point of the three chains in the hinge; the Cartesian coordinates of this bead are thus (0,0,0). One long axis of the Fc domain and the chain of beads connected to the Fc are fixed on the z axis (Fig. 2 a). The orientations of the symmetrical axes of the two Fab ellipsoids connect with the line connecting the hinge minibeads to the Fab.
FIGURE 2.

Scheme for the construction of an intact antibody model. (a) A model with a 4-4-8 hinge arrangement. (b) The position of the center of the symmetrical axis of the ellipsoid is represented as the Cartesian coordinates (xc,yc,zc) and two sphericalpolar angles (θ and ϕ). θ is the angle subtended by the main particle axis and the z axis, ϕ is the angle subtended by the projection of the main particle axis on the xy plane and the x axis.
Hinge flexibility is introduced by adoption of the Y-shaped linker, as the orientation of the arms and body of the Y can be altered. Hinge flexibility is explored by a Monte Carlo procedure to alter the orientation of the continuous line defining the Fab major axis, which connects the Fab to the chain of minibeads in the arm of the Y and thence to the central hinge bead at (0,0,0). Usually the Fc would be anchored in a fixed position, with one long axis along the z axis and the Fab domains moved relative to that. Changes in Fab domain position are reported as sphericopolar angles (θ and ϕ) (see Fig. 2 b) of the major axis of each Fab.
The Monte Carlo procedure generates uniform random values for the θ and φ angles of each Fab. The ϕ value is uniformly distributed in the (0, 2π) range and θ is defined by constraining cosθ as a random number distributed uniformly in (−1, 1). In this way, the whole spatial arrangement allowed for the two Fab ellipsoids could be observed. The second step of the simulation rejects any conformations resulting in bead overlap. Therefore, the simulation generates angles to cover all possible conformations, from the most opened to the most closed, that the two Fab ellipsoids could adopt with respect to the Fc plane. As in any Monte Carlo simulation, the larger the number of conformations generated the greater the possibility that suitable models exist. Here we suggest use of >100 (i.e., 100+) conformations to explore the available “conformational space”.
In the models being analyzed, antibody conformation is constrained by hinge size and geometry. Changes in antibody conformation are achieved by alteration of hinge bead numbers and/or the sphericopolar angles defining each main Fab axis. Bead coordinates for “test” conformations were generated using the algorithm MONTESUB (14), which rejects any conformations resulting in bead overlap and produces output in a form usable by HYDROSUB for calculation of the solution properties of the models, and enables calculation of the corresponding universal shape functions by SOLPRO (23,26).
Hydrodynamic properties of the hinge
The size and number of the hinge beads in each model are assigned by the user, and for convenience all the beads in the hinge have been set at the same size (radius 1.8 Å) following Garcia de la Torre et al. (27). Bead-shell modeling was developed on the basis that the shape of the macromolecular surface is fundamentally important in determining macromolecular hydrodynamics. Therefore, in adopting the bead-shell approach to assessing antibody conformation it seems appropriate to consider that even for human IgG3 the hinge is a relatively small part of the intact molecule and possibly contributes only marginally to the molecule's hydrodynamic properties. Consequently, the modeling has been undertaken on models with “frictionless hinges”, in which the hinge beads are not included in the determination of the frictional properties of the intact molecule, which are thus determined by the relative orientation of the Fab and Fc domains. However, the hinge beads are included as solid-body elements in the calculation of the geometric properties, radius of gyration, and volume.
The validity of this approach has been examined by comparing the average change in the parameter values calculated for models of identical three-dimensional orientation that included just two Fabs and one Fc (the “notional” hinge model), and models that included two Fabs, one Fc, and a “frictionally active” hinge (the “geometric” hinge model), using the same molecular weight for both types of model. For the experimentally measurable parameters the average difference was found to be 0.51% for sedimentation coefficient, 0.06% for radius of gyration, and 0.48% for intrinsic viscosity. Similar differences were found for the derived universal shape parameters as follows: 0.44% for the Perrin function, 0.28% for the reduced radius of gyration function, and 0.27% for the viscosity increment. If allowance is made for the difference in molecular weight between the “notional” hinge and geometric hinge models, the average difference in the experimentally measurable parameters was 8.08% for the sedimentation coefficient, 0.06% for the radius of gyration, and 8.78% for the intrinsic viscosity, whereas for the corresponding universal shape parameters, because they are size-independent, the average differences are the same as before, namely, 0.44% for the Perrin function, 0.28% for the reduced radius of gyration function, and 0.27% for the viscosity increment. It can be seen that the average differences in the computed values of the universal shape parameters remain the same for both the notional and geometric hinge models, both when hinge mass is ignored and when it is included, and are within the 1–2% uncertainty that is associated with calculation of the hydrodynamic coefficients. The equivalence of the shape functions for frictionless hinge and frictionally active hinge models indicates that adopting frictionless hinge models is reasonable. It also further demonstrates the usefulness of the universal shape functions and validates their use in conformational analysis. The pronounced change in calculated values of sedimentation coefficient and intrinsic viscosity on inclusion of hinge mass is consistent with expectation, given that both properties have a dependence on molecular mass.
The bead model approximation and experimental errors
Comparing different modeling strategies, Carrasco and Garcia de la Torre (28) earlier demonstrated that calculation of the hydrodynamic properties of bead-shell models could reach an accuracy of 1–2% based on comparison with those equivalent structures—smooth ellipsoids—whose hydrodynamic properties can be calculated exactly.
After assessing how different sources of uncertainty contribute to the experimental estimation of translational friction, Errington and Rowe (29) suggested an error of ∼3% for experimentally determined sedimentation coefficients. Consequently, the Perrin function, calculated through the translational friction ratio that is directly related to the sedimentation coefficient (see Eqs.1 and 2), is considered to include an uncertainty of ∼3%. The measurement of intrinsic viscosity of protein solutions is reviewed in Harding (19). Typically, the error associated with the measurement varies according to the type of viscometer employed. Relative viscosities can be measured to ∼2% with the pressure-imbalance type of viscometer (19). Taking into account additional errors in concentration measurement, an error of 5% was assigned for the viscosity increment ν. The experimental determination of the radius of gyration has at least 2% error. The G function depends on 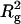 , so an ∼3% error margin is expected.
, so an ∼3% error margin is expected.
The maximum dimension Dmax can be computed from small-angle x-ray scattering curves by a Fourier cosine transformation. The numerical calculations carried out by Muller et al. (30) suggested that the precision with which Dmax can be computed from the experimental scattering curves with noise is comparable to the precision with which other particle parameters, such as the volume and molecular weight, can be determined. Unfortunately, until now there has been no objective work carried out to evaluate the uncertainty in determination of the maximum dimension. Principally, for a globular protein, an uncertainty of 5% is considered in determination of Dmax.
Finally, it has to be understood that the modeling depends critically on an appropriate value being assigned for δapp based on shape and hydrodynamic information on the domains. Wrong assignment of this value will lead to systematic errors throughout.
Despite these sources of error, this does not limit the utility of our proposed crystallohydrodynamic approach in assessing the average spatial orientation of the domains of multidomain proteins. To illustrate its application, we consider the human IgG3 antibody subclass.
Human IgG3
Human IgG3 is formed late during the immune response (31) and is very efficient in inducing complement activation (31,32) and interacting with FcR and to induce opsonophagocytosis (31). Structurally, IgG3 is unique with a hinge four times the length of the other human IgG subclasses (33) and the hinge is coded by four exons with short introns in between (34). Interestingly, the complement activation of IgG3 is enhanced by shortening the hinge to that of IgG1 by deleting three hinge exons (35). The chimeric IgG3 antibody under study is a wild-type antibody molecule with specificity for the hapten NIP.
The hinge region of an IgG molecule can be divided into three discrete structural regions: the upper, middle, and lower hinge regions (36). The structural hinge of native IgG3 is composed of a 12-amino-acid upper hinge stretching from the C-terminal end of CH1 to the first hinge cysteine, a 50-amino-acid middle hinge stretching from the first to the last cysteine in the hinge, and an 8-amino-acid lower hinge stretching from the last cysteine in the hinge to Gly-237 in CH2 (37), whereas the “genetic” IgG3 hinge is encoded by the 62 amino acids in the upper and middle hinge regions (2). In this classification, the middle hinge contains ∼4 times as many amino acids as the upper hinge. The table shows that only the middle hinge contains cysteine residues, which introduces disulphide linkages into the hinge, thus keeping the two amino acid chains together in this region. There are no cysteines in the upper hinge, so it is likely that the amino acid chains will “separate” in the upper-hinge region, allowing the two Fabs to adopt orientations unsymmetrical with each other and the Fc.
TABLE 2.
Hinge sequences of the chimeric human IgG3
| IgG type | C-terminal CH1 | Upper hinge | Middle hinge | Lower hinge |
|---|---|---|---|---|
| IgG3 | VDKRV | ELKTPLGDTTHT | CPRCP(EPKSCDPPPCPRCP)3 | APELLGGP |
The hinge length of IgG3 has been somewhat controversial, and its conformation could well affect the flexibility of the Fab arm of the molecule. Electron micrographs obtained by Pumphrey (38) suggested that the main body of the IgG3 hinge was ∼90 Å by electron microscopy. Gregory et al. (39) found that using “primitive” bead models with hinge lengths of either 90 Å or 75 Å could give good agreement with the experimental data. These hinge distances are consistent with immunoelectron microscopy studies carried out by our collaborators using immunoelectron microscopy, which revealed a value of 80 ± 23 Å for the mean hinge distance of an IgG3 molecule in immune complex (40).
EXPERIMENTAL
All experimental determinations were performed in phosphate-buffered saline (pH 7.4, I = 0.16 M). The production and purification of the chimeric human IgG3 wild-type molecule has been previously reported (41,42). Measurements were undertaken to determine the monodispersity, sedimentation coefficient, intrinsic viscosity, and radius of gyration of the chimeric human IgG3. The sedimentation coefficient of the molecule was obtained from sedimentation velocity studies in a Beckman Optima XLA running at 40,000 rpm after sedimentation by absorption at 280 nm on solutions up to 1 mg/ml concentration. Data were analyzed by the least-squares g*(s) method as implemented in SEDFIT (43), and the sedimentation coefficient was corrected to standard conditions (density and viscosity of water at 20°C) to give 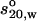 . g*(s) profiles also confirmed the monodispersity and aggregate-free nature of the solutions (Fig. 3), important for the subsequent interpretation of the x-ray scattering and viscosity data.
. g*(s) profiles also confirmed the monodispersity and aggregate-free nature of the solutions (Fig. 3), important for the subsequent interpretation of the x-ray scattering and viscosity data.
FIGURE 3.

Sedimentation velocity, g*(s) analysis, of the chimeric human IgG3 sample using a multi-Gaussian fit of the g*(s) distributions from SEDFIT at a loading concentration of 0.7 mg/ml. The diamonds represent the raw data. g*(s) is the apparent (e.g., not corrected for diffusion or nonideality) distribution of sedimentation coefficients. The peak maximum corresponds to sT,b = 5.92 S at temperature T and in buffer b.
The SAXS data was obtained at Station 2.1 at the Synchrotron Radiation Source (SRS, Daresbury, UK), employing camera lengths of 1.0 m (to cover a Q-range of 0.038 < Q < 0.32 Å−1) and 4.3 m (for 0.008 < Q < 0.18 Å−1). The detector was calibrated with wet rat tail collagen and silver behenate. Data were collected at 6 mg/mL on the 1-m camera in 60-s time frames and at 1.1 mg/mL on the 4.3-m camera in 10-s frames. Using the standard Daresbury software package XOTOKO (44), the data were then normalized to the intensity of the incident beam, radially averaged, and corrected for the detector response. The total scattering intensity from each of the time frames was determined to check for beam-induced aggregation in the sample; those frames showing increasing counts were excluded from further analysis as the increase was considered to be due to radiation-induced aggregation.
After subtracting the buffer contribution, to correct for the interparticle interaction effects in the low-angle region of the high-concentration measurement, the low-angle regions of the low-concentration data were scaled to merge with the high-angle region of the high-concentration data using SigmaPlot (Systat Software, Richmond, CA). The distance distribution function p(r) and the maximum dimension (Dmax) were obtained by using GNOM (45). The radii of gyration (Rg) were determined using the Guinier approximation (46) with data from the low-angle region, and from transformation of the entire scattering profile using GNOM.
The intrinsic viscosity [η] had already been measured previously by “pressure imbalance differential viscometry” using a Viscotek (Basingstoke, U.K.) instrument (47). In this method (see, for example, Harding (19), and Haney (48,49)), the relative viscosity ηr is measured from the pressure difference between solvent and solution flow in capillaries, and then the intrinsic viscosity is estimated from the Solomon-Ciuta equation:
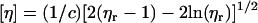 |
(5) |
the concentration c being recorded using an on-line refractometer. This method of measuring intrinsic viscosity, along with Eq. 5, requires only relatively small amounts of material: an injection volume of 100 μl with a loading concentration of 0.5–1.0 mg/ml, minimizing aggregation phenomena (which can be removed by an on-line column).
The partial specific volume of the molecule was calculated using SEDNTERP (50,51), as were the density and viscosity of the buffer.
RESULTS AND DISCUSSION
Following the scheme in Fig. 1, the solution properties of the molecule were determined experimentally, universal shape functions were derived from the experimentally determined quantities, and then >100 bead-shell models of differing domain orientation were constructed for each hinge arrangement (using MONTESUB), covering a representative range of possible orientations. A unique match was then sought. Experimental values for the sedimentation coefficient, 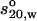 , intrinsic viscosity, [η], radius of gyration, Rg, and maximum particle dimension, Dmax for IgG3 are shown in Table 3, along with the related universal shape functions. Calculation of these functions includes the effect of hydration; as for the reduced radius of gyration G, the approximation v = vs is used. The objective of the modeling was to reproduce the universal shape parameters to determine whether a unique model would emerge.
, intrinsic viscosity, [η], radius of gyration, Rg, and maximum particle dimension, Dmax for IgG3 are shown in Table 3, along with the related universal shape functions. Calculation of these functions includes the effect of hydration; as for the reduced radius of gyration G, the approximation v = vs is used. The objective of the modeling was to reproduce the universal shape parameters to determine whether a unique model would emerge.
TABLE 3.
Experimental hydrodynamic properties and universal shape functions for the chimeric IgG3 wild-type antibody
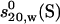 |
P (δ = 0.59) | Rg (Å) | G hydrated | Dmax (Å) | [η]* (mL/g) | ν* |
|---|---|---|---|---|---|---|
| 6.11 ± 0.02 | 1.44 (1.40–1.48) | 71.6 | 2.72 (2.64–2.80) | 195 (185–205) | 9.9 | 7.53 (7.15–7.91) |
Data are adapted from Longman et al. (47). Numbers in parentheses represent ranges, with a 3% error for P values, 3% for G, 5% for Dmax, and 5% for ν, where T,b means at temperature T and buffer b.
Generation of candidate models
Shape parameters for >100 representative bead models covering the entire range of possible domain orientations for a given hinge arrangement of 5-5-20 (described later), were determined, with a representative subset shown in Table 4.
TABLE 4.
Summary of hydrodynamic properties and spatial arrangements for 20 of the models with a 5-5-20 hinge bead arrangement
| Model | P | ν | G | Dmax (Å) | 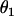 |
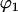 |
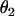 |
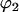 |
Fab-Fab angle |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.40 | 7.78 | 3.08 | 243 | 31.6 | 253.6 | 125.1 | 212.9 | 99.5 |
| 2 | 1.38 | 7.49 | 2.94 | 238 | 39.1 | 154.8 | 124.4 | 135.7 | 86.9 |
| 3 | 1.30 | 5.86 | 2.07 | 195 | 86.9 | 174.3 | 127.8 | 136.0 | 54.1 |
| 4 | 1.42 | 7.64 | 2.79 | 188 | 94.0 | 165.3 | 97.9 | 43.9 | 120.4 |
| 5 | 1.22 | 4.88 | 1.45 | 154 | 126.5 | 94.4 | 141.3 | 195.6 | 68.4 |
| 6 | 1.38 | 7.51 | 2.94 | 238 | 127.9 | 307.0 | 40.1 | 348.3 | 95 |
| 7 | 1.38 | 7.07 | 2.57 | 190 | 92.8 | 215.5 | 96.8 | 135.4 | 79.8 |
| 8 | 1.49 | 9.33 | 3.80 | 225 | 65.6 | 176.3 | 57.4 | 4.6 | 122.4 |
| 9 | 1.36 | 6.53 | 2.25 | 182 | 111.9 | 243.6 | 99.4 | 326.8 | 80.2 |
| 10 | 1.41 | 7.72 | 2.91 | 215 | 127.0 | 303.6 | 69.6 | 133.0 | 161.5 |
| 11 | 1.40 | 7.71 | 2.97 | 225 | 57.4 | 336.9 | 133.0 | 117.3 | 147.3 |
| 12 | 1.37 | 7.31 | 2.87 | 230 | 52.3 | 82.1 | 109.4 | 118.4 | 66.5 |
| 13 | 1.36 | 7.28 | 2.87 | 239 | 137.7 | 91.4 | 38.9 | 86.9 | 98.8 |
| 14 | 1.46 | 8.71 | 3.45 | 233 | 95.8 | 152.5 | 47.3 | 28.7 | 118.4 |
| 15 | 1.36 | 7.00 | 2.59 | 211 | 72.6 | 167.9 | 139.7 | 292.3 | 125.2 |
| 16 | 1.40 | 7.79 | 3.09 | 236 | 43.9 | 68.3 | 105.7 | 17.0 | 77.1 |
| 17 | 1.41 | 8.19 | 3.30 | 249 | 126.0 | 299.4 | 9.6 | 255.6 | 118.9 |
| 18 | 1.41 | 7.40 | 2.70 | 196 | 111.9 | 243.6 | 99.4 | 326.8 | 80.2 |
| 19 | 1.31 | 5.70 | 1.81 | 157 | 123.9 | 280.9 | 121.4 | 26.3 | 84 |
| 20 | 1.45 | 9.00 | 3.78 | 231 | 51.6 | 22.2 | 51.9 | 272.3 | 80 |
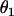 and
and 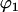 are the sphericopolar angles of Fab1,
are the sphericopolar angles of Fab1, 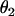 and
and 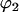 are the sphericopolar angles of Fab2. Values in bold are the modeling parameters that match the experimental parameters in Table3. For clarity, we only show 20 models here, within which we found two models matching all parameters (Models 4 and 18), although we have assessed >100 models in total.
are the sphericopolar angles of Fab2. Values in bold are the modeling parameters that match the experimental parameters in Table3. For clarity, we only show 20 models here, within which we found two models matching all parameters (Models 4 and 18), although we have assessed >100 models in total.
The domains and hinge coordinates are configured by appropriate entry of initial input parameters into MONTESUB. The “join” bead in the hinge was positioned at (0,0,0) in the reference Cartesian coordinate system used, and a long axis (or minor axis) of the oblate ellipsoid representing the Fc was positioned along the z axis, below the xy plane. Each prolate ellipsoid representing a Fab was positioned above the xy plane and defined by the Cartesian coordinates x,y,z of the center of the main axis of the ellipsoid. The orientation of the major axis of a Fab is defined by two angles: θ is the angle between the major axis of the Fab and the z axis and ϕ is the angle between the projection of the major Fab axis in the xy plane and the x axis (Fig. 2 b).
To incorporate the unique features of the structural hinge of IgG3 into the modeling strategy, the combined middle hinge and upper hinge was modeled as a Y shape, such that the middle hinge, the body of the Y shape, was four times longer than each branch of the upper hinge, the arms of the Y. The middle hinge was represented as a single linear chain of beads (allowing for disulphide linkages in this region), whereas the upper hinge was modeled as two linear chains (in the absence of disulphide linkages), each connecting a Fab to the middle hinge. This arrangement can be represented by a three-number index, U-U-M, where U is the number of beads in each branch of the upper hinge and M is the number of beads in the middle hinge. Using identically sized beads for the genetic hinge region, typical bead arrangements could be 2-2-8, 3-3-12, 4-4-16 etc.
Using the U-U-M bead arrangement described above, models were constructed with the following bead compositions: 4-4-16, 5-5-20, 6-6-24, and 7-7-28, all with 1.8-Å bead radius. Models that included hinges with these arrangements reproduced at least two of the universal shape functions within the accepted uncertainty when the angles θ and ϕ were varied. No other hinge construction with the U-U-M composition could reproduce even two of the universal shape functions. Therefore, for bead-shell models with frictionless hinges the hinge region is effectively described by this bead composition.
Comparison with experiment: selection of the appropriate model(s)
In Table 4, we have marked in bold font all the modeled values for P, ν, and G that give matches with the experimentally determined values, allowing for experimental error. Of all the candidate models examined, two were found to give matches for all three universal shape functions. Details of these two models are given in Tables 5 and 6, in which the “projection on z axis” is the projection on the z axis of a line connecting the outermost point of each ellipsoid body (Fab and Fc) with the central hinge bead located at (0, 0, 0) in the reference Cartesian coordinate system; and the “hinge distance between Fab and Fc” is the arithmetic difference along the z axis of the linking beads in the hinge between each Fab and the Fc. A negative value for the projection on the z axis indicates that the ellipsoid body in question lies below the xy plane, which for the Fab means that it is bent toward the Fc; this orientation could also be deduced by noting that θ > 90°, a condition which positions the main axis of the Fab below the xy plane.
TABLE 5.
Model 4 for IgG3 wild-type antibody with hinge designed as 5-5-20
| Model 4 | θ (°) | ϕ (°) | Projection on z axis (Å) | Hinge distance between Fab and Fc (Å) | Angle between Fabs (°) | Dmax (Å) |
|---|---|---|---|---|---|---|
| Fc arm | 90.0 | 0 | −73.8 | — | ||
| Fab1 arm | 97.9 | 43.9 | −2.72 | 71.08 | 120.4 | 188 |
| Fab2 arm | 94.0 | 165.3 | −1.38 | 72.42 |
TABLE 6.
Model 18 for IgG3 wild-type antibody with hinge designed as 6-6-24
| Model 18 | θ (°) | ϕ (°) | Projection on z axis (Å) | Hinge distance between Fab and Fc (Å) | Angle between Fabs (°) | Dmax (Å) |
|---|---|---|---|---|---|---|
| Fc arm | 90.0 | 0 | −88.2 | — | ||
| Fab1 arm | 111.9 | 243.6 | −8.73 | 79.47 | 80.2 | 196 |
| Fab2 arm | 99.4 | 326.8 | −3.82 | 84.38 |
To establish which of these two representative models is most likely the Dmax parameter (maximum dimension of the scattering particle) derived from the distance distribution function can be used as a final discriminator. The experimentally obtained value was 195 Å (Fig. 4), which is more closely approximated by Model 18 (Dmax = 196 Å) than Model 4 (Dmax = 188 Å), so the final model shown in Fig. 5 is Model 18.
FIGURE 4.

Distance function p(r) of the chimeric human IgG3. The maximum dimension at 195 Å is denoted by Dmax. See text for details.
FIGURE 5.

Different views of a model (Model 18) for the chimeric IgG3 wild-type. The Fc domains are at the bottom of the model linked by a bent hinge to the two Fab domains.
In this model, the Fab arms are seen to bend toward the Fc but due to the existence of the extended hinge, the Fab domains are positioned away from that part of the Fc domain. This is consistent with IgG3 being the most efficient complement activator among the human IgG subclasses, as in this arrangement the Fabs would not obstruct access to the C1q binding site.
We are not saying that this is the actual solution structure of IgG3 but that the model shown appears to best represent the four measured parameters (P, ν, G, and Dmax), bearing in mind that it is a time-averaged model because of the putative flexibility of the molecule.
It is possible to further interpret the p(r) distribution in terms of the positions of the maxima, but this will be the topic of a future comparative study on IgG immunoglobulins.
CONCLUDING REMARKS
In this study, we have proposed a systematic routine for the evaluation of domain orientation in multidomain proteins in dilute solution, minimizing the uniqueness problem to the lowest degree and avoiding the ad hoc approaches to hydration and model selection. The procedure appears to have provided a plausible solution conformation for human IgG3, showing a good solvent exposure of the N-terminal part of Fc where the C1q binding site is situated, consistent with an efficient complement activation of wild-type IgG3. The basis for application of the procedure to other multidomain structures in solution appears to have been laid, provided adequate information is known about the domains themselves and allowance for particle solvation and flexibility are made.
Acknowledgments
We thank the United Kingdom Engineering and Physical Science Research Council for supporting this work, and the Council for the Central Laboratory of the Research Councils for access to the Synchrotron Radiation Source, Daresbury, UK. Work at the University of Murcia and a predoctoral fellowship to A. Ortega were supported by grant BQU2003-04517 to M.E.C. We thank Nicole Marheineke at Viscotek (Basingstoke, UK) for help with the viscosity measurements.
References
- 1.Sandin, S., L. Ofverstedt, A. Wikstrom, O. Wrange, and U. Skoglund. 2004. Structure and flexibility of individual immunoglobulin G molecules in solution. Structure. 12:409–415. [DOI] [PubMed] [Google Scholar]
- 2.Bongini, L., D. Fanelli, F. Piazza, P. De Los Rios, S. Sandin, and U. Skoglund. 2005. Dynamics of antibodies from cryo-eletron tomography. Biophys. Chem. 115:235–240. [DOI] [PubMed] [Google Scholar]
- 3.Carrasco, B., J. Garcia de la Torre, K. G. Davis, S. Jones, D. Athwal, C. Walters, D. R. Burton, and S. E. Harding. 2001. Crystallohydrodynamics for solving the hydration problem for multi-domain proteins: open physiological conformations for human IgG. Biophys. Chem. 93:181–196. [DOI] [PubMed] [Google Scholar]
- 4.Longman, E., K. Kreusel, S. B. Tendler, I. Fiebrig, K. King, J. Adair, P. O'Shea, A. Ortega, J. Garcia de la Torre, and S. E. Harding. 2003. Estimating domain orientation of two human antibody IgG4 chimeras by crystallohydrodynamics. Eur. Biophys. J. 32:503–510. [DOI] [PubMed] [Google Scholar]
- 5.Harding, S. E., E. Longman, B. Carrasco, A. Ortega, and J. Garcia de la Torre. 2004. Studying antibody conformations by ultracentrifugation and hydrodynamic modeling. In Antibody Engineering: Methods and Protocols. B. K. C. Lo, editor. Humana Press, Totowa, NJ. 93–113. [DOI] [PubMed]
- 6.Sun, Z., A. Almogren, P. B. Furtado, B. Chowdhury, M. A. Kerr, and S. J. Perkins. 2005. Semi-extended solution structure of human myeloma immunoglobulin D determined by constrained X-ray scattering. J. Mol. Biol. 353:155–173. [DOI] [PubMed] [Google Scholar]
- 7.Furtado, P. B., P. W. Whitty, A. Robertson, J. T. Eaton, A. Almogren, M. A. Kerr, J. M. Woof, and S. J. Perkins. 2004. Solution structure determination of monomeric human IgA2 by X-ray and neutron scattering, analytical ultracentrifugation and constrained modelling: a comparison with monomeric human IgA1. J. Mol. Biol. 338:921–941. [DOI] [PubMed] [Google Scholar]
- 8.Aslam, M., J. M. Guthridge, B. K. Hack, R. J. Quigg, V. M. Holers, and S. J. Perkins. 2003. The extended multidomain solution structures of the complement protein Crry and its chimeric conjugate Crry-Ig by scattering, analytical ultracentrifugation and constrained modelling: implications for function and therapy. J. Mol. Biol. 329:525–550. [DOI] [PubMed] [Google Scholar]
- 9.Boehm, M. K., J. M. Woof, M. A. Kerr, and S. J. Perkins. 1999. The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by X-ray and neutron solution scattering and homology modelling. J. Mol. Biol. 286:1421–1447. [DOI] [PubMed] [Google Scholar]
- 10.Halle, B., and M. Davidovic. 2003. Biomolecular hydration: from water dynamics to hydrodynamics. Proc. Natl. Acad. Sci. USA. 100:12135–12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oncley, J. L. 1941. Evidence from physical chemistry regarding the size and shape of protein molecules from ultracentrifugation, diffusion, viscosity, dielectric dispersion and double refraction of flow. Ann. N. Y. Acad. Sci. 41:121–150. [Google Scholar]
- 12.Scheraga, H. A., and L. Mandelkern. 1953. Consideration of the hydrodynamic properties of proteins. J. Am. Chem. Soc. 75:179–184. [Google Scholar]
- 13.Tanford, C. 1961. Physical Chemistry of Macromolecules. Wiley, NewYork.
- 14.Garcia de la Torre, J., H. E. Perez Sanchez, A. Ortega, J. G. Hernandez, M. X. Fernandes, F. G. Diaz, and M. C. Lopez Martinez. 2003. Calculation of the solution properties of flexible macromolecules: methods and applications. Eur. Biophys. J. 32:477–486. [DOI] [PubMed] [Google Scholar]
- 15.Garcia de la Torre, J. 1989. Hydrodynamic properties of macromolecular assemblies. In Dynamic Properties of Biomolecular Assemblies. S. E. Harding and A. J. Rowe, editors. The Royal Society of Chemistry, Cambridge, U.K. 3–31.
- 16.Davis, K. G., M. Glennie, S. E. Harding, and D. R. Burton. 1990. A model for the solution conformation of rat IgE. Biochem. Soc. Trans. 18:935–936. [DOI] [PubMed] [Google Scholar]
- 17.Finney, J. L. 1996. Overview lecture. Hydration processes in biological and macromolecular systems. Faraday Discuss. Chem. Soc. 103:1–18. [DOI] [PubMed] [Google Scholar]
- 18.Garcia de la Torre, J., and B. Carrasco. 2002. Hydrodynamic properties of rigid macromolecules composed of ellipsoidal and cylindrical subunits. Biopolymers. 63:163–167. [DOI] [PubMed] [Google Scholar]
- 19.Harding, S. E. 1997. The intrinsic viscosity of biological macromolecules. Progress in measurement, interpretation and application to structure in dilution solution. Prog. Biophys. Mol. Biol. 68:207–262. [DOI] [PubMed] [Google Scholar]
- 20.Harding, S. E. 1987. A general method for modelling macromolecular shape in solution. A graphic (II-G) intersection procedure for triaxial ellipsoids. Biophys. J. 51:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carrasco, B., S. E. Harding, and J. Garcia de la Torre. 1998. Bead modeling using HYDRO and SOLPRO of the conformation of multisubunit proteins: sunflower and grape-seed 11S globulins. Biophys. Chem. 74:127–133. [DOI] [PubMed] [Google Scholar]
- 22.Perkins, S. J. 2001. X-ray and neutron scattering analyses of hydration shells: a molecular interpretation based on sequence predictions and modelling fits. Biophys. Chem. 93:129–139. [DOI] [PubMed] [Google Scholar]
- 23.Garcia de la Torre, J., S. E. Harding, and B. Carrasco. 1999. Calculation of NMR relaxation, covolume and scattering of properties of bead models using the SOLPRO computer program. Eur. Biophys. J. 28:119–132. [DOI] [PubMed] [Google Scholar]
- 24.Volkov, V. V., V. A. Lapuk, R. L. Kayushina, E. V. Shtykova, E. Y. Varlamova, M. Malfois, and D. I. Svergun. 2003. Low-resolution structure of immunoglobulins IgG, IgM and rheumatoid factor IgM-RF from solution X-ray scattering data. J. Appl. Crystallogr. 36:503–508. [Google Scholar]
- 25.Semenyuk, A. V., and D. I. Svergun. 1991. GNOM-a program package for small-angle scattering data processing. J. Appl. Crystallogr. 24:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia de la Torre, J., B. Carrasco, and S. E. Harding. 1997. SOLPRO: theory and computer program for the prediction of SOLution PROperties of rigid macromolecules and bioparticles. Eur. Biophys. J. 25:361–372. [DOI] [PubMed] [Google Scholar]
- 27.Garcia de la Torre, J., M. L. Huertas, and B. Carrasco. 2000. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 78:719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carrasco, B., J. Garcia de la Torre, O. Byron, D. King, C. Walters, S. Jones, and S. E. Harding. 1999. Novel size-independent modeling of the dilute solution conformation of the immunoglobulin IgG Fab' domain using SOLPRO and ELLIPS. Biophys. J. 77:2902–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Errington, N., and A. J. Rowe. 2003. Probing conformation and conformational change in proteins is optically undertaken in relative mode. Eur. J. Biochem. 32:511–517. [DOI] [PubMed] [Google Scholar]
- 30.Muller, J. J., P. W. Schmidt, and G. Damaschun. 1980. Determination of the largest particle dimension by direct Fourier cosine transformation of experimental small-angle X-ray scattering data. J. Appl. Crystallogr. 13:280–283. [Google Scholar]
- 31.Næss, L. M., T. Aarvak, A. Aase, F. Oftung, E. A. Høiby, R. H. Sandin, and T. E. Michaelsen. 1999. Human IgG subclass responses in relation to serum bactericidal and opsonic activities after immunisation with three doses of the Norwegian serogroup B meningococcal outer membrane vesicle vaccine. Vaccine. 17:754–764. [DOI] [PubMed] [Google Scholar]
- 32.Garred, P., T. E. Michaelsen, and A. Aase. 1989. The IgG subclass pattern of complement activation depends on epitope density, antibody and complement concentration. Scand. J. Immunol. 30:379–382. [DOI] [PubMed] [Google Scholar]
- 33.Michaelsen, T. E., B. Frangione, and E. C. Franklin. 1977. Primary structure of the “hinge” region of the human IgG3. Probable quadruplication of a 15 amino acid residue basic unit. J. Biol. Chem. 252:883–889. [PubMed] [Google Scholar]
- 34.Huck, S., P. Fort, D. H. Crawford, M. P. Lefranc, and G. Lefranc. 1986. Sequence of a human immunoglobulin γ 3 heavy chain constant region gene: comparison with the other human C γ genes. Nucleic Acids Res. 14:1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michaelsen, T. E., A. Aase, C. Westbye, and I. Sandlie. 1990. Enhancement of complement activation and cytolysis of human IgG3 by deletion of hinge exons. Scand. J. Immunol. 32:517–528. [DOI] [PubMed] [Google Scholar]
- 36.Burton, D. R. 1985. Immunoglobulin G: functional sites. Mol. Immunol. 22:161–206. [DOI] [PubMed] [Google Scholar]
- 37.Roux, K. H., L. Strelets, O. H. Brekke, I. Sandlie, and T. E. Michaelsen. 1998. Comparisons of the ability of human IgG3 hinge mutants, IgM, IgE, and IgA2, to form small immune complexes: a role for flexibility and geometry. J. Immunol. 161:4083–4090. [PubMed] [Google Scholar]
- 38.Pumphrey, R. 1986. Computer models of the human immunoglobulins:shape and segmental flexibility. Immunol. Today. 7:174–178. [DOI] [PubMed] [Google Scholar]
- 39.Gregory, L., K. G. Davis, B. Sheth, J. Boyd, R. Jefferis, C. Nave, and D. R. Burton. 1987. The solution conformations of the subclasses of human IgG deduced from sedimentation and small angle X-ray scattering studies. Mol. Immunol. 24:821–829. [DOI] [PubMed] [Google Scholar]
- 40.Roux, K. H., L. Strelets, and T. E. Michaelsen. 1997. Flexibility of human IgG subclasses. J. Immunol. 159:3372–3382. [PubMed] [Google Scholar]
- 41.Michaelsen, T. E., O. H. Brekke, A. Aase, R. H. Sandin, B. Bremnes, and I. Sandlie. 1994. One disulfide bond in front of the second heavy chain constant region is necessary and sufficient for effector function of human IgG3 without a genetic hinge. Proc. Natl. Acad. Sci. USA. 91:9243–9247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brekke, O. H., T. E. Michaelsen, R. Sandin, and I. Sandlie. 1993. Activation of complement by an IgG molecule without a genetic hinge. Nature. 363:628–630. [DOI] [PubMed] [Google Scholar]
- 43.Schuck, P. 2000. Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78:1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boulin, C. J., R. Kempf, A. Gabriel, and M. H. J. Koch. 1988. Data acquisition systems for linear and area x-ray detectors using delay-line readout. Nucl. Instrum. Methods. Phys. Res. A 269:312–320. [Google Scholar]
- 45.Svergun, D. I. 1992. Determination of the regularisation parameter in indirect transform using perceptual criteria. J. Appl. Crystallogr. 25:495–503. [Google Scholar]
- 46.Guinier, A., and G. Fournet. 1995. Small Angle Scattering of X-Rays. Wiley, New York.
- 47.Longman, E., N. Marheineke, and S. E. Harding. 2006. Identifying differences in solution conformations of two chimeric IgG3 antibodies through triple detection SEC. LC GC N. Am. January. http://www.lcgcmag.com/lcgc/article/articleDetail.jsp?id=283454&pageID=3.
- 48.Haney, M. A. 1985. A new differential viscometer. Part One. Am. Lab. 17:41–56. [Google Scholar]
- 49.Haney, M. A. 1985. A new differential viscometer.Part Two. Am. Lab. 17:116–126. [Google Scholar]
- 50.Laue, T. M., B. D. Shah, T. M. Ridgeway, and S. L. Pelletier. 1992. Computer-aided interpretation of analytical sedimentation data for proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science. S. E. Harding, A. J. Rowe, and J. C. Horton, editors. Royal Society of Chemistry, Cambridge, U.K. 90–125.
- 51.Lebowitz, J., M. S. Lewis, and P. Schuck. 2002. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11:2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]