

Abstract
This article presents theoretical analysis and experimental data for the use of resonant waveguide grating (RWG) biosensors to characterize stimulation-mediated cell responses including signaling. The biosensor is capable of detecting redistribution of cellular contents in both directions that are perpendicular and parallel to the sensor surface. This capability relies on online monitoring cell responses with multiple optical output parameters, including the changes in incident angle and the shape of the resonant peaks. Although the changes in peak shape are mainly contributed to stimulation-modulated inhomogeneous redistribution of cellular contents parallel to the sensor surface, the shift in incident angle primarily reflects the stimulation-triggered dynamic mass redistribution (DMR) perpendicular to the sensor surface. The optical signatures are obtained and used to characterize several cellular processes including cell adhesion and spreading, detachment and signaling by trypsinization, and signaling through either epidermal growth factor receptor or bradykinin B2 receptor. A mathematical model is developed to link the bradykinin-mediated DMR signals to the dynamic relocation of intracellular proteins and the receptor internalization during B2 receptor signaling cycle. This model takes the form of a set of nonlinear, ordinary differential equations that describe the changes in four different states of B2 receptors, diffusion of proteins and receptor-protein complexes, and the DMR responses. Classical analysis shows that the system converges to a unique optical signature, whose dynamics (amplitudes, transition time, and kinetics) is dependent on the bradykinin signal input, and consistent with those observed using the RWG biosensors. This study provides fundamentals for probing living cells with the RWG biosensors, in general, optical biosensors.
INTRODUCTION
The ability of examining living cells in their native and physiological relevant context is crucial to understand the biological functions of cellular targets, and to the success of drug discovery and development. Although more complex and less specific than biochemical assays, cell-based assays that monitor the activities and health of living cells have gained popularity in drug discovery and development, because they have distinct advantages of extracting functional information that would otherwise be lost with biochemical assays and of being able to facilitate the measurements of mode of action, pathway activation, toxicity, and phenotypic responses of cells mediated by exogenous stimuli. Most of the cell-based assays measure a specific cellular event, ranging from second-messenger generation, to the translocation of a particular target tagged with a fluorescent label, to the expression of a reporter gene, and to the alteration of a particular phenotype (1–3). However, current cell-based assays require more manipulations (e.g., overexpression of targets with and without a readout tag) than biochemical assays; and such manipulations could pose significant issues to the cellular physiology of the targets of interest (4,5). Thus, a cell-based assay that is able to provide a noninvasive and continuous record of cellular activity with high sensitivity would be desired.
Optical biosensors that employ evanescent waves have seen widespread utility in both basic and applied research (6,7). These biosensors including surface plasmon resonance (SPR) and resonant waveguide grating (RGW) are mainly used to determine the affinities and kinetics of target analytes in a sample binding to the biological receptors immobilized on the sensor surface. A rapid growing interest in this field is to probe the activities of living cells, such as cell adhesion and spreading, toxicity, and proliferation (8–12). Recently, we had applied RWG biosensors to investigate cytoskeleton modulation (13), and cell signaling mediated through epidermal growth factor (EGF) receptor (14) or a G-protein-coupled receptor (GPCR) bradykinin B2 receptor (15). These studies had led to development of a novel label-free cell-based assay, termed mass redistribution cell assay technologies (MRCAT). The core of MRCAT is to use optical biosensors to online monitor the stimulation-mediated dynamic mass redistribution (DMR) within the bottom portion of cells. When measuring the dynamic changes in incident angle or resonant wavelength, the DMR signal is primarily resulted from the redistribution of cellular contents that occurs perpendicular to the sensor surface (14,15). The DMR signal represents a novel quantifiable cellular readout for studying cell activities including signaling and its network interactions. Because stimulation could lead to dynamic redistribution of cellular contents in three-dimension, monitoring the cell responses with the changes in incident angle or wavelength may not be sufficient for cell sensing with the biosensors. Here we introduce multiple optical readouts for cell sensing using the RWG biosensors, and present theoretical analysis and experimental data with special focus on the sensitivities of these optical readouts to the nature of dynamic mass redistribution.
MATERIALS AND METHODS
Reagents
EGF, trypsin, dimethyl sulfoxide (DMSO), and vincristine were purchased from Sigma Chemical (St. Louis, MO). AG1478 was obtained from Tocris Chemical (St. Louis, MO). Texas red-labeled phalloidin (TR-phalloidin), and Live/Dead cell viability reagent kit for animal cells were obtained from Molecular Probes (Eugene, OR). Bradykinin and SLIGLR-amide were obtained from Bachem Biosciences (King of Prussia, PA). Corning Epic 96well biosensor microplates were obtained from Corning (Corning, NY), and cleaned by exposure to high intensity UV light (UVO-cleaner, Jelight, Laguna Hills, CA) for 6 min before use.
Cell culture
Human epidermoid carcinoma A431 cells and Chinese hamster ovary (CHO-K1) were obtained from American Type Cell Culture (Manassas, VA). A431 were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 4.5 g/l glucose, 2 mM glutamine, and antibiotics. Unless specifically mentioned, ∼3–7.5 x 104 cells suspended in 200 μl the DMEM medium containing 10% FBS were placed in each well of a 96well biosensor microplate, and were cultured at 37°C under air/5% CO2 until ∼95% confluency was reached (∼2–4 days). For trypsin studies, cells in microplate were washed twice with 1× HBSS (1× regular Hank's balanced salt solution, 20 mM Hepes-KOH, pH 7.0), and maintained with 100 μl HBSS buffer. For EGF, SLIGLR-amide and bradyinin studies, cells in microplate were subject to starvation using DMEM without any FBS for ∼20 h, and then washed twice with HBSS, and maintained with 100 μl HBSS. After washing, the sensor microplate containing cells was placed into an arrayed angular interrogation system. For cell adhesion and spreading, each biosensor was initially covered with a 100 μl DMEM medium in the absence and presence of 200 nM vincristine. After reaching a stable resonant peak as well as a steady angular shift, 1 × 105 cells suspended in 100 μl DMEM containing 10% FBS were subsequently added into each well.
CHO-K1 cells were grown in Kaighn's modification of Ham's F12 medium (F-12K) supplemented with 10% FBS, 2 mM L-glutamine, 3 mg/ml sodium bicarbonate, and antibiotics. For cell confluency studies, different initial seeding numbers of cells suspended in 200 μl the F-12K medium containing 10% FBS were placed in each well of a sensor microplate. After cultured at 37°C under air/5% CO2 for ∼2 days, the resonant peaks were collected and analyzed. The intensity of each biosensor with distinct cell confluency was fine-tuned for reliable peak-at-half-maximum (PWHM) calculation. For DMSO studies, 100 μl 36% DMSO in 1× HBSS was applied to each biosensor covered with cells of ∼80% confluency in 1× HBSS (100 μl), and the resonant peaks were recorded at specific times during the treatment.
Optical biosensor measurements
Corning Epic angular interrogation system with transverse magnetic or p-polarized TM0 mode was used for all studies. The details of both instrument and assay protocol were previously described (13,14). New features were introduced to collect the kinetics of cell responses with multiple optical parameters. Briefly, the sensor microplate with and without cultured cells were placed into the system and equilibrated for certain time until a steady state was achieved. After continuous monitoring for half an hour to make sure no obvious drifting or changes in the resonant peak occurred, a compound solution was introduced into each well. Cell responses were then continuously recorded for a certain time. The effect of the “vehicle” (i.e., HBSS in most cases) was also monitored in parallel to eliminate any artifacts. One response unit for the shift in incident angle, obtained as a change in pixel of the central position of the resonant band of each sensor recorded with a charge-coupled device camera with the current system, was found to correspond to ∼5.82 × 10−4 refractive index changes in the cover medium (15).
Fluorescence imaging
After the assay with the biosensors, cells in the sensor microplates were directly stained with the Live/Dead reagent kit, or stained with TR-phalloidin after fixed with 4% paraformaldehyde, permeabilized in 0.2% Triton, and subsequently blocked with 1% bovine serum albumin. After final washes and mounting, cells were examined with a 40× objective using a Zeiss Axioplan fluorescence microscope.
Data analysis
The cell confluency was characterized by light microscopy imaging and quantified with Metamorph image analysis software (Universal Imaging, Downingtown, PA). At least three replicates were carried out for each measurement. The graphs presented in the figures were representative from at least three independent measurements.
THEORY OF MRCAT
Fig. 1 showed the principle of MRCAT. Cells are directly cultured onto the surface of a RWG biosensor. Exogenous signals mediate the activation of specific cell signaling, in many cases resulting in dynamic redistribution of cellular contents (16–18), equivalent to dynamic mass redistribution. When occurring within the sensing volume (i.e., penetration depth of the evanescent wave), the DMR can be manifested and thus monitored in real time by a RWG biosensor—a label free technology that is sensitive to change in local refractive index in the vicinity of the sensor surface (19). Because of its ability for multiparameter measurements, the biosensor has potential to provide high information content for cell sensing. These parameters include the angular shift (one of the most common outputs), the intensity, the PWHM, and area of the resonant peaks. In addition, because of the unique design of our angular interrogation system that uses a light beam of ∼200 × 3000 μm to illuminate each sensor, the resonant band image of each sensor can provide additional useful information regarding to the uniformity of cell states (e.g., density and adhesion degree) as well as the homogeneity of cell responses for cells located at distinct locations across the entire sensor (14). The following sections are meant to address the theoretical considerations for the MRCAT.
FIGURE 1.

The principle of RWG biosensor for sensing living cells. Cells are directly cultured onto the surface of a RWG biosensor. The mass redistribution within the bottom portion of cells, mediated by stimulus such as GPCR agonists or EGFR ligands, is directly measured with the biosensor. RWG biosensor utilizes an optical beam with an appropriate angular content to illuminate a waveguide film in which a grating structure is embedded. When this beam is reflected by the sensor surface, the resonant angle dominates in the output beam. The mass redistribution within the sensing volume alters the incident angle.
Sensor configurations and detection schemes
The RWG biosensor exploits the evanescent wave that is generated by the resonant coupling of light into a waveguide via a diffraction grating (19). The RWG biosensor typically consists of a substrate layer, a waveguide film wherein a grating structure is embedded, a medium, and an adlayer (i.e., a layer of adherent cells) (Fig.1). The guided light can be viewed as one or more mode(s) of light that all have directions of propagation parallel with the waveguide, due to the confinement by total internal reflection at the substrate-film and medium-film interfaces. The waveguide has higher refractive index value than its surrounding media. Because the guided light mode has a transverse amplitude profile that covers all layers, the effective refractive index N of each mode is a weighed sum of the refractive indices of all layers (19):
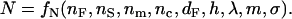 |
(1) |
Here, nF, ns, nm, and nc is refractive index of the waveguide, the substrate, the medium, and the adlayer of cells, respectively. dF and h is the effective thickness of the film, and the height of the cell, respectively. λ is the vacuum wavelength of the light used. m = 0, 1, 2, … is the mode number; and σ is the mode-type number that equals 0 for transverse electric (TE), or s-polarized, and 1 for transverse magnetic (TM), or p-polarized modes.
The weighing depends on the mode's distribution of power among all the layers. The effective indices of each mode N can be determined by simply measuring the incident angle of a beam on a diffraction grating incorporated in the waveguide, as governed by the following equation (20):
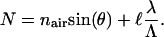 |
(2) |
Here θ is the maximum efficiency coupling angle. ℓ is the diffraction order, and 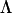 is the grating period. nair is the refractive index of air and equals 1.
is the grating period. nair is the refractive index of air and equals 1.
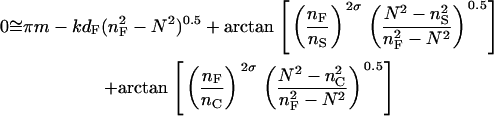
Optical sensing of adherent cells is unique and quite challenging, because of the nature of cells interacting with surfaces and the complexity of cell structure and functions. Some types of cells are known to adhere to a surface, primarily through three types of contacts: focal contacts, close contacts, and extracellular matrix (ECM) contacts (21–24). The focal contacts are narrow regions of an adhere cell membrane (e.g., 0.2 μm × 10 μm) that come within 10–15 nm of the substrate surface. The close contacts refer to regions of the cell membrane separated from the substrate of 1–50 nm, whereas the ECM contacts designate regions of the cell membrane separated from the substrate by 100 nm or more. Thus, one can imagine that the sensor is still able to sense the medium, even when the cell confluency is high (∼95%). However, it is known that living cells contain ∼70% water, and most of intracellular bio-macromolecules are highly organized by the matrices of filament networks (13,25) and spatially restricted to appropriate sites in mammalian cells (26,27). Furthermore, the height of the cells is typically beyond the wavelength of incident light (here λ = 830 nm), and the penetration depth is generally much smaller than the height of cells. Thus, the biosensor for cell sensing can be viewed as a three-layer configuration: the substrate, the waveguide, and the cell layer.
At the single cell level, it is obvious that there are differences in local refractive index from one area (i.e., focal contact) to another area (i.e., noncontact area) (24). However, because the RWG biosensor used gives rise to a relatively poor lateral resolution due to the long propagation of the guided light within the waveguide film, the signals measured represent an average response of a cluster of cells, considering the high cell confluency used for most of the assays. Therefore, to simplify the numerical analysis, the thickness of the bottom portion of cells probed by the sensor is assumed to be approximately constant as determined by the penetration depth. Because the refractive indices of the substrate and the waveguide as well as the thickness of the waveguide are known, it is possible from a measurement of the effective refractive index, N, to deduce the local refractive index of the cells probed and its changes mediated by stimulation.
Vertical mass movement
The value of effective refractive index N can be calculated numerically from the mode equation for a given mode of a three-layer waveguide (19):
 |
(3) |
Here, the wave vector k = 2π/λ.
The guided light modes propagate parallel to the surface of a plane waveguide, thus creating an electromagnetic field (i.e., an evanescent wave) extending into low-refractive index mediums surrounding both sides of the film with a characteristic of exponential decaying. The amplitude (Em) of the evanescent wave decays exponentially with increasing distance z from the interface toward the cell layer or the substrate (19):
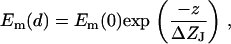 |
(4) |
with:
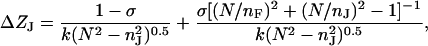 |
(5) |
is the penetration depth of the evanescent tail of the waveguide mode that extends into the cell (J = c) or substrate (J = s). Based on the configuration of the biosensors used and the dimensions of cells, the penetration depth of the TM0 mode used here is ∼133 nm, meaning that we only monitored the bottom portion of the cells.
The translocation of proteins and/or molecular assemblies is common to many cell responses triggered by exogenous signals. The translocation enables the precise control in the amplitude, duration, and kinetics of cell signaling through a specific target (28–32). In addition, in some cases exogenous stimuli could also cause changes in cell status such as the adhesion degree and cytoskeletal structure. When such changes occur within the sensing volume, the mode index N is altered due to the interaction between the cell and the evanescent tail.
For the redistribution of cellular contents in a direction that is perpendicular to the sensor surface but parallel to the evanescent tail of the guided modes (referred as vertical mass redistribution), we can divide the bottom portion of adherent cells into multiple equal-spaced and homogenous thin layers, assuming that the degree and configurations of adhesion are similar for cells being probed with the light beam at a given time. Each layer has its own refractive index ni and is away from the sensor surface by a distance of zi (Fig. 2). The thickness of each layer is denoted as d. All layers could be considered to have equal volume, because the unit area A is considered to be constant and determined by the spatial resolution of the optical biosensor, which is limited to the physical size of incident light beam as well as the propagation length of the guided wave in the waveguide. The refractive index of a given volume within cells is largely determined by the concentrations of biomolecules, mainly proteins (33,34):
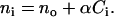 |
(6) |
FIGURE 2.

A three-layer configuration for detecting the stimulation-mediated vertical mass redistribution within the sensing volume. The bottom portion of cells is viewed to consist of multiple equal-spaced and homogenous thin layers, each layer has its own refractive index ni, protein concentration Ci, distance Zi (away from the sensor surface). A grating with a periodicity of Λ is embedded with the waveguide film with a refractive index of nF and a thickness of dF. The waveguide film is deposited on the top surface of a substrate with a refractive index of ns.
Here no is the index of the solvent, which is constant and approximately equals to water in cells. α is the specific refraction increment, and is ∼0.0018/100 ml/g for protein, and 0.0016/100 ml/g for other solutes found in cells such as sodium (33). Ci is the concentration of solutes (in g/100 ml) in the layer i. Although the specific refraction increments are similar for proteins and other solutes, proteins primarily account for the refractive index of each layer because their concentrations in terms of weight per volume are considerably greater than other solutes (34). Thus, the refractive index changes Δni of the homogeneous layer i approximately form a piecewise continuous function:
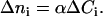 |
(7) |
Considering the exponential decaying nature of the evanescent wave (Eq. 4), the weighed index change Δnc within the sensing volume should be the integration of Δni with a weighed factor exp(−zi/ΔZc). Thus, we have:
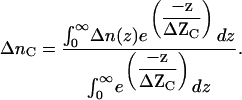 |
(8) |
Integrated from z = 0 to z= 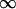 , after substituting Eq. 7 for
, after substituting Eq. 7 for 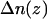 and rearrangement, we have:
and rearrangement, we have:
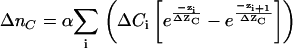 |
(9) |
Because in most cases Δnc is a small portion of the refractive index of the cells sensed by the biosensors (generally <20%) (13), thus to first order the change in effective refractive index is,
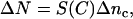 |
(10) |
where S(C) is the sensitivity to the cells:
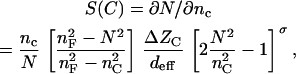 |
(11) |
with deff being the effective waveguide thickness given by:
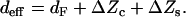 |
(12) |
Inserting Eqs. 9 and 11 into Eq. 10, we obtain, for the detected signal:
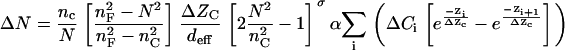 |
(13) |
Equation 13 suggests: i), changes of the effective refractive index, thus the optical signature relating to the shift in incident angle measured, is primarily sensitive to the vertical mass redistribution within the sensing volume (this type of signal was previously referred to as DMR (13–15)); ii), changes of the effective refractive index is directly a function of changes in protein concentration due to protein relocation, rather than ion mobilization such as Ca2+ influx and Ca2+ flux, mediated by a stimulation; iii), the relocation of a target or complex of certain mass near the sensor surface contributes more to the overall response than those further away from the surface; iv), the optical signature is an integrated signal that is a sum of contributions from mass redistribution occurring at different distances away from the sensor surface. Because of the complex nature of cell signaling, the activation of distinct cell signaling mediated through different targets might result in similar overall DMR signal. Because of “a priori” known ensemble of targets and inhibitors for a signaling process, it is possible to make a selective search for the cell signaling activated by the target, based on the analysis of the modulation profiles of these inhibitors on the optical response (14,35). The effect of an inhibitor on the optical responses, in terms of the overall dynamics, the kinetics, and the amplitude of the response, is an indication of whether the biomolecule with which the inhibitor interacts is involved in the signaling or not.
Horizontal mass movement
As discussed above, the shift in incident angle or resonant wavelength is largely determined by vertical mass redistribution within the sensing volume when cells respond to stimulation. Because of the poor lateral resolution of the biosensor, the lateral mass redistribution may be difficult to be resolved by these shifts. This section examines the possibility of measuring the horizontal mass movement with the resonant spectrum of a waveguide mode.
Using a zigzag wave model (36), Horvath et al. (37) have described a theory for explaining the relation between the surface inhomogeneity and the shape of the resonant peak of a given mode. After the guided modes propagating in the planar waveguide have passed a full zigzag, the phase difference between the ordinary wave and the twice-reflected wave is a function of the χ wave-vector component of a guided mode (β) for a given waveguide structure and fixed polarization and wavelength. Determined by self-consistency criteria, the β-value of a mth guided mode can be calculated numerically from the mode equation (37):
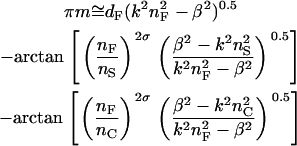 |
(14) |
Here, the mode propagation direction in the waveguide is χ.
The coupled light intensity, I(θ), as a function of the incident angle θ, is governed by:
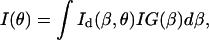 |
(15) |
where Id(β,θ) is the intensity distribution of the first-order diffraction:
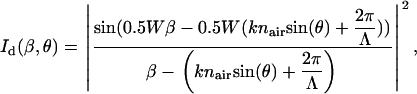 |
(16) |
and IG(β) is the intensity of the coupled light after the nth section that is proportional to the absolute square of the amplitude of the light G(β),
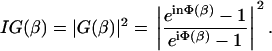 |
(17) |
Here i is the imaginary unit, and G(β) is the amplitude or intensity of the light. Assuming that the whole grating length is illuminated (as in this study), the coupling length W equals to the grating length, the total number, n, of the full zigzags can be calculated:
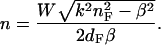 |
(18) |
Because of the finite width of the grating and laser beam, the diffracted light can be described using a plane wave distribution when illuminating the grating under angle θo with a plane wave. This leads to a peak at βo with a PWHM of ∼2π/W, calculated from the optical uncertainty principle, when no any adlayer exists. Modeling using these equations led to interesting findings, which suggest that the shape of the resonant peaks (or spectra) carries valuable information about the lateral inhomogeneity of mass distribution (36). Such lateral inhomogeneity, as exampled in Fig. 3, does not strongly perturb the cell refractive index, but significantly alters the shape of the resonant peaks.
FIGURE 3.

The phase shift as a function of asymmetrically lateral redistribution of cellular contents mediated by stimulation. The guided light, propagating in the planar waveguide, is viewed as zigzag waves. The inhomogeneity of lateral mass distribution within the sensing volume results in broadening, and even splitting of the resonant peak of a given mode.
It is well known that certain exogenous signals could lead to significant asymmetric lateral mass redistribution at the levels of both single cell and multiple cells (38–40). For example, distinct populations of cells could respond heterogeneously to compounds that are toxic to cells, while a single adherent cell could undergo uneven distribution of certain cellular targets or molecular assemblies during some cellular processes such as cell migration and invasion. We hypothesized that when occurring, the asymmetric lateral mass redistribution could also result in the change in the fine structure and shape of the resonant peaks.
RESULTS AND DISCUSSIONS
Multiple optical output parameters were recorded in real time and in parallel for several well-studied cell responses and processes, including adhesion and spreading, detachment, as well as signaling of cells through EGFR or bradykinin B2 receptor. These optical readouts include the shift in incident angle, as well as three parameters defining the shape of the resonant peaks: intensity, PWHM, and area. Because of its high sensitivity and information content, only TM0 mode was used for all data collection and analysis.
The shape of the TM0 peak
As shown in Fig. 4 A, the shape and position of the TM0 peak for cultured CHO cells was found to be dependent on the cell confluency. As the cell confluency increases, the resonant peak shifts toward the direction of high incident angles. The PWHM value, a parameter defining the peak shape, also exhibited a dependence on the cell confluency (Fig. 4 B). The PWHM value reached its maximum at the cell confluency of ∼50%; the maximum PWHM was ∼35% higher than those in the absence or presence of cell monolayer with high density above 75%. It is worthy noting that these data were obtained after cells became fully spread after ∼2 days culturing in a growth medium.
FIGURE 4.

The resonant peak (A) and the PWHM (B) of TM0 mode as a function of CHO cell confluency.
The TM0 peak of adherent CHO cells was also found to be sensitive to DMSO—a toxic compound when high doses are used. As shown in Fig. 5 A, the shape and position of the TM0 peak exhibited dynamic changes when proliferating cells, obtained in 10% FBS, was treated with 18% DMSO. After treating with DMSO, both the intensity and area of the peak increases throughout the time monitored (∼3 h). However, the peak position (i.e., the incident angle) showed dynamic characteristics: the incident angle initially shifts toward an increase in mass (e.g., 25 min), and then a decrease in mass (e.g., 40 and 120 min). Similarly, the peak shape initially became broadened and showed a complicate fine structure (e.g., 25 min), and eventually became narrow (e.g., 40 and 120 min). The appearance of complicated peak structures has been used as an implication of large-scale irregular inhomogeneity of mass at or near the sensor surface (37), thus indicating that during certain period after treatment with DMSO, the biosensor senses an increase in surface inhomogeneity. The live/dead staining pattern of CHO cells, obtained 25 min after the DMSO treatment, showed that there were mixed populations of cells: viable, dead, and affected cells (Fig. 5 B), implying that the CHO cells seem to respond heterogeneously to the DMSO treatment. These results suggested that the shape of the TM0 peak is useful to examine the heterogeneous lateral mass distribution within the sensing volume.
FIGURE 5.

The effect of DMSO on adherent CHO cells. (A) The dynamic effect of 18% DMSO on the shape of the TM0 peak at a confluency of ∼80%. (B) Live/Dead staining pattern of CHO cells after treated with 18% DMSO for 25 min. The bar represents 60 μm.
Cell adhesion and spreading
The adhesion and spreading of cells at surfaces were well studied using optical imaging techniques (41–43), as well as optical biosensors such as SPR (44) and RWG (8,9). Cells start to interact with a surface by initial contact or attachment where cells generally retain the round shape they possessed in suspension. Subsequently, attached cells undergo morphological changes known as spreading—a process that the cells increase their area in contact with the surface. Both attachment and spreading are dependent on the nature of the surface and of the medium in which the cells are suspended. Because cell adhesion and spreading obviously leads to both vertical and horizontal mass redistribution within the sensing volume, we first characterized the adhesion and spreading of A431 cells in 5% FBS in the absence and presence of vincristine. Vincristine is a plant alkaloid that inhibits microtubule assembly by binding to tubulin (45).
Fig. 6 summarized the optical signatures of the adhesion and spreading of A431 cells in 5% FBS in the absence and presence of vincristine at room temperature (25°C). In the absence of vincristine, the shift in incident angle exhibited three major phases (Fig. 6 A). Following the addition of cell solution (the point a), there is an immediate and rapid increased signal (as indicated by the arrow b), which probably resulted from three events: the increased bulk index from the addition of the cell solution, the immobilization of serum proteins onto the sensor surface, and the sedimentation of cells and subsequent contact of cells with the surface. Afterwards, a prolonged increased signal occurred (as indicated by the arrow c), indicating the slow process of cell spreading at room temperature. Ultimately a saturated level was reached (as indicated by the arrow d). The saturated level (16.8 ± 0.6 unit, n = 3) was much lower than those of fully spread cells at similar density obtained at 37°C (22.6 ± 1.0 unit, n = 3). On the other hand, the normalized PWHM value was also found to be dynamic with distinct characteristics (Fig. 6 B). After the cell solution was added, the PWHM value started to increase. About 20 min later, the PWHM began to decay back to its original level within ∼2 h, followed by a slowly continuous increase until it reached a plateau. The PWHM at the endpoint was ∼25% higher than that at the starting point, suggested that the cells were still not fully spread, even after 20 h assaying with the biosensor under ambient condition. This was confirmed by light microscopy images (data not shown). These results suggested that i), at room temperature A431 cells seem not be able to reach optimal degree of adhesion; ii), the cells interact with the surface through multiple steps and each has its own characteristics; and iii), the spreading step clearly increases the mass within the sensing volume, which means increased contact of the cell with the surface.
FIGURE 6.

Real-time monitoring the adhesion and spreading of A431 cells in the absence and presence of vincristine using the shift in incident angle (A), or the PWHM of the TM0 mode (B).
The presence of 100 nM vincristine significantly altered the optical signatures. The presence of vincristine suppressed not only both of the initial and total responses, but also reduced the kinetics of cell spreading (Fig. 6 A). The total change in incident angle in the presence of vincristine was ∼20% less than that in the absence of vincristine. Interestingly vincristine also altered the dynamic features of the PWHM value (Fig. 6 B). Unlike that in the absence of vincristine, the PWHM initially decreased and remained low for ∼3 h, and subsequently increased until it reached a plateau, indicating that vincristine primarily affects the initial steps during the cell adhesion and spreading processes. These results suggested that the biosensor is not only able to provide insights for the interaction of cells with the surfaces, but also to differentiate compounds for their ability to alter cell adhesion and spreading processes.
Cell detachment
Trypsin is a pancreatic serine protease with substrate specificity based upon positively charged lysine and arginine side chains (46,47). Because of its ability for orderly and unambiguous cleavage of protein molecules, trypsin is widely used to detach adherent cells from the surface of a cell culture vessel. Thus, we characterized the optical signatures of cell detachment by trypsinization.
Fig. 7 presented the DMR responses of quiescent A431 cells of ∼95% confluency mediated by trypsin at different doses between 0.03% and 1.96%. Treatment of A431 cells with trypsin at different doses triggered a similar DMR signal that consists of three phases: a phase with increased signal (termed as positive-DMR, or P-DMR), a transition phase, and a phase with decreased signal (termed as negative-DMR, or N-DMR). As the concentration of trypsin increases, the amplitude of the P-DMR decreases whereas the amplitude of the N-DMR increases. The increased N-DMR seems to correlate well with the increased detachment of cells from the sensor surface, as confirmed by light microscopy imaging (data not shown). Compared to the total signal (25.4 ± 1.3 unit, n = 10) measured for cells of ∼95% confluency (cultured under normal conditions), the maximum value of the amplitude of the N-DMR phase (18.2 ± 0.7 unit, n = 3), measured ∼2.5 h after trypsinization using 1.96% trypsin, was smaller. This was probably due to the fact that some protein molecules or domains still remain on the surface after trypsinization, meaning that the cell detachment mediated by trypsinization is primarily resulted from the breakdown of the “anchorage” protein molecules that facilitate the cell adherence. The relatively slow kinetics in the N-DMR phase observed seems due to the un-optimal trypsinization conditions, because the detachment process was monitored at room temperature, and HBSS contains Ca2+ and Mg2+.
FIGURE 7.

The dynamic mass redistribution of A431 cells mediated by trypsin at different doses. The final concentrations of trypsin are indicated in the graph. The arrow indicates the time when a trypsin solution is introduced.
Because trypsin of high doses led to complicated signatures for the other three parameters defining the peak shape, we mainly focused on the cell responses mediated by trypsin at low doses between 0.005% and 0.04% (Fig. 8). Results showed that similar to high doses, trypsin at these low doses also mediated similar DMR responses, as implicated by the shift in incident angle (Fig. 8 A). In addition, both the PWHM and the intensity of the resonant peak also exhibited a dynamic change in a dose-dependent manner. After trypsin was introduced, the PWHM value started to increase and subsequently decreased back to the original level (Fig. 8 B), whereas both the peak intensity (Fig. 8 C) and area (data not shown) showed a dynamic change that is opposite to the PWHM. However, all three parameters posed similar kinetics to that of the incident angle change. These results suggested that trypsinization triggers dynamic mass redistribution in both vertical and horizontal directions.
FIGURE 8.

Optical signatures of A431 cells induced by trypsin at low doses (A–C), or SLIGLR-amide (D). (A) The shift in incident angle. (B) The normalized PWHM. (C) The intensity of the TM0 peak. (D) The shift in incident angle mediated by SLIGLR-amide at different doses. The arrows indicate the time when a solution is added.
Interestingly, when the dose-dependent change in the N-DMR amplitude seems well correlated with the fact that trypsin causes cell detachment, the appearance of the P-DMR phase suggested that the trypsin treatment might result in cell signaling, which, in turn, contributes to the DMR signal. The pretreatment of quiescent A431 cells with 10 μM AG1478—a potent and selective EGF receptor tyrosine kinase inhibitor—partially attenuated both the P-DMR and N-DMR mediated by trypsin (data not shown), suggesting that EGF receptor could be a downstream target of trypsinization (48). EGFR is known to be a common downstream target of numerous mitogenic GPCRs (49–51) and trypsin is a natural agonist of protease-activated receptors (PARs) (52). Thus we reasoned that the unique optical signature mediated by trypsin at low doses indicates the activation of endogenous PARs in A431. At high doses, trypsin results in cell detachment, which dominates the optical signals obtained. However, at low doses trypsin is not sufficient to result in significant cell detachment. Instead, trypsin could activate endogenous PARs that lead to typical Gq-signaling. To test that, conventional Fluo-3 assays were used to measure the effect of trypsin on intracellular Ca2+ level. Results showed that at doses below 0.03% trypsin dose-dependently induced a rapid and transient increase in intracellular Ca2+ (data not shown), suggesting that trypsin mediates a classical Gq-signaling in A431 cells. Since A431 is known to endogenously express PAR2 receptor (53,54), SLIGLR-amide, a PAR2-activating peptide without the cleavage activity of trypsin, was used to stimulate the cells. As shown in Fig. 8 D, SLIGLR-amide dose-dependently triggered a DMR signal that is similar to those induced by trypsin at low doses, suggesting that the optical signature induced by trypsin at low doses is primarily originated from the Gq-signaling.
Epidermal growth factor receptor signaling
Previously, we had characterized a unique optical signature for EGF receptor signaling in A431 cells mediated by EGF using the RWG biosensors (14). Rich information had been obtained through analysis of the modulation of the EGF-induced DMR signals by a variety of known modulators. Results showed that the DMR in quiescent A431 cells mediated by EGF required EGFR tyrosine kinase activity, actin polymerization, and dynamin activity, and mainly proceed through MEK (MAPK/ERK kinase). Here we further characterized the optical signatures of EGFR signaling mediated by EGF using parallel multiparameter measurements. As showed in Fig. 9 A, stimulation of quiescent A431 cells with EGF at appropriate doses triggered almost identical DMR signatures to those previously reported (14). The cell responses, as manifested by the angular shift, induced by high doses of EGF were particularly interesting. When stimulated with high doses of EGF above 32 nM, a novel phase of DMR signal was observed for quiescent A431 cells. Besides an initial rapid P-DMR with increased signal followed by a short transition phase and a long decay N-DMR with decreased signal, there is a partial recovery phase with increased signal before the cells ultimately reach a plateau, which exhibits a similar level to those induced by either 16 nM or 32 nM. One possibility is that after stimulated with high doses of EGF the cells underwent a detachment process (14,17), followed by a partial reattachment process.
FIGURE 9.

The responses of quiescent A431 cells to EGF stimulation. (A) The dynamic shift in incident angle as a function of time. (B) The normalized PWHM as a function of time. (C and D) Staining pattern of actin filaments with TR-phalloid: (C) untreated A431; (D) A431 treated with 16nM EGF for 15 min. The bar represents 40 μM.
Because EGF mediates asymmetric lateral redistribution of certain cellular targets such as PI3K that is important for EGF-induced cell migration (38–40), parameters defining the shape of the resonant peak were monitored in parallel. However, all three parameters seem to remain constant after stimulated with EGF at different doses (Fig. 9 B; only the PWHM was presented), suggesting that there is no detectable inhomogeneity of lateral mass distribution within the bottom portion of cells mediated by EGF. This is contradictory to the staining pattern of actin filaments with TR-phalloidin (Fig. 9, C and D). These images showed that EGF mediated significant rearrangement of actin filaments in lateral dimensions. Because the EGF stimulation is known to lead to the cell rounding and detachment (17), it is conceivable that the inability to detect any inhomogeneity of lateral mass redistribution triggered by EGF indicates that the asymmetric redistribution might mainly occur outside the sensing volume.
Bradykinin B2 receptor signaling
Bradykinin B2 receptor is a G-protein-coupled receptor, and accounts for most of the physiological and pathophysiological action of bradykinin (BK) (55,56). Bradykinin appears to act as a mediator of a wide variety of physiological and pathophysiological responses including mitogenic and antimitogenic effects. A431 cells endogenously express bradykinin B2 receptor, but not B1 receptor (57). Previously we had studied the optical signatures of quiescent A431 cells in response to BK stimulation, and found that A431 cells responded to BK stimulation with dynamic mass redistribution; its kinetics, amplitudes, and duration depend on the cell culture conditions, the dose of BK, and the cellular context (15). Here quiescent A431 cells, obtained through culturing using the medium without any serum for at least 20 h, were used. As shown in Fig. 10, BK stimulation of quiescent A431 cells leads to an optical signature that is similar to those induced by SLIGLR-amide or trypsin at low doses. BK stimulation of quiescent A431 cells resulted in a rapid increase in PWHM, followed by a slow decay back to the original level, while the peak intensity gave rise to a dynamic response that is inverse to those of the PWHM and the angular shift. Furthermore, the changes in both parameters are also BK dose-dependent, and their dynamics and kinetics exhibited similarity to the DMR signals previously reported (15). These results suggested that compared to EGF-mediated cell responses, BK stimulation leads to more significant asymmetric redistribution of cellular contents, which, in turn, increases the inhomogeneity of lateral mass distribution within the bottom portion of cells.
FIGURE 10.

Optical signatures of quiescent A431 cells mediated through bradykinin B2 receptor by bradykinin: the PWHM (A) and the intensity (B) of the resonant peak of the TM0 mode. The arrows indicate the time when a bradykinin solution is added.
MATHEMATICAL MODELING
Dynamic redistribution of cellular contents is common to many cellular processes including the signaling through G-protein-coupled receptors in response to stimulation. Monitoring such redistribution has provided insights into GPCR signaling and a powerful means for GPCR screens (58,59). For example, direct visualization of the relocation of β2-adrenoceptor-GFP conjugates after agonist stimulation (16) initiated interest in this process as a direct screening strategy (58). In this section, we developed a mathematic model of the dynamics of the coupling between B2 receptor activation and the relocation of intracellular proteins (recruitment to the cell plasma membrane) and activation receptors (internalization to endosomes).
Upon bradykinin stimulation, B2 receptor becomes activated, which, in turn, leads the activation of its coupled heterotrimeric G-proteins composed of α-, β-, and γ-subunits. The Gαq- and Gβγ-subunits dissociate, and each triggers its own downstream signaling pathway(s). Meanwhile, GPCR kinase (GRK) is recruited to the activated receptor at the plasma membrane, and phosphorylates the carboxy terminus of the receptor. Afterwards, many intracellular proteins including protein kinase C (60) and β-arrestin (16) rapidly translocates from the cytoplasm to the activated receptors at the plasma membrane. Interestingly, β-arrestin binds the phosphorylated receptor, and acts as an adaptor protein to interact with other signaling proteins to form a signaling complex, which eventually translocates to clathrin-coated pits at the cell surface where the receptor is internalized in clathrin-coated vesicles. Finally some GPCRs dissociate from β-arrestin at or near the plasma membrane and recycle rapidly to the cell surface, whereas others remain associated with arrestin and traffic into endocytic vesicles.
For numerical analysis, a simplified signaling cycle of Gq-coupled receptors was used to predict a quantitative relationship between ligand binding and the DMR response. The model is:
 |
(19) |
R is a homogenous, and monovalent population of surface receptors (no./cell), L is the extracellular ligand concentration introduced at time t = 0 (assumed constant for t > 0), and LR is the number of ligand-receptor complexes (no./cell) that form with an association rate constant of kf and are lost with a dissociation rate constant of kr. R* is the number of phosphorylated receptors with a rate constant of kp. The R*, complexed with ligands, initiate the translocation of cytoplasmic proteins to the cell membrane and interact with the receptor to form complexes. The complexes eventually internalize with a rate constant of kin to give internalized complexes (Rin) into vesicles or endosomes in the cell. Some receptors dissociate from β-arrestin at or near the cell surface and recycle to the cell surface with a rate constant of kre. To achieve steady state, the rate of formation of internalized complexes R* should be equal to the rate of loss, thus kp = kin. We have assumed that internalization occurs only after the formation of R*.
The characteristics of this model are as follows: i), the adherent cell adopt an ellipsoid geometry with a height of h and a radius of r; ii), the bottom surface region of the cell is in close contact with the sensor surface, and can be considered as being continuously flat but with an average separation distance of ∼20 nm from the sensor surface; iii), a receptor at the cell surface binds only one ligand, but with multiple intracellular proteins a; iv), there are two classes of receptors: phosphorylated and unphosphorylated, after ligand binding; v), only phosphorylated receptors, together with other regulatory proteins, are internalized; vi), there are two classes of proteins or molecular assemblies in the cell: relocated and nonrelocated. During the relocation processes, except for the relocated biomolecules, the rest of the cell remains static; vii), the receptor activation at the cell surface triggers bidirectional relocation of cytosolic proteins: the proteins at the top half of the cell move toward the top cell membrane surface, and the proteins at the bottom half of the cell move toward the bottom cell membrane surface. Only the latter relocation event contributes to the DMR signals measured, because of the short penetration depth of the biosensor; viii), the relocation of proteins or molecular assemblies is governed by diffusion; ix), the receptors are randomly distributed at the cell surface; and x), both the total number of relocated proteins in the cell and the receptors at the cell surface is conserved.
The total number of receptors, Rtot, is assumed constant. Hence we have, according to the conservation law,
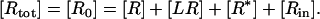 |
(20) |
Changes in the four states of the receptors are given by:
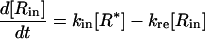 |
(21) |
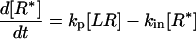 |
(22) |
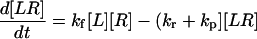 |
(23) |
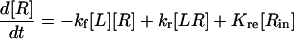 |
(24) |
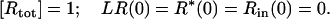 |
(25) |
For protein translocation and receptor internalization, considering the array of equal-spaced layer within a cell (Fig. 2), the change in the concentration of biomolecules in the ith layer, Ci = Ci(x,t), i = 0,1,2,…,n, is governed by the rate of formation of phosphorylated receptors that are capable of interacting with the intracellular proteins, the rate of formation of internalized complexes, as well as the cytoplasmic diffusion of proteins or protein complexes, with an effective diffusion coefficient Ds,
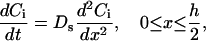 |
(26) |
where h denotes the height of a cell, and x is mapped for each layer individually to the interval (0, h/2). The diffusivity of molecules or molecular assemblies in the cytosol Ds can be estimated based on measurements made in intact eukaryotic cells following the diffusion of proteins of various molecular weights (61,62), The value of Ds is observed to be 0.5–2 × 10−8 cm2s−1, relatively insensitive to molecular size. Because of the small dimension of a cell as well as the small penetration depth of the RWG biosensors used, the diffusion time, for either the intracellular proteins reaching the cell surface or the internalized complexes moving out of the sensing volume, is estimated to be within seconds. Compared to the dynamics of the optical responses observed, the diffusion of molecules or molecular assemblies can be considered as a secondary factor. Thus, the net change in protein concentration within the sensing volume is the sum of protein accumulation near the cell surface and the internalization of receptor complexes. For the net change in protein concentration adjacent to the cell surface, we have:
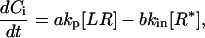 |
(27) |
where a is the number of proteins interacting with an activated receptor, where b is the number of proteins internalizing together with the receptor.
Following stimulation with bradykinin, fluorescent protein kinase C isoforms, initially randomly distributed in the cytoplasm of the cell, translocated and enriched near the cell surface (<200 nm) (16). Thus we reasoned that the net change in protein concentration among different layers near the cell surface (zi < 200 nm) is same, we have:
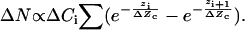 |
(28) |
This simplified model neglects several events in GPCR signaling, including i), changes in the oligomerization state of the receptors; ii), the fate of ligand beside binding, such as degradation, internalization with the receptor through endocytosis or diffusion into the cytoplasm of the cells; iii), morphological changes of the cell; iv), the conversion of the affinity state of ligand-receptor complex; v), receptor upregulation after stimulation; vi), both de novo receptor synthesis and the degradation of internalized receptors are negligible throughout the assay (∼1 h); vii), the constitutive endocytosis; viii), the rate of membrane synthesis; and ix), the compartmentalization of cell surface receptors.
Table 1 shows the model parameters. Using the simplified model, numerical analysis showed that in the physiological range from 1 to 128 × 10−9 M, the variation in bradykinin signal input results in a clearly dose-dependent dynamics in the different states of receptors (Fig. 11). Upon bradykinin stimulation, all four states of receptors eventually reach steady state, each with distinct characteristics. Interestingly, the model predicts that at each bradykinin concentration, the rate of formation of phosphorylated receptors is faster than that of internalized complexes, and the total number of phosphorylated receptors is typically greater than that of internalized receptors.
TABLE 1.
Model parameters
| Parameter | Definition | Value | Reference |
|---|---|---|---|
| Optical | |||
| α | Refraction increment of proteins | 0.0018/100 ml/g | (33,34) |
| ι | Diffraction order | −1 | |
| λ | Illumination wavelength | 830 nm | |
| σ | Mode type number | 1 | (19) |
| ∧ | Grating period | 500 nm | |
| dF | Thickness of waveguide film | 150 nm | |
| dad | Thickness of cell layer | 3000 nm | |
| deff | Effective waveguide thickness | 517 nm* | |
| ΔZc | Penetration depth into the medium | 133 nm* | |
| ΔZs | Penetration depth into the substrate | 234 nm* | |
| W | Grating length | 3000 nm | |
| m | Mode number | 0 | |
| N | Effective index | 1.685* | |
| nc | Refractive index of cell | 1.372 | (33,34) |
| nair | Refractive index of air | 1.00 | |
| nm | Refractive index of medium | 1.328 | |
| nF | Refractive index of waveguide film | 2.37 | |
| ns | Refractive index of substrate | 1.51 | |
| Cell dimension | |||
| d | Imaginary thickness of a layer | ||
| h | Height | 3000 nm | |
| r | Radius | 5000 nm | |
| Signaling | |||
| a | No. of proteins interacted with the receptor | 3-4 | (59,60) |
| b | No. of proteins internalized with the receptor | 2-3 | (59) |
| Rtot | Total number of receptors per cell | 2 × 104 | (50,63) |
| Ptot | Total number of translocated proteins per cell | 2 × 104 | (63) |
| Ds | Diffusion constant of proteins or assemblies | 0.5–2 × 10−8 cm2s−1 | (61,62) |
| κf | Association rate constant | 1.3 × 105 M−1 s−1 | (64) |
| κr | Dissociation rate constant | 1.6 × 10−4 s−1 | (64) |
| κp | Rate of receptor phosphorylation | 3.3 × 10−3 s−1 | (65) |
| κin | Rate of receptor internalization | 3.3 × 10−3 s−1 | (65) |
| κre | Rate of receptor recycling | 3.3 × 10−3 s−1 | (65) |
| L | Ligand concentration | 1–128 nM |
The values were calculated based on the three-layer waveguide configuration.
FIGURE 11.

The dynamics of four different states of receptors in response to different bradykinin signal input, as predicted by the model. (a) The number of free receptors at the cell surface; (b) the number of receptor-ligand complexes at the cell surface; (c) the number phosphorylated receptors at the cell surface; and (d) the number of internalized receptor complexes. The total number of receptors was normalized to 1.
The change in effective index ΔNc is found to be sensitive to a number of parameters, including the total number of receptors, the total number of translocated proteins from cytosol to the cell membrane, and the ligand concentration. Based on the typical Gq-signaling cycle through a Gq-coupled receptor, it is reasonable to assume that each phosphorylated receptor recruits an adaptor protein which, in turn, recruits and interacts with the other three proteins to form complexes, and all proteins have a molecular weight of ∼80 kDa. Although the size of internalized complexes (i.e., vesicles) is much bigger (typically 100–400 nm in diameter) than individual proteins, the internalized process can be approximately viewed as an inverse process to the recruitment, assuming that the loss in membranes due to internalization is balanced by the rearrangement/synthesis of membranes. Because some portions of recruited proteins stay at/near the cell surface to fulfill their functions throughout the signaling cycle, the loss in mass due to the internalization is assumed to be one receptor associated with three proteins. Using Eqs. 27 and 28, numerical analysis showed that bradykinin stimulation leads to a dose-dependent ΔNc response. The ΔNc is normalized to the unit measured using the RWG biosensor system. As shown in Fig. 12 A, the optical signatures predicted exhibit interesting characteristics; its amplitude, overall dynamics, and kinetics are similar to those observed using the biosensors for the DMR responses mediated by bradykinin, SLIGLR-amide, or trypsin at low doses. The amplitudes of the P-DMR phases also clearly exhibit a dose-dependence (Fig. 12 B). Both types of calculations, based on either the amplitude at a given time after stimulation (250 s) or the maximum amplitude, gave rise to similar EC50 (42 and 24 nM, respectively). These results suggested that the protein translocation and receptor internalization are two primary resources for the DMR signatures observed for Gq-coupled receptor signaling (15).
FIGURE 12.

The DMR signals mediated by bradykinin, as predicted by the model. (A) The responses as a function of time and bradykinin concentration. (B) Two types of amplitudes of the P-DMR phase, obtained at a given time (250 s) after stimulation or the maximum level, as a function of bradykinin concentrations. The change in effective index of the cell layer is calculated and normalized to the unit measured using the present optical sensor system.
CONCLUSIONS
Conventional cell-based assays typically measure a specific cell response (e.g., Ca2+ mobilization, or reporter gene expression) or a change in location of a tagged molecule in the signaling cascade mediated through a particular target. In most cases the target-mediated signaling dominates the cellular response measured, although compounds that activate other endogenous targets might also lead to similar response. Therefore, the signaling specificity is mostly conserved. On the other hand, for optical biosensor-based cell assays the signaling specificity is believed to be largely problematic, because i), these assays typically do not use specific labeled target, ii), the cell signaling is a complicated process and depends on the cellular context (i.e., cell status and types); and iii), the optical signature obtained is an integrated response, meaning that distinct types of stimulation could lead to similar optical signature. For example, two completely distinct stimuli: EGF (a natural agonist of EGF receptor) and methyl-β-cyclodextrin (a cholesterol-depletion reagent), had been found to mediate similar optical signatures (14,35). Although it appears that the DMR signals are not specific in these cases, signaling pathway analysis, based on the modulation profiles with a panel of well-known modulators using the biosensors, suggests that both stimulations lead to similar but not identical cellular signaling events. The EGF-mediated DMR signal in quiescent A431 cells is primarily linked to the Ras/mitogen-activated protein (MAP) kinase pathway (14), while the DMR signals induced by cholesterol depletion are primarily linked to the transactivation of EGF receptor, and involve multiple signaling pathways including MAPK, PKC, and phosphatidylinositol 3-kinase (35). Furthermore, the similarity of the optical responses in A431 cells mediated by bradykinin, SLIGLR-amide, or trypsin at low doses suggested that these agonists trigger similar cell signaling event (e.g., Gq-mediated signaling), as confirmed by conventional Ca2+ mobilization assays. These studies suggest that the activation of a particular signaling pathway (i.e., Gq-signaling or EGFR signaling) could lead to a unique type of optical signature. Interestingly, although same family receptors (e.g., Gq-coupled receptors B2 and PAR2) may lead to the same type of optical signature, distinct members may have its own fine characteristics (i.e., the amplitudes and the kinetics of DMR responses), due to its own signaling network interaction. Based on our previous studies on the signaling of EGFR and B2 receptor, as well as the cellular function of cholesterol, we believe that the RWG biosensor is well suited for analyzing signaling pathway and its network interaction, and is able to provide rich information content for monitoring cell activities.
In this article, we further characterized the optical biosensors for probing cell activities, based on theoretical, experimental, and numerical analysis. Theoretical analysis revealed that the optical signatures measured are integrated responses and can be used as novel readouts for examining cells in their native environments without the need of labels. Several cell responses and processes including adhesion and spreading, detachment, and cell signaling through EGFR and bradykinin B2 receptor had been investigated systematically. Parallel and kinetic measurements of multiple optical output parameters led to identification of unique signatures for stimulation-mediated dynamic mass redistribution in both vertical and lateral dimensions within the bottom portion of cells. Cell adhesion and spreading had been found to involve multiple steps; vincristine was found to be able to modulate the cell adhesion and spreading by interfering with its initial steps. Trypsinization not only led to cell detachment from the sensor surface, but also mediated cell signaling, which probably involves EGFR transactivation and Gq-mediated signaling. Unexpectedly, EGF did not trigger obvious asymmetric lateral mass redistribution, at least within the bottom portion of cells. On the other hand, the activation of B2 receptor in A431 induced by bradykinin triggered both vertical and lateral mass redistribution. Numerical analysis provides a link between the DMR signal and the signaling cycle of a Gq-couple receptor. This study highlighted the great potentials of the RWG biosensors for sensing living cells.
Acknowledgments
The authors appreciate valuable suggestions from anonymous reviewers.
References
- 1.Blake, R. A. 2001. Cellular screening assays using fluorescence microscopy. Curr. Opin. Pharmacol. 1:533–538. [DOI] [PubMed] [Google Scholar]
- 2.Taylor, D. L., E. S. Woo, and K. A. Giuliano. 2001. Real-time molecular and cellular analysis: the new frontier of drug discovery. Curr. Opin. Biotechnol. 12:75–81. [DOI] [PubMed] [Google Scholar]
- 3.Abraham, V., D. L. Taylor, and J. R. Haskins. 2004. High content screening applied to large-scale cell biology. Trends Biotechnol. 22:15–22. [DOI] [PubMed] [Google Scholar]
- 4.Gribbon, P., and A. Sewing. 2003. Fluorescence readouts in HTS: no gain without pain? Drug Discov. Today. 8:1035–1043. [DOI] [PubMed] [Google Scholar]
- 5.Balla, T., and P. Varnai. 2002. Visualizing cellular phosphoinositide pools with GFP-fused protein-modules. Sci. STKE. 125:pI3. [DOI] [PubMed] [Google Scholar]
- 6.Cooper, M. A. 2002. Optical biosensors in drug discovery. Nat. Rev. Drug Discov. 1:515–528. [DOI] [PubMed] [Google Scholar]
- 7.Rich, R. L., L. A. Hoth, K. F. Geoghegan, T. A. Brown, P. K. LeMotte, S. P. Simons, P. Hensley, and D. G. Myszka. 2002. Kinetic analysis of estrogen receptor/ligand interactions. Proc. Natl. Acad. Sci. USA. 99:8562–8567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramsden, J. J., S.-Y. Li, J. E. Prenosil, and E. Heinzle. 1994. Kinetics of adhesion and spreading of animal cells. Biotechnol. Bioeng. 43:939–945. [DOI] [PubMed] [Google Scholar]
- 9.Horath, R., H. C. Pedersen, N. Skivesen, D. Selmeczi, and N. B. Larsen. 2005. Monitoring of living cell attachment and spreading using reverse symmetry waveguide sensing. Appl. Phys. Lett. 86. In press.
- 10.Voros, J., R. Graf, G. L. Kenausis, A. Bruinink, J. Mayer, M. Textor, E. Wintermantel, and N. D. Spencer. 2000. Feasibility study of an online toxicological sensor based on the optical waveguide technique. Biosens. Bioelectron. 15:423–429. [DOI] [PubMed] [Google Scholar]
- 11.Quinn, J. G., S. O'Neill, A. Doyle, C. McAtamney, D. Diamond, B. D. MacCraith, and R. O'Kennedy. 2000. Development and application of surface plasmon resonance-based biosensors for the detection of cell-ligand interactions. Anal. Biochem. 281:135–143. [DOI] [PubMed] [Google Scholar]
- 12.Hide, M., T. Tsutsui, H. Sato, T. Nishimura, K. Morimoto, S. Yamamoto, and K. Yoshizato. 2002. Real-time analysis of ligand-induced cell surface and intracellular reactions of living mast cells using a surface plasmon resonance-based biosensor. Anal. Biochem. 302:28–37. [DOI] [PubMed] [Google Scholar]
- 13.Fang, Y., A. M. Ferrie, and G. Li. 2005. Probing cytoskeleton modulation by optical biosensors. FEBS Lett. 579:4175–4180. [DOI] [PubMed] [Google Scholar]
- 14.Fang, Y., A. M. Ferrie, N. H. Fontaine, and P. K. Yuen. 2005. Characteristics of dynamic mass redistribution of EGF receptor signaling in living cells measured with label free optical biosensors. Anal. Chem. 77:5720–5725. [DOI] [PubMed] [Google Scholar]
- 15.Fang, Y., G. Li, and J. Peng. 2005. Optical biosensor provides insights for bradykinin B2 receptor signaling in A431 cells. FEBS Lett. 579:6365–6374. [DOI] [PubMed] [Google Scholar]
- 16.Barak, L. S., S. S. G. Ferguson, J. Zhang, C. Martenson, T. Meyer, and M. G. Caron. 1997. Internal trafficking and surface mobility of a functionally intact β2-adrenergic receptor-green fluorescent protein conjugate. Mol. Pharmacol. 51:177–184. [DOI] [PubMed] [Google Scholar]
- 17.Lu, Z., G. Jiang, P. Blume-Jensen, and T. Hunter. 2001. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol. 21:4016–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu, C.-D., and T. K. Kerppola. 2003. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 21:539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiefenthaler, K., and W. Lukosz. 1989. Sensitivity of grating couplers as integrated-optical chemical sensors. J. Opt. Soc. Am. B. 6:209–220. [Google Scholar]
- 20.Kunz, R. E. 1997. Miniature integrated optical modules for chemical and biochemical sensing. Sens. Actuators B. 38–39:13–28. [Google Scholar]
- 21.Pierres, A., P. Eymeric, E. Baloche, D. Touchard, A.-M. Benoliel, and P. Bongrand. 2003. Cell membrane alignment along adhesive surfaces: contribution of active and passive cell processes. Biophys. J. 84:2058–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burridge, K., K. Fath, T. Kelly, G. Nuckolls, and C. Turner. 1988. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu. Rev. Cell Biol. 4:487–525. [DOI] [PubMed] [Google Scholar]
- 23.Chen, W. T., and S. J. Singer. 1982. Immunoelectron microscopic studies of the sites of cell-substratum and cell-cell contacts in cultured fibroblasts. J. Cell Biol. 95:205–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burmeister, J. S., L. A. Olivier, W. M. Reichert, and G. A. Truskey. 1998. Application of total internal reflection fluorescence microscopy to study cell adhesion to biomaterials. Biomaterials. 19:307–325. [DOI] [PubMed] [Google Scholar]
- 25.Hudder, A., L. Nathanson, and M. P. Deutscher. 2003. Organization of mammalian cytoplasm. Mol. Cell. Biol. 23:9318–9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato, T. K., M. Overduin, and S. D. Emr. 2001. Location, location, location: membrane targeting directed by PX domains. Science. 294:1881–1885. [DOI] [PubMed] [Google Scholar]
- 27.Kholodenko, B. N. 2003. Four-dimensional organization of protein kinase signaling cascades: the roles of diffusion, endocytosis and molecular motors. J. Exp. Biol. 206:2073–2082. [DOI] [PubMed] [Google Scholar]
- 28.Wiley, H. S. 2003. Trafficking of the ErbB receptors and its influence on signaling. Exp. Cell Res. 284:78–88. [DOI] [PubMed] [Google Scholar]
- 29.Schnapp, B. J. 2003. Trafficking of signaling modules by kinesin motors. J. Cell Sci. 116:2125–2135. [DOI] [PubMed] [Google Scholar]
- 30.Wehrman, T. S., C. L. Casipit, N. M. Gewertz, and H. M. Blau. 2005. Enzymatic detection of protein translocation. Nat. Methods. 2:521–527. [DOI] [PubMed] [Google Scholar]
- 31.Tan, C. M., A. E. Brady, H. H. Nickols, Q. Wang, and L. E. Limbird. 2004. Membrane trafficking of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 44:559–609. [DOI] [PubMed] [Google Scholar]
- 32.Lefkowitz, R. J., and E. J. Whalen. 2004. β-arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol. 16:162–168. [DOI] [PubMed] [Google Scholar]
- 33.Barer, R., and S. Joseph. 1954. Refractometry of living cells. Part I. Basic principles. Q. J. Microsc. Sci. 95:399–423. [Google Scholar]
- 34.Beuthan, J., O. Minet, J. Helfmann, M. Herrig, and G. Muller. 1996. The spatial variation of the refractive index in biological cells. Phys. Med. Biol. 41:369–382. [DOI] [PubMed] [Google Scholar]
- 35.Fang, Y., A. M. Ferrie, and G. Li. 2006. Cellular functions of cholesterol probed with optical biosensors. Biochim. Biophys. Acta. 1763:254–261. [DOI] [PubMed] [Google Scholar]
- 36.Tien, P. K. 1977. Integrated optics and new wave phenomena in optical waveguides. Rev. Mod. Phys. 49:361–420. [Google Scholar]
- 37.Horvath, R., J. Voros, R. Graf, G. Fricsovszky, M. Textor, L. R. Lindvold, N. D. Spencer, and E. Papp. 2001. Effect of patterns and inhomogeneities on the surface of waveguides used for optical waveguide lightmode spectroscopy applications. Appl. Phys. B. 72:441–447. [Google Scholar]
- 38.Vicente-Manzanares, M., D. J. Webb, and A. R. Horwitz. 2005. Cell migration at a glance. J. Cell Sci. 118:4917–4919. [DOI] [PubMed] [Google Scholar]
- 39.Zhao, M., A. Dick, J. V. Forrester, and C. D. McCaig. 1999. Electric field-directed cell motility involves up-regulated expression and asymmetric redistribution of the epidermal growth factor receptors and is enhanced by fibronectin and laminin. Mol. Biol. Cell. 10:1259–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eddy, R. J., L. M. Pierini, and F. R. Maxfield. 2002. Microtubule asymmetry during neutrophil polarization and migration. Mol. Biol. Cell. 13:4470–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braun, D., and F. Fromherz. 1997. Fluorescence interface-contrast microscopy of cell adhesion on oxidized silicon. Appl. Phys. A. 65:341–348. [Google Scholar]
- 42.Bereiter-Hahn, J., C. H. Fox, and B. Thoell. 1979. Quantitative reflection contrast microscopy of living cells. J. Cell Biol. 82:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Truskey, G. A., J. S. Burmeister, E. Grapa, and W. M. Reichert. 1992. Total internal reflection fluorescence microscopy (TIRFM) II. Topographical mapping of relative cell/substratum separation distances. J. Cell Sci. 103:491–499. [DOI] [PubMed] [Google Scholar]
- 44.Giebel, K.-F., C. Bechinger, S. Herminghaus, M. Riedel, P. Leiderer, U. Weiland, and M. Bastmeyer. 1999. Imaging of cell/substrate contacts of living cells with surface plasmon resonance microscopy. Biophys. J. 76:509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lobert, S., B. Vulevic, and J. J. Correia. 1996. Interaction of vinca alkaloids with tubulin: a comparison of vinblastine, vincristine and vinorelbine. Biochemistry. 35:6806–6814. [DOI] [PubMed] [Google Scholar]
- 46.Brown, W., and F. Wold. 1973. Alkyl isocyanates as active-site-specific reagents for serine proteases. Reaction properties. Biochemistry. 12:828–834. [DOI] [PubMed] [Google Scholar]
- 47.Brown, W., and F. Wold. 1973. Alkyl isocyanates as active-site-specific reagents for serine protease. Identification of the active-site serine as the site of reaction. Biochemistry. 12:835–840. [DOI] [PubMed] [Google Scholar]
- 48.Darmoul, D., V. Gratio, H. Devaud, and M. Laburthe. 2004. Protease-activated receptor 2 in colon cancer. Trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J. Biol. Chem. 279:20927–20934. [DOI] [PubMed] [Google Scholar]
- 49.Prenzel, N., E. Zwick, H. Daub, M. Leserer, R. Abraham, C. Wallasch, and A. Ullrich. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 402:884–888. [DOI] [PubMed] [Google Scholar]
- 50.Schafer, B., A. Gschwind, and A. Ullrich. 2004. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor to promote migration and invasion. Oncogene. 23:991–999. [DOI] [PubMed] [Google Scholar]
- 51.Wetzker, R., and F.-D. Bohmer. 2003. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell Biol. 4:651–657. [DOI] [PubMed] [Google Scholar]
- 52.Macfarlane, S. R., M. J. Seatter, T. Kanke, G. D. Hunter, and R. Plevin. 2001. Proteinase-activated receptors. Pharmacol. Rev. 53:245–282. [PubMed] [Google Scholar]
- 53.Iwakiri, K., M. Ghazizadeh, E. Jin, M. Fujiwara, T. Takemura, S.-I. Takezaki, S. Kawana, S. Yasuoka, and O. Kawanami. 2004. Human airway trypsin-like protease induce PAR-2 mediated IL-8 release in psoriasis vulgaris. J. Invest. Dermatol. 122:937–944. [DOI] [PubMed] [Google Scholar]
- 54.van Baal, J., J. de Widt, N. Divecha, and W. J. van Blitterswijk. 2005. Translocation of diacylglycerol kinase θ from cytosol to plasma membrane in response to activation of G protein-coupled receptors and protein kinase C. J. Biol. Chem. 280:9870–9878. [DOI] [PubMed] [Google Scholar]
- 55.Marceau, F., and D. Regoli. 2004. Bradykinin receptor ligands: therapeutic perspectives. Nat. Rev. Drug Discov. 3:845–852. [DOI] [PubMed] [Google Scholar]
- 56.Leeb-Lundberg, L. M. F., F. Marceau, W. Muller-Esterl, D. J. Pettibone, and B. L. Zuraw. 2005. International union of pharmacology. XLV. Classification of the kinin receptor family: from molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 57:27–77. [DOI] [PubMed] [Google Scholar]
- 57.Liebmann, C., A. Graness, B. Ludwig, A. Adomeit, A. Boehmer, F.-D. Boehmer, B. Nurnberg, and R. Wetzker. 1996. Dual bradykinin B2 receptor signalling in A431 human epidermoid carcinoma cells: activation of protein kinase C is counteracted by a Gs-mediated stimulation of the cyclic AMP pathway. Biochem. J. 313:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milligan, G. 2003. High-content assays for ligand regulation of G-protein-coupled receptors. Drug Discov. Today. 8:579–585. [DOI] [PubMed] [Google Scholar]
- 59.Lefkowitz, R. J., and E. J. Whalen. 2004. β-arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol. 16:162–168. [DOI] [PubMed] [Google Scholar]
- 60.van Baal, J., J. de Widt, N. Divecha, and W. J. van Blitterswijk. 2005. Translocation of diacylglycerol kinase θ from cytosol to plasma membrane in response to activation of G protein-coupled receptors and protein kinase C. J. Biol. Chem. 280:9870–9878. [DOI] [PubMed] [Google Scholar]
- 61.Jacobson, K., and J. Wojcieszyn. 1984. Translational mobility of substances within the cytoplasmic matrix. Proc. Natl. Acad. Sci. USA. 81:6747–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haugh, J. M., and D. A. Lauffenburger. 1998. Analysis of receptor internalization as a mechanism for modulating signal transduction. J. Theor. Biol. 195:187–218. [DOI] [PubMed] [Google Scholar]
- 63.Clement, F., D. Monniaux, J. Stark, K. Hardy, J. C. Thalabard, S. Franks, and D. Claude. 2001. Mathematical model of FSH-induced cAMP production in ovarian follicles. Am. J. Physiol. Endocrinol. Metab. 281:E35–E53. [DOI] [PubMed] [Google Scholar]
- 64.Luo, S. F., C. T. Tsai, W. B. Wu, S. L. Pan, Y. J. Tsai, and C. M. Yang. 1996. Pharmacological and functional characterization of bradykinin receptors in canine cultured tracheal epithelial cells. Br. J. Pharmacol. 119:439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoffman, J. F., J. J. Linderman, and G. M. Omann. 1996. Receptor up-regulation, internalization, and interconverting receptor states. 271:18394–18404. [DOI] [PubMed]