

Abstract
Angiopoietin-like protein 3 (angptl3), a member of the vascular endothelial growth factor family, was shown to play an important role in regulating lipid metabolism. To elucidate the mechanism by which PPARβ represses angptl3 promoter activity, reporter constructs were prepared and transfection analysis carried out. PPARβ repressed angptl3-Luc promoter activity and activation of PPARβ by L-165041, a PPARβ-specific ligand, increased the extent of repression. The repression by L-165041 was lost in angptl3-Luc plasmids having a deleted or mutated LXRα binding site (DR4). PPARβL405R, deficient in RXRα binding, had no effect on angptl3-Luc promoter activity. PPARβ did not repress the activity of GAL4-LXRα which activates of GAL4DBD TK-Luc independent of RXR. Addition of RXRα completely abolished the repression of angptl3-Luc activity by PPARβ. Mammalian two-hybrid analysis revealed that PPARβ ligand binding enhanced the dissociation of the LXRα-RXRα heterodimer. Gel shift assays also indicated that PPARβ ligand binding increased dissociation of LXRα/RXRα binding to a DR4 oligonucleotide probe; addition of RXRα restored the binding lost by addition of PPARβ. Collectively, these results suggest that the binding of PPARβ-specific ligand enhances the affinity between RXRα and activated PPARβ and thus may regulate angptl3 gene expression through a DR4 element by competing with LXRα for RXRα.
Keywords: PPAR, PPARβ/δ, Triglyceride, Angiopoietin, LXR, RXR
Abbreviations: angptl3, mouse angiopoietin-like 3; LXR, liver X receptor; RXR, retinoid X receptor; PPAR, peroxisome proliferator-activated receptor; FXR, farnesoid-X-receptor; VLDL, very low density lipoprotein; HF, high fat
1. Introduction
The peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily. Three major isoforms of PPARs (α, β/δ, and γ) have been identified (Dreyer et al., 1992; Kliewer et al., 1994), each of which forms an obligate heterodimer with retinoid X receptor (RXRs) (Kliewer et al., 1992a,b). PPARβ and PPARγ control lipid catabolism and storage and are the targets for hyperlipidemic and anti-type 2 diabetes drugs, respectively (Berger et al., 2005; Desvergne et al., 2004). While PPARα and PPARγ function primarily through control of gene expression in liver and adipose tissues, respectively, expression of PPARβ is more ubiquitous and thus elucidating its physiological function is more complicated.
PPARβ appears to have a role in fatty acid oxidation in skeletal muscle and adipose tissue (Fredenrich and Grimaldi, 2005) and in skin homeostasis (Burdick et al., 2006; Di-Poi et al., 2004). Studies using selective agonists suggest that PPARβ regulates serum lipid levels. The PPARβ agonist L-165041 was found to raise plasma cholesterol concentrations associated with HDL particles in insulin resistant db/db mice, while having no effect on either glucose or triglycerides (Leibowitz et al., 2000). L-165041 also reduced LPL activity in white adipose tissue. In addition, a potent and selective PPARβ agonist, GW501516, produced a dose-dependent rise in serum HDL-cholesterol concomitant with a reduction in levels of small-density LDL, fasting triglycerides, and fasting insulin in genetically obese rhesus monkeys (Oliver et al., 2001). However, the mechanism for regulation of serum lipids by PPARβ remains unknown.
Recently, PPARβ-null mice, placed on a high fat (HF) diet, were found to be hypertriglyceridemic as a result of elevated levels of serum triglycerides associated with very low density lipoprotein (VLDL) and this increase was not due to increased triglyceride synthesis in liver (Akiyama et al., 2004). Interestingly, hepatic expression of the genes encoding angiopoietin-like 3 (angptl3) was significantly increased in PPARβ-null mice on a HF diet (Akiyama et al., 2004). angptl3 is a member of a family of secreted growth factors. angptl3-mutant KK/San mice had low levels of plasma lipids (Koishi et al., 2002) and over-expression of angptl3 elicited a rapid increase in circulating levels of plasma cholesterol, triglycerides, and non-esterified fatty acids (Koishi et al., 2002).
PPARs are known to function not only as positive regulators of transcription but also as negative regulators (De Vos et al., 1996; Delerive et al., 1999). However, the mechanisms of transcriptional repression by PPARs remains less clear. While the repression mechanisms by members of other steroid hormone receptor super family have been reported as: (1) binding to specific negative response elements, as is the case for the thyroid hormone receptor on a number of specific genes (Bodenner et al., 1991; Hollenberg et al., 1995), (2) interfering through protein–protein interactions or direct competition for DNA binding, with positively acting transcription factors such as c-jun or C/EBP (Stein and Yang, 1995; Yang-Yen et al., 1990), and (3) by competition for limiting co-factors such as CREB binding protein (CBP)/p300 (Kamei et al., 1996).
In a previous study, the increased expression of angptl3 observed in PPARβ-null mice suggested the possibility that PPARβ represses promoter activity of the angptl3 gene by a direct or indirect mechanism. Therefore, the angptl3 promoter may be a useful model to study the mechanism of negative regulation of gene expression by PPARβ. The present study revealed that PPARβ represses angptl3 promoter activity in a ligand-dependent manner through an LXRα binding site.
2. Materials and methods
2.1. Reagents
T0901317 and chenodeoxycholic acid were purchased from Cayman Chemical and Sigma–Aldrich, respectively. Merck Research Laboratories (Rahway, NJ) generously provided L-165041. 9-Cis-retinoic acid was purchased from Wako Pure Chemical Industries (Japan).
2.2. Construction of plasmids
The expression vector encoding mouse PPARβ was kindly provided by Walter Wahli (Universite de Lausanne, Lausanne, Switzerland). The human RXRα and FXR expression vectors were obtained from Ronald M. Evans (The Salk Institute for Biological Studies, La Jolla, CA). Construction of the reporter PPRE × 3-TK-LUC in which the luciferase gene is under the control of the herpes simplex virus thymidine kinase promoter and three PPREs, was previously described (Kliewer et al., 1992a,b).
The transcription start site of mouse angptl3 was determined in an earlier report (Kaplan et al., 2003). The sequence of the promoter region is found in the public mouse genomic sequence database (GeneBank accession numbers NT 039280). The − 1691 (D5), 552 (D4), 309 (D3), and 96 (D2)/− 1 fragments from the transcriptional start site of the mouse angptl3 promoter containing KpnI and MluI sites in the 5′ - and 3′ -end of the primers were amplified by PCR and cloned into the luciferase reporter vector, pGL3 basic (Promega, Madison, WI). Point-mutations were introduced into the DR4 site in the D4 constructs by PCR-based, site-directed mutagenesis using the following two primer pairs, Mut1; 5′ -GGGTTACATTCATGCAAGTTAGCACAGCTTAATG-3′ and 5′ -CTTGCAT-GAATGTAACCCTCCCCCATGTTGAGTT-3′ (mutations in the distal DR4 are underlined), Mut2; 5′ -TGGGGGAGGATTACATTCGTGCAAGTTAGCACA-GC-3′ and 5′ -AATGTAATCCTCCCCCATGTTGAGTTAGA-3′ (mutations in the proximal DR4 site are underlined). Replacement of a single amino acid in the mouse PPARβ and human RXRα proteins was created by PCR-based, site-directed mutagenesis using the following two primer pairs, PPARβC93S; 5′ -TGCGAGGGGAGCAAGGGCTTCTTCCGCCGGACAATC-3′ and 5′ -GCC-CTTGCTCCCCTCGCACGCGTGGAC-3′, PPARβ L394R; 5′ -AGCCAG-TACCGCTTCCCCAAGCTGCTGCAGAAGAT-3′ and 5′ -GGGGAAGCGGT-ACTGGCTGTCAGGGTGGTT-3′, PPARβL405R; 5′ -ATGGCAGACCGCCG-GCAGCTGGTCACTGAGCATGCCCA-3′ and 5′ -CTGCCGGCGGTCTGC-CATCTTCTGCAGCAG-3′ (mutations in the mouse PPARβ are underlined), RXRα Y397A; 5′ -AAGGTCGCTGCGTCCTTGGAGGCCTACTGCAAGCA-3′ and 5′ -CAAGGACGCAGCGACCTTCTCCCTCAGCGCCTCCA-3′, RXRαK417A; 5′ -TTCGCTGCGCTCTTGCTCCGCCTGCCGGCT-3′ and 5′ -GAGCAAGAGCGCAGCGAACCTTCCCGGCTGCTC-3′ (mutations in the human RXRα are underlined). These mutants were characterized in earlier reports (Juge-Aubry et al., 1995; Vivat-Hannah et al., 2003).
The GAL4DBD-TK and TK Luc plasmids were provided by David D. Moore (Baylor College of Medicine, Houston, TX). The complete open reading frame (ORF) of mouse LXRα was amplified by PCR from a mouse liver cDNA library by using gene-specific primers containing BamHI (5′) and XbaI (3′) sites, and cloned into the pBIND Vector (Promega). Mammalian two-hybrid analysis was performed by use of the CheckMateTM Mammalian Two-Hybrid System kit (Promega, Madison, WI). The complete open reading frame (ORF) of human RXRα was amplified by PCR from the human RXRα expression vector described above by using gene-specific primers containing XbaI (5′) and KpnI (3′) sites, and cloned into pACT vector. The complete PCR-amplified product was subjected to DNA sequencing to verify the absence of errors.
2.3. Cell culture and transient transfection assays
HepG2 cell lines were cultured at 37 °C in Dulbecco’s modified Eagle’s medium (Biosource, Camarillo, CA) containing 10% fetal bovine serum (Gemini, Woodland, CA) and 100 units/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA). Cells were seeded in 24-well tissue culture plates and grown to 90–95% confluency. For transfections, 300 ng/well pGL3 angptl3-Luc constructs and 20 ng/well phRL/TK (Promega) were transfected. For cotransfections, 300 ng/well of the PPARβ (pSG5-PPARβ) and RXRα (pSG5-RXRα) expression plasmids were used. All transfections were performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. The medium was changed 6 h after transfection to fresh medium including each ligand at concentrations described in the figure legends and after 48 h, the cells were harvested. Luciferase activity was measure by the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. HEK 293 cells were also cultured and transfected as similar conditions.
2.4. Western blotting
Cell extracts from HEK 293 cells were preparated by directly addition of 2 × sample buffer (125 mM Tris–HCl, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue) to cells after washing with PBS. Total cell extract proteins were assayed by use of the BCA protein assay (Pierce Chemical Co., Rockford, IL) and 5 μg was subjected to electrophoresis on a 10% SDS-polyacrylamide gel, transferred to Immobilon-P membranes (Millipore, Bedford, Mass.) and probed according to the manufacturer’s recommendations with anti-PPARβ (N20; Santa Cruz Biotechnology, Inc. Santa Cruz, CA). An enhanced chemiluminescence detection system was used to visualize immunoreactive proteins (Amersham, Inc., Arlington Heights, IL).
2.5. In vitro translation and electrophoretic mobility shift assay
Murine PPARβ, LXRα and human RXRα proteins were synthesized in vitro by the TNT-coupled transcription/translation system kit (Promega) with 1 μg of pSG5-PPARβ, pcDNA-LXRα and pSG5-hRXRα. An oligonucleotide (obtained from SIGMA Genosys) consensus DR4 element as probe was synthesized with the following sequence: 5′ -GGAGGGTTACATTCGTGCAAG-3′ along with an oligonucleotide of complementary sequence. The oligonucleotides were mixed (50 ng/μl final concentration) and denatured by heating to 95 °C for 10 min in 0.1 M Tris–HCl, 50 mM MgCl2 (pH 7.9) and allowed to anneal by slowly cooling to room temperature. The annealed oligonucleotides were end-labeled with [γ-32P]ATP using T4 polynucleotide kinase according to the supplier’s (New England Biolabs, Ipswich, MA) instructions. Each sample after in vitro translation reaction was added in the components as volume decreased in figure legends. After addition of 1 μM L-165041, a 20-min at room temperature was incubated and then 20,000 cpm of the labeled DR4 probe was added, and the incubation was continued for a further 20 min. The conditions of electrophoresis were described previously (Sinal et al., 2001).
3.Results
3.1. PPARβ represses the promoter activity of angptl3
To examine whether PPARβ represses angptl3 promoter activity, a reporter plasmid (angptl3-Luc) including a promoter region of angptl3 (− 1700/− 1 bp) was cotransfected onto HepG2 cells with a PPARβ expression plasmid, pSG-PPARβ (Fig. 1). The angptl3-Luc activity was suppressed by PPARβ by 63% and the extent of suppression was slightly increased by the addition of L-165041, a PPARβ-specific ligand (52% without PPARβ). A search for transcription factor binding sites in the region (− 1700/− 1 bp) of the angptl3 gene revealed the presence of putative sites for HNF-1α, LXRα, NF-κB and C/EBP. Earlier studies revealed that LXRα is a major regulator of hepatic angptl3 gene expression (Inaba et al., 2003; Kaplan et al., 2003). Interestingly, a typical direct repeat 1 (DR1), PPAR-response element (PPRE) was not observed in the region. These results indicate that PPARβ is capable of repressing activity of the angptl3 promoter independent of a PPRE.
Fig. 1.
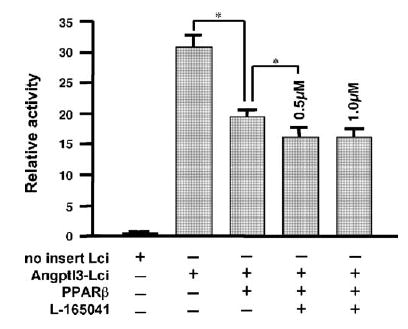
Effect of PPARβ expression on promoter activity of the angptl3 gene. HepG2 cells were transfected with the pSG-PPARβ vector and the angptl3-Luc plasmid that includes the region from − 1 to − 1700 bp from angptl3. Following 6 h after transfection, the medium was changed to fresh medium including L-165041 (0.5 or 1.0 μM) and 48 h after transfection, the cells were harvested and luciferase activity measured. *p < 0.05.
3.2. PPARβ represses the angptl3 promoter activity through a DR4, LXRα binding site
To identify the cis-element responsible for ligand-dependent repression by PPARβ, reporter constructs with serially deletion of 5′ -flanking DNA of angptl3 were prepared. The addition of L-165041 repressed the activity of the luciferase reporter constructs D5, D4 and D3, whereas construct D2, lacking the LXR-binding site (direct repeat 4 element, DR4), had dramatically decreased basal activity without L-165041 and was not repressed by L-165041 (Fig. 2A). The activity of luciferase plasmids D5 and D4, that included the LXR-binding site (DR4), were markedly induced by the addition of T0901317, an LXRα-specific ligand (Fig. 2B). The induced activities were substantially decreased by the addition of L-165041 as compared with the activity, which was shown in Figs. 1 and 2A, in the absence of LXRα ligand. The luciferase construct D2, lacking the DR4 motif, completely lost induction of activity by T0901317 and repression by L-165041 but also showed a marked decrease of luciferase activity to nearly background levels (Fig. 2A and B). Thus, it is possible that loss of repression of the D2 construct by L-165041 is due to background promoter activity, although activity of the D2 construct is sufficiently higher than that of the luciferase vector without the promoter. To exclude this possibility, a point mutation was introduced into the DR4 element in the D4 construct (Fig. 2C). The relative inhibition of T0901317-induced luciferase activity by PPARβ + L-165041 was wild-type (D4); 82%, Mut-1; 51% and Mut-2; 24%. The degree of repression by PPARβ was correlated with T0901317-induced activity. Further, the luciferase activity induced by T0901317 was repressed in a pSG-PPARβ plasmid concentration-dependent manner (Fig. 2D); 60 or 94% inhibition was observed with 25 ng of the pSG-PPARβ expression construct DNA with or without 1 μM L-165041, respectively. These results suggest that PPARβ represses angptl3 promoter activity through the DR4 LXR binding site and LXR ligand activation. Further, activation of PPARβ by L-165041 additively enhances the repression of promoter activity activated by LXRα ligand.
Fig. 2.
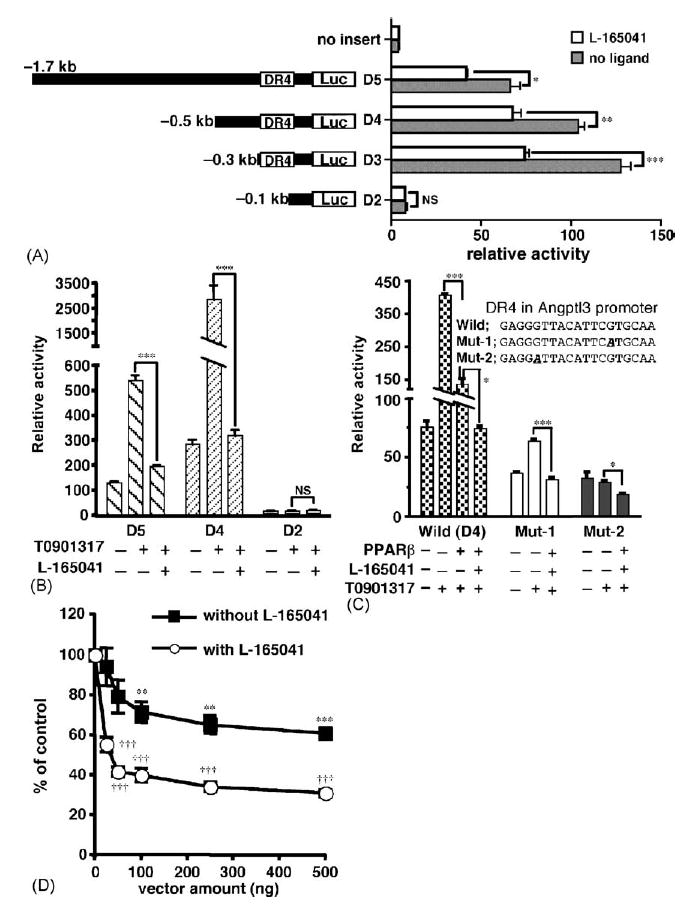
PPARβ indirectly represses the promoter activity of angptl3 by impairing LXRα-signaling. (A) The repression of PPARβ is mediated by a DR4 element in the mouse angptl3 promoter. HepG2 cells were transfected with the pSG-PPARβ vector and the reporter plasmids containing serially deleted angptl3-Luc plasmid. After transfection, the cells were treated with or without 1 μM L-165041 before harvest. (B) PPARβ efficiently repressed induction of the angptl3 promoter by the LXRα ligand. HepG2 cells were transfected with the pSG-PPARβ vector and each deletion mutant. Cells were treated with or without 1 μML-165041 and 100 nM T0901317. (C) HepG2 cells were transfected with the reporter plasmids (D4 plasmid in A) containing a point-mutation of the DR4 sequence in the angptl3 promoter. The experimental conditions are as described in (B). (D) PPARβ concentration-dependent repression of induction of the angptl3 promoter by the LXRα ligand. HepG2 cells were transfected with pSG-PPARβ vector (25, 50, 100, 250 and 500 ng) and D4 deletion mutant. Cells were treated with or without 1 μM L-165041 and 100 nM T0901317. The results are displayed as % values of control without pSG-PPARβ vector. Significant differences were as compared to control. *p < 0.05; **p < 0.01; ***p < 0.001; †††p < 0.001; NS, no significant difference.
3.3. A PPARβ mutant lacking RXRα binding does not suppress angptl3 promoter activity
In an earlier report, point mutations were created in the human PPARα changing codon 122 (within the P box region) from a Cys to Ser or codon 433 (within the heptad repeat region) from a Leu to Arg (Juge-Aubry et al., 1995). The hPPARαC122S was able to form a heterodimer with RXRα but did not activate transcriptional activity by the addition of specific ligand, while hPPARαL433R could neither heterodimerize with RXRα nor be activated by ligand. The amino acid residue replaced in the hPPARα is conserved in mouse PPARβ (Fig. 3A). Therefore, to elucidate the mechanism for repression of the angptl3 promoter by PPARβ, these mutants were introduced into the mouse PPARβ cDNA. First, the post-translational stability for the mutated proteins was examined. By use of a PPARβ antibody that specifically reacts with PPARβ, all mutants transfected to HEK 293 cells were found to be expressed at similar levels (Fig. 3a and b). The luciferase activity, under the thymidine kinase promoter control, including the PPRE reporter plasmid, was markedly induced by addition of L-165041 with PPARβ wild-type or L394R (used as a control for the heptad repeat region) constructs but not with PPARβC93S or PPARβL405R, similar to the earlier report (Fig. 3C left) (Juge-Aubry et al., 1995). PPARβ wild-type, PPARβC93S and PPARβL394R effectively repressed the angptl3 promoter activity induced by T0901317. Contrary to these mutants, PPARβL405R, lacking binding with RXRα, had no effect on angptl3 promoter activity (Fig. 3C right).
Fig. 3.
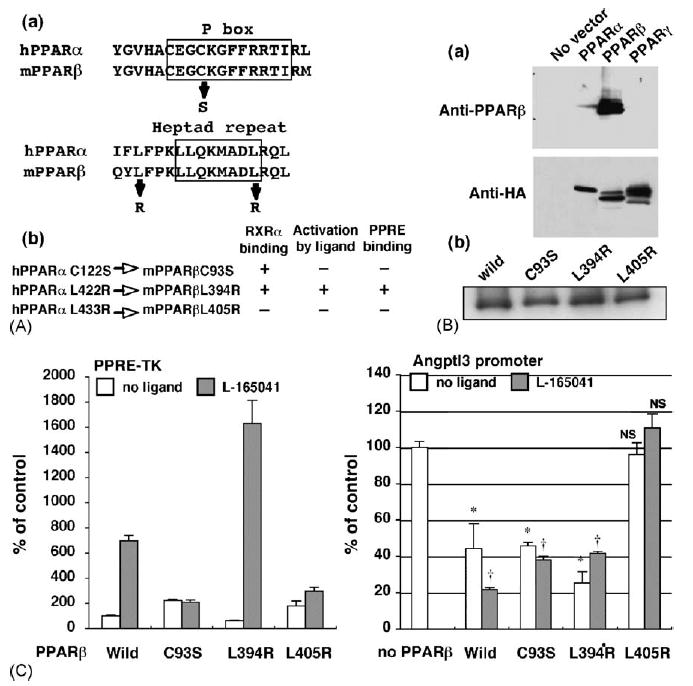
PPARβ mutant lacking RXRα binding capacity has no effect on the angptl3 promoter activity. (A-a) The human PPARα sequences of the P box region and heptad repeat motif were aligned with mouse PPARα. The substituted amino acids are shown as arrows. (A-b) Characterization of each PPARβ point mutant. The amino residues mutated in human PPARα protein were completely conserved in mouse PPARβ. These mutants were characterised in an earler report (Juge-Aubry et al., 1995). (B-a) Each PPAR isoform expression vector containing C terminal HA epitop tag was transfected to HEK293 cells. Western blot using the cell lysates was performed by PPARβ (upper panel) or HA (lower panel) antibody. (B-b) The same levels of full-length PPARβ wild-type and each mutant protein expression were observed after transient expression in HEK293 cells as assayed by Western blotting. (C) The effect of mutanted PPARβ on the PPRE-TK (left) and angptl3 (right) promoter activity. HepG2 cells were transfected with the angptl3 promoter (D4 mutant as shown in Fig. 2A) or thymidine kinase promoter including the PPRE sequence plasmids. Cells were treated with or without 1 μM L-165041 and 100 nM T0901317 (when the angptl3 promoter was used). The results are displayed as % values of PPARβ wild-type vector without L-165041 (PPRE-TK) or without L-165041 and PPARβ vector (angpyl promoter). Significant differences from control (without PPARβ): *p < 0.001; †p < 0.001; NS, no significant difference.
3.4. Limited RXRα is responsible for the repression of angptl3-Luc activity by PPARβ
Recently, it was reported that PPARα suppresses SREBP-1c promoter activity through reduction of LXRα/RXRα heterodimer formation (Ide et al., 2003; Yoshikawa et al., 2003).
To assess the role of RXRα in the repression effect of PPARβ on the angptl3 promoter, an RXRα expression vector was cotransfected with the PPARβ vector (Fig. 4A and B). RXRα completely abolished repression of angptl3-Luc activity by PPARβ either with or without L-165041 or T0901317. Interestingly, the addition of RXRαinduced luciferase activity in the presence of PPARβ + L-165041. A variant mouse RXRα, RXRαY402A was reported to have enhanced homodimerization and weak heterodimerization with the thyroid hormone receptor (TR), Vitamin D receptor (VDR) and PPARγ (Vivat-Hannah et al., 2003). Therefore, the human RXRαY397A and RXRαK417A (as a control) was examined to determine its effect on repression of angptl3-Luc activity by PPARβ. Cotransfection of wild-type RXRα or RXRαK417A completely restored the angptl3-Luc activity repressed by PPARβ; the activity was only partially restored by RXRαY397A by 53% (Fig. 4C). Formation of the RXR/LXR heterodimer is required for efficient binding to the DR4 element (Ide et al., 2003; Yoshikawa et al., 2003). To examine whether PPARβ represses the activity by modulating binding of LXRα to the DR4 element, the LXRα ORF was fused to the yeast transcription factor GAL4 to produce a DR4- or constitutive LXRα-independent system and the transcriptional activity was measured with a reporter plasmid controlled by the GAL4 DNA binding element (Fig. 4D). GAL4-LXRα significantly induced the activity after addition of T0901317. However, the repression by PPARβ, either with and without L-165041, was completely lost (Fig. 4D).
Fig. 4.
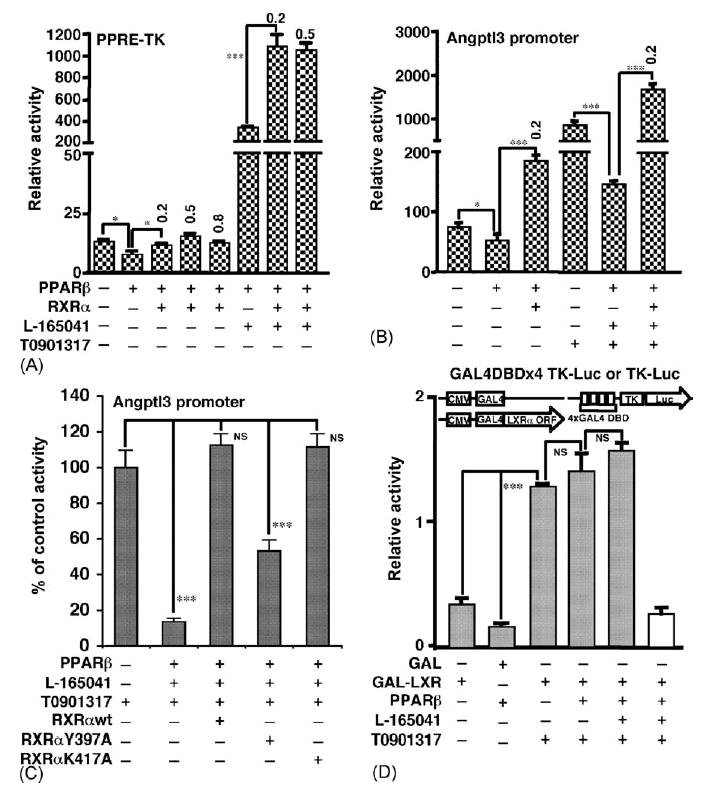
RXRα expression attenuates repression of the angptl3 promoter by PPARβ. Two reporter genes were used: (A) PPRE-TK as shown in Fig. 3C, and (B) the angptl3 promoter (D4 mutant as shown in Fig. 2A). The amount (μg) of RXRα plasmid is shown in the figure (0.2 or 0.5 μg). (C) RXRα with a low heterodimerization potential with PPARs has impaired attenuation of the angptl3 promoter. RXRαY397A and RXRαK417A were characterized in an earlier report (Vivat-Hannah et al., 2003). The amount of RXRα, RXRαY397A and RXRαK417A added was 0.05 μg. The D4 mutant of the angptl3 promoter was used as a reporter plasmid. The results are displayed as % values of control without PPARβ vector. (D) PPAPβ has no effect on the transcriptional activity of the GAL4-fused LXRα. HepG2 cells were transfected with the GAL4DBDx4 TK-luciferase and GAL4-fused LXRα plasmids without or with PPARβ and cells were treated without or with 1 μM L-165041 and 100 nM T0901317. TK-luciferase that does not include GAL4DBD (shown as white bar) and GAL4 plasmids, were used as negative control. *p < 0.05; ***p < 0.001; NS, no significant difference.
3.5. PPARβ ligand binding enhances the dissociation of the LXRα -RXRα heterodimer
The present results revealed that L-165041 enhanced repression of angptl3-Luc activity by PPARβ. To assess the effect of ligand on heterodimerization of LXRα and RXRα, mammalian two-hybrid analysis was performed (Fig. 5). In this experiment, HEK 293 cells that do not express any PPAR subtypes, were used in order to eliminate any potential interference by endogenous PPAR expression (Guardiola-Diaz et al., 1999). Indeed, cotransfection of VP16 (herpes simplex virus VP16 activation domain)-fused RXRα and GAL4-fused LXRα plasmids activated the luciferase activity resulting from the GAL4DBDx4 TK plasmid, indicating LXRα-RXRα heterodimerization in HEK293 cells; VP16 plasmid without the RXRα ORF was inactive (Fig. 5A). Under conditions of LXRα-RXRα heterodimerization, the addition of PPARβ without L-165041 decreased the luciferase activity and the extent of suppression was increased by the addition of L-165041 (Fig. 5B). In contrast to PPARβ, the addition of PPARβL405R, showing no heterodimerization with RXRα (Fig. 3C), had no effect on luciferase activity with or without L-16504 (Fig. 5B). Therefore, the decrease of luciferase activity by L-165041 suggests disassociation of GAL4-LXRα and VP16-RXRα by competition of VP16-RXRα with ligand-bound PPARβ. These results suggest that the L-165041 may increase the affinity of PPARβ for RXRα. Furthermore, the disassociation of LXRα-RXRα heterodimers by another type II nuclear receptor was compared with PPARβ using the mammalian two-hybrid system (Fig. 5C). Farnesoid X Receptor (FXR) is also a partner receptor for RXRα. FXR without ligand did not lead to a decrease in luciferase activity although PPARβ without lig-and led to a decrease in luciferase activity in a dose-dependent manner. While FXR transfected in the presence of the specific ligand, chenodeoxycholic acid, could significantly decrease the activity in a dose-dependent manner. However, the degree of activity decreased by FXR was clearly lower as compared with the liganded PPARβ. C/EBPα, a basic region-leucine zipper type transcription factor, showing no heterodimerization with RXRα as a negative control had no effect on luciferase activity. These results suggest that each nuclear receptor may have a different affinity for RXRα.
Fig. 5.
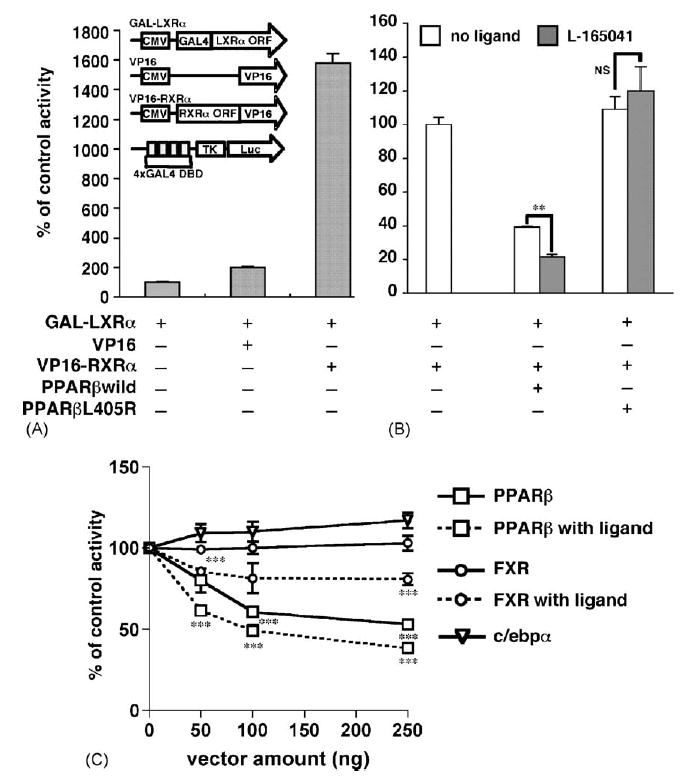
Ligand-binding PPARβ enhances the dissociation of LXRα-RXRα heterodimers in the mammalian two-hybrid system. (A) HEK293 cells were transfected with 200 ng of GAL4-fused LXRα, VP16-fused RXRα and GAL4DBDx4 TK-luciferase plasmids. TK-luciferase that does not include GAL4DBD (shown as a white bar) and VP16 without RXRα ORF plasmids were used as negative controls. The results are displayed as % values of GAL4-LXRα + TK-luciferase activity. (B) PPARβ and PPARβL405R mutants shown in Fig. 3 were additively transfected using the experimental conditions described in (A) and HEK293 cells were treated without or with 1 μM L-165041. The results were displayed as % values of GAL4-LXRα + VP16-RXRα + GAL4DBDx4 TK-luciferase activity. (C) PPARβ and FXR show a different dose–response for dissociation of LXRα-RXRα heterodimers. PPARβ, FXR and C/EBPα (as a negative control) were additively transfected in a dose dependent manner using the experimental conditions described in (A) and HEK293 cells were treated without or with 1 μM L-165041 or 50 μM chenodeoxycholic acid. The results are displayed as % values of GAL4-LXRα + VP16-RXRα + GAL4DBDx4 TK-luciferase activity. **p < 0.01; ***p < 0.001; NS, no significant difference.
3.6. PPARβ interferes with LXRα /RXRα binding to the DR4 element in the angptl3 promoter
To elucidate the role of PPARβ in LXRα/RXRα binding, electrophoretic mobility shift assays using a DR4 probe derived from the mouse angptl3 promoter sequence were performed (Fig. 6). The PPARβ/RXRα heterodimer could not directly bind to the DR4 but the addition of PPARβ to the reaction components including LXRα and RXRα clearly decreased the extent of DR4-LXRα/RXRα binding in a concentration-dependent manner. This decreased binding was additively enhanced by the addition of L-165041 (Fig. 6A). To examine whether limited RXRα alters the degree of binding of the DR4 to LXRα/RXRα, the amount of RXRα was increased in the presence of PPARβ and L-165041. After addition of RXRα, the DR4-LXRα/RXRα complex that was decreased in the presence of PPARβ and L-165041, was almost completely restored (Fig. 6B). The addition of 9-cis-retinoic acid had no effect on the binding restored by RXRα (data not shown). These results suggest that PPARβ decreases LXRα/RXRα binding to the DR4 element by competing with LXRα for RXRα.
Fig. 6.
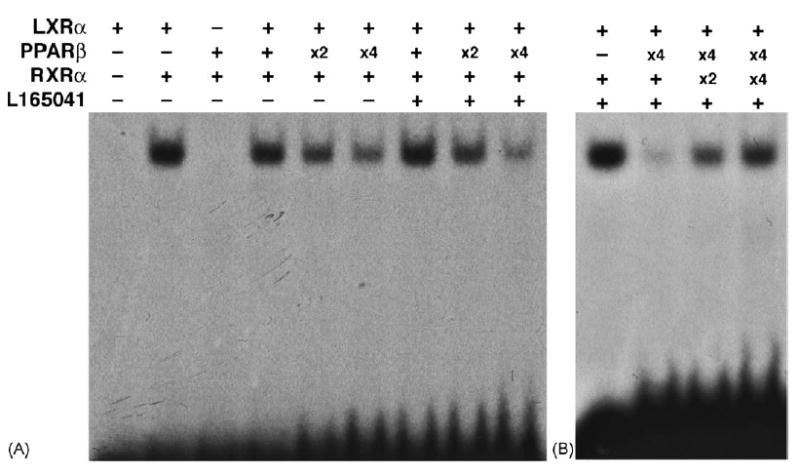
PPARβ interferes with LXRα/RXRα binding to theDR4 probe, derived from the angptl3 promoter, by competing with LXRα for RXRα. (A) Electrophoretic mobility shift assays were carried out by using 32P-labeled DR4 oligonucletide. The components containing 0.5 μg each of PPARβ, LXRα and RXRα, produced by in vitro translation, were incubated with or without 1 μg L-165041 under conditions described in Section 2. The amount of PPARβ after in vitro translation was serially increased 2 (X2)- or 4 (X4)-fold higher than the initial amount (0.5 μg). (B) LXRα/RXRα binding decreased by PPARβ is attenuated in an RXRα amount-dependent manner. The amount of RXRα after in vitro translation was serially increased 2 (X2)- or 4 (X4)-fold higher than the initial amount (0.5 μl).
4. Discussion
PPARs are well established as positive regulators of transcription. However, only a few studies have demonstrated repression of transcription by PPARs, notably PPARγ, although the mechanism of repression is less clear that with positive gene activation. BRL49653, a PPARγ ligand, decreased leptin mRNA levels in rat adipose in a dose-dependent manner and the reporter construct including 5′ -upstream region of leptin was repressed by BRL49653 + PPARγ (De Vos et al., 1996; Hollenberg et al., 1997). The results obtained by use of reporter constructs with serially deletion of 5′ -flanking DNA of leptin suggested that the repression by PPARγ is due to, at least in part, to functional antagonism of liganded PPARγ on C/EBPα trans-activation through a C/EBP( consensus sequence containing in the leptin promoter. However, the precise mechanism for the antagonism by PPARγ such as direct or indirect interaction between two transcription factors remains unclear. Further, the glucagon gene promoter is strongly activated by PAX6 as the transcription factor expressed early in pancreatic development defining endocrine cell lineages (St-Onge et al., 1997). PPARγ, expressed in glucagon-producing α-cells of the endocrine pancreas, repressed expression of the glucagon gene. Therefore, PPARγ directly binds to Pax6 and represses the PAX6-dependent transcriptional activity (Schinner et al., 2002). These studies suggest a repression mechanism by PPARs as interfering, through protein–protein interactions or direct competition for DNA binding, with positively acting transcription factors.
In the present study, PPARβ repressed the mouse angptl3 romoter activity in vitro, even though a PPAR response element was not found in the 5′ -upstream region or introns of angpt13. Therefore, studies were undertaken to determine whether PPARβ directly or indirectly participates in repression of the angptl3 promoter. LXRα is the only known positive regulator of this gene (Inaba et al., 2003; Kaplan et al., 2003). The present study showed that the LXRα DR4 binding site located in the angptl3 promoter mediates repression by PPARβ. This repression is completely abolished by the addition of RXRα and increased by addition of a PPARβ-specific ligand.
A series of single mutations in PPARβ or RXRα were created to exclude the possibility of artifacts due to the nature of the transient transfection assays. The present data revealed that PPARβ 405R, corresponding to hPPARαL433R, which could neither heterodimerize with RXRα nor be activated by ligand, had no effect on angptl3 promoter activity. Nevertheless, a control mutation, PPARβL394R, located only 11 amino acids from the 405 Leu residue (but out of heptad repeat region) could repress the angptl3 promoter activity similar to the wild-type PPARβ. Interestingly, activation of a PPRE-TK luciferase reporter by PPARαL394R with L-165041 was actually higher than that of PPARβ. Furthermore, the involvement of RXRα in repression by PPARβ was also examined using mutants for RXRα. RXRαK417A could attenuate angptl3 promoter activity repressed by PPARβ similar to wild-type RXRα while RXRαY397A showed impaired attenuation. RXRαY397A did not completely lose its ability to attenuate the promoter activity suggesting that it may retain the potential for heterodimerization with PPARβ. The results of mammalian two-hybrid analysis between mouse RXRαY402A and PPARγ indicated that this variant could weakly heterodimerize with RXRα (Vivat-Hannah et al., 2003). Thus, the results obtained with PPARβ and RXRα mutants suggest that repression of the mouse angptl3 promoter by PPARβ is mediated by RXRα competition between PPARβ and LXRα resulting in decreased activation of the angptl3 through DR4 binding.
The experiments with these mutants cannot exclude the confounding effects of endogenous PPARβ or RXRα. Therefore, a GAL4-LXRα chimera system was used to determine possible DR4-independent effects of PPARα. The use of a GAL4-LXRα system allowed for a more direct assessment of PPARβ contribution to DR4 binding by LXRα. In this system, RXRα is dispensable for the binding of LXRα to the DR4 site. In contrast to results with the angptl3 promoter, the repression by PPARβ was completely lost in this system. The results of electrophoretic mobility shift assays directly demonstrated that PPARβ decreases the binding of LXRα/RXRα to the DR4 and that the binding decreased in the presence of PPARβ + L-165041, was restored in a RXRα concentration-dependent manner. Recently, it was demonstrated that PPARα and PPARγ in a dose-dependent manner inhibited SREBP-1c, a known LXRα target gene (Ide et al., 2003; Yoshikawa et al., 2003). Similar to the RXRα attenuation of the inhibition by PPARβ of the angptl3 promoter luciferase construct PPARα and PPARγ inhibited LXRα/RXRα binding to the LXRα binding site in the SREBP1c promoter (Ide et al., 2003; Yoshikawa et al., 2003). The present results suggest that PPARβ competes with LXRα for limited cellular RXRα leading to lower formation of the transcriptionally competent LXRα/RXRα heterodimer and its binding to the mouse angptl3 promoter DR4 element.
The reporter activity repressed in the presence of PPARβ + L-165041 was significantly elevated by the presence of RXRα. In addition, 9-cis-retinoic acid had no effect on LXRα/RXRα binding to the DR4 element thus suggesting that heterodimerization with RXRα not only facilitates binding to the DR4 element but also contributes to the additive effect of LXRα on transcriptional activity. Indeed, recent studies suggested the possibility of additive effects of RXRα. RXR promotes the dissociation of a corepressor from its heterodimeric partner, thyroid hormone receptor (Li et al., 2002). Liganded Vitamin D receptor allosterically modifies RXR in the absence of RXR ligand and as result of this modification, heterodimerized RXR acquired the capacity to recruit coactivators (Bettoun et al., 2003). However, it is not clear whether heterodimerization with RXRα causes the same effect for LXRα.
Ligand-occupied nuclear receptors generally activate transcription by recruiting coactivators (Glass and Rosenfeld, 2000). On the other hand, unliganded receptors actively repress transcription through nuclear receptor corepressors (Glass and Rosenfeld, 2000; Hu et al., 2003). Indeed, the specific ligand for PPARβ was reported to modulate the association with repressor. The nuclear receptor corepressor (NCoR) was isolated as a specific repressor of unliganded PPARβ. NCoR strongly interacts with the ligand-binding domain of PPARβ and a PPARβ-specific ligand antagonized PPARβ–NCoR interactions (Krogsdam et al., 2002). These studies suggest that ligand binding to nuclear receptors allows modulation of co-associated proteins with nuclear receptors probably through modulating conformational changes.
The present study provides new insights into the effect of ligand on PPARβ. The PPARβ ligand L-165041 enhanced repression of the angptl3 promoter and the ligand effect was independent of direct transcriptional activation by PPARβ as revealed by studies with the PPARβC93S and PPARβL394R variants. Further, ligand-bound PPARβ more efficiently represses the promoter activity activated by LXRα ligand as compared with non-LXRα ligand and effectively competed with the LXRα-RXRα heterodimer and LXRα binding to the angptl3 DR4. The results of the mammalian two hybrid system showed that the addition of L-165041 leads to enhance disassociation between LXRα and RXRα in the presence of PPARβ. Interestingly, another type II nuclear receptor, FXR promoted disassociation only by the addition of the FXR ligand chemodeoxycholic acid; there was no effect in the absence of ligand. These data suggest that the affinity of unliganded FXR for RXRα is the lower than that of LXR for RXRα, while liganded FXR is capable of disassociating the heterodimerization of LXR due to its enhanced affinity for RXRα. Thus, type II nuclear receptor activation by a specific ligand not only activates transcription but also may produce conformational changes that leads to a higher affinity for RXRα As support for this notion, 1,25-dihydroxyvitamin D3 also enhances heterodimerization of the vitamin D3 receptor with RXRα (Cheskis and Freedman, 1994, 1996).
It remains unclear whether the mechanism involving RXR competition occurs in vivo. Recently, gene expression profiles generated from livers of PPARα ligand Wy-14,643-treated wild-type and PPARα-null mice were compared with the profiling data from the wild-type and LXRα/LXRβ-null mouse liver after exposure to the LXR ligand T0901317 (Anderson et al., 2004). The results demonstrated that only one of 64 genes constitutively regulated by PPARα was repressed by T0901317 treatment, suggesting that PPARα and LXRα activate an overlapping set of genes. This is in contrast to our proposal and an earlier study (Ide et al., 2003). The in vivo appearance of RXR competition appears to be due to different affinities for RXR or different expression levels of competitor receptors. Thus, the discrepancy could be as fellows. Ide et al. carried out a series of in vivo studies using livers from fasted mice (Ide et al., 2003). This condition actually induces the levels of hepatic PPARα. The angptl3 mRNA was decreased only in livers from high fat fed mice (Akiyama et al., 2004). Since other reports demonstrated that fatty acid metabolites directly bind to PPARβ (Fyffe et al., 2006; Krey et al., 1997; Xu et al., 1999) or triglyceride-rich very low-density lipoprotein sufficiently activates PPARβ (Chawla et al., 2003; Lee et al., 2006), PPARβ in mice fed a high diet may be more active and have a higher affinity for RXR as compared with PPARβ expressed in mice fed normal low-fat diets. Therefore, the in vivo appearance of RXR competition appears to be conditionally limited. More studies are needed to elucidate whether the present mechanism is also functional in vivo.
Since RXR is also a partner for other class II nuclear receptors, the RXR competition model is not likely to be specific for PPARβ. Although PPARα is expressed in liver, it is unclear whether hepatic PPARα and PPARβ cooperate in repression of the angptl3 gene. However, each class II nuclear receptor may have a different affinity for RXRα. Receptors having a high affinity appear to be predisposed to potential RXR competition as compared to those with lower RXR affinity. Indeed, the present study revealed that liganded FXR is likely to have less affinity for RXRα than PPARβ. In an earlier report, activation of PPARγ by the ligands ciglitazone and troglitazone could decrease thyroid hormone receptor-mediated type X collagen gene expression that was restored upon overexpression of RXR (Wang et al., 2005). Therefore, the PPARs may have a higher affinity for RXR and cause RXR competition as compared to other RXR partners such as thyroid hormone receptor or FXR.
In summary, activated PPARβ represses mouse angptl3 promoter activity by interfering with binding of LXR to DR4 as presented in Fig. 4. The present results also suggest the possibility that increased expression or activation of PPARβ indirectly interferes the function of other class II nuclear receptor. The interference may partially contribute to the effects of PPARβ-specific ligands as previously suggested (Leibowitz et al., 2000; Oliver et al., 2001) (Fig. 7).
Fig. 7.
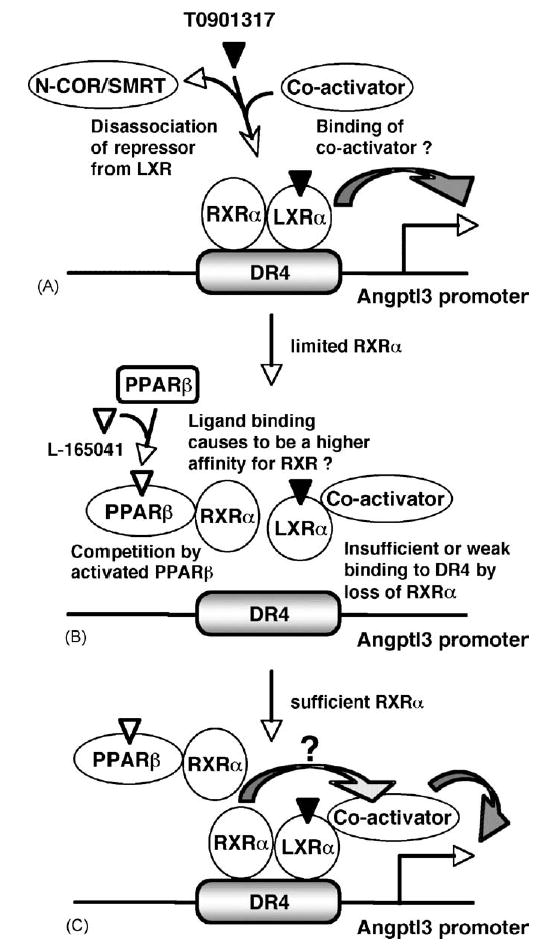
A simplified repression model of angptl3 promoter activity by PPARβ. (A) LXRα regulates the angptl3 promoter activity (Inaba et al., 2003; Kaplan et al., 2003). RXRα facilitates binding of type 2 nuclear receptors to their response elements. Activation by LXRα ligand disassociates repressors N-COR/SMART (Hu et al., 2003) and promotes association with co-activator (Mangelsdorf and Evans, 1995). (B) PPARβ is also a partner for RXRα. Therefore, activated PPARβ interfers the binding of LXRα to the LXR response element DR4 in the angptl3 promoter. L165041-activated PPARβ may result in higher affinity binding to RXRα. (C) Increased cellular levels of RXRα overcomes the inhibition by PPARβ leading to sufficient LXRα/RXRα for binding to the DR4. Thus, RXRα not only facilitates binding to the DR4 element but also may contribute to the additive effect of LXRα-transcriptional activity through an unknown pathway.
Acknowledgments
K.M. was supported by a postdoctoral fellowship from the Japanese Society for the Promotion of Science. This research was supported in part by the Intramural Research Program of the National Cancer Institute.
References
- Akiyama TE, Lambert G, Nicol CJ, Matsusue K, Peters JM, Brewer HB, Jr, Gonzalez FJ. Peroxisome proliferator-activated receptor {beta}/{delta} regulates very low density lipoprotein production and catabolism in mice on a western diet. J Biol Chem. 2004;279:20874–20881. doi: 10.1074/jbc.M312802200. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Dunn C, Laughter A, Yoon L, Swanson C, Stulnig TM, Steffensen KR, Chandraratna RA, Gustafsson JA, Corton JC. Overlapping transcriptional programs regulated by the nuclear receptors peroxisome proliferator-activated receptor alpha, retinoid X receptor, and liver X receptor in mouse liver. Mol Pharmacol. 2004;66:1440–1452. doi: 10.1124/mol.104.005496. [DOI] [PubMed] [Google Scholar]
- Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–251. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Bettoun DJ, Burris TP, Houck KA, Buck DW, 2nd, Stayrook KR, Khalifa B, Lu J, Chin WW, Nagpal S. Retinoid X receptor is a nonsilent major contributor to vitamin D receptor-mediated transcriptional activation. Mol Endocrinol. 2003;17:2320–2328. doi: 10.1210/me.2003-0148. [DOI] [PubMed] [Google Scholar]
- Bodenner DL, Mroczynski MA, Weintraub BD, Radovick S, Wondis-ford FE. A detailed functional and structural analysis of a major thyroid hormone inhibitory element in the human thyrotropin beta-subunit gene. J Biol Chem. 1991;266:21666–21673. [PubMed] [Google Scholar]
- Burdick AD, Kim DJ, Peraza MA, Gonzalez FJ, Peters JM. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell Signal. 2006;18:9–20. doi: 10.1016/j.cellsig.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Chawla A, Lee CH, Barak Y, He W, Rosenfeld J, Liao D, Han J, Kang H, Evans RM. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci USA. 2003;100:1268–1273. doi: 10.1073/pnas.0337331100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheskis B, Freedman LP. Ligand modulates the conversion of DNA-bound vitamin D3 receptor (VDR) homodimers into VDR-retinoid X receptor heterodimers. Mol Cell Biol. 1994;14:3329–3338. doi: 10.1128/mcb.14.5.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheskis B, Freedman LP. Modulation of nuclear receptor interactions by ligands: kinetic analysis using surface plasmon resonance. Biochemistry. 1996;35:3309–3318. doi: 10.1021/bi952283r. [DOI] [PubMed] [Google Scholar]
- De Vos P, Lefebvre AM, Miller SG, Guerre-Millo M, Wong K, Saladin R, Hamann LG, Staels B, Briggs MR, Auwerx J. Thiazolidinediones repress ob gene expression in rodents via activation of peroxisome proliferator-activated receptor gamma. J Clin Invest. 1996;98:1004–1009. doi: 10.1172/JCI118860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Michalik L, Wahli W. Be fit or be sick: peroxisome proliferator-activated receptors are down the road. Mol Endocrinol. 2004;18:1321–1332. doi: 10.1210/me.2004-0088. [DOI] [PubMed] [Google Scholar]
- Di-Poi N, Michalik L, Desvergne B, Wahli W. Functions of peroxisome proliferator-activated receptors (PPAR) in skin homeostasis. Lipids. 2004;39:1093–1099. doi: 10.1007/s11745-004-1335-y. [DOI] [PubMed] [Google Scholar]
- Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- Fredenrich A, Grimaldi PA. PPAR delta: an uncompletely known nuclear receptor. Diab Metab. 2005;31:23–27. doi: 10.1016/s1262-3636(07)70162-3. [DOI] [PubMed] [Google Scholar]
- Fyffe SA, Alphey MS, Buetow L, Smith TK, Ferguson MA, Sorensen MD, Bjorkling F, Hunter WN. Recombinant human PPAR-beta/delta ligand-binding domain is locked in an activated conformation by endogenous fatty acids. J Mol Biol. 2006;356:1005–1013. doi: 10.1016/j.jmb.2005.12.047. [DOI] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- Guardiola-Diaz HM, Rehnmark S, Usuda N, Albrektsen T, Feltkamp D, Gustafsson JA, Alexson SE. Rat peroxisome proliferator-activated receptors and brown adipose tissue function during cold acclimatization. J Biol Chem. 1999;274:23368–23377. doi: 10.1074/jbc.274.33.23368. [DOI] [PubMed] [Google Scholar]
- Hollenberg AN, Monden T, Flynn TR, Boers ME, Cohen O, Wond-isford FE. The human thyrotropin-releasing hormone gene is regulated by thyroid hormone through two distinct classes of negative thyroid hormone response elements. Mol Endocrinol. 1995;9:540–550. doi: 10.1210/mend.9.5.7565802. [DOI] [PubMed] [Google Scholar]
- Hollenberg AN, Susulic VS, Madura JP, Zhang B, Moller DE, Tontonoz P, Sarraf P, Spiegelman BM, Lowell BB. Functional antagonism between CCAAT/Enhancer binding protein-alpha and peroxisome proliferator-activated receptor-gamma on the leptin promoter. J Biol Chem. 1997;272:5283–5290. doi: 10.1074/jbc.272.8.5283. [DOI] [PubMed] [Google Scholar]
- Hu X, Li S, Wu J, Xia C, Lala DS. Liver X receptors interact with corepressors to regulate gene expression. Mol Endocrinol. 2003;17:1019–1026. doi: 10.1210/me.2002-0399. [DOI] [PubMed] [Google Scholar]
- Ide T, Shimano H, Yoshikawa T, Yahagi N, Amemiya-Kudo M, Mat-suzaka T, Nakakuki M, Yatoh S, Iizuka Y, Tomita S, Ohashi K, Takahashi A, Sone H, Gotoda T, Osuga J, Ishibashi S, Yamada N. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. II LXRs suppress lipid degradation gene promoters through inhibition of PPAR signaling. Mol Endocrinol. 2003;17:1255–1267. doi: 10.1210/me.2002-0191. [DOI] [PubMed] [Google Scholar]
- Inaba T, Matsuda M, Shimamura M, Takei N, Terasaka N, Ando Y, Yasumo H, Koishi R, Makishima M, Shimomura I. Angiopoietin-like protein 3 mediates hypertriglyceridemia induced by the liver X receptor. J Biol Chem. 2003;278:21344–21351. doi: 10.1074/jbc.M213202200. [DOI] [PubMed] [Google Scholar]
- Juge-Aubry CE, Gorla-Bajszczak A, Pernin A, Lemberger T, Wahli W, Burger AG, Meier CA. Peroxisome proliferator-activated receptor mediates cross-talk with thyroid hormone receptor by competition for retinoid X receptor. Possible role of a leucine zipper-like heptad repeat. J Biol Chem. 1995;270:18117–18122. doi: 10.1074/jbc.270.30.18117. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kaplan R, Zhang T, Hernandez M, Gan FX, Wright SD, Waters MG, Cai TQ. Regulation of the angiopoietin-like protein 3 gene by LXR. J Lipid Res. 2003;44:136–143. doi: 10.1194/jlr.m200367-jlr200. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Man-gelsdorf DJ, Umesono K, Evans RM. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature. 1992a;355:446–449. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature. 1992b;358:771–774. doi: 10.1038/358771a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koishi R, Ando Y, Ono M, Shimamura M, Yasumo H, Fujiwara T, Horikoshi H, Furukawa H. Angptl3 regulates lipid metabolism in mice. Nat Genet. 2002;30:151–157. doi: 10.1038/ng814. [DOI] [PubMed] [Google Scholar]
- Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, Wahli W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol Endocrinol. 1997;11:779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- Krogsdam AM, Nielsen CA, Neve S, Holst D, Helledie T, Thomsen B, Bendixen C, Mandrup S, Kristiansen K. Nuclear receptor corepressor-dependent repression of peroxisome-proliferator-activated receptor delta-mediated transactivation. Biochem J. 2002;363:157–165. doi: 10.1042/0264-6021:3630157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Kang K, Mehl IR, Nofsinger R, Alaynick WA, Chong LW, Rosenfeld JM, Evans RM. Peroxisome proliferator-activated receptor delta promotes very low-density lipoprotein-derived fatty acid catabolism in the macrophage. Proc Natl Acad Sci USA. 2006;103:2434–2439. doi: 10.1073/pnas.0510815103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz MD, Fievet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J. Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett. 2000;473:333–336. doi: 10.1016/s0014-5793(00)01554-4. [DOI] [PubMed] [Google Scholar]
- Li D, Li T, Wang F, Tian H, Samuels HH. Functional evidence for retinoid X receptor (RXR) as a nonsilent partner in the thyroid hormone receptor/RXR heterodimer. Mol Cell Biol. 2002;22:5782–5792. doi: 10.1128/MCB.22.16.5782-5792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinner S, Dellas C, Schroder M, Heinlein CA, Chang C, Fischer J, Knepel W. Repression of glucagon gene transcription by peroxisome proliferator-activated receptor gamma through inhibition of Pax6 transcriptional activity. J Biol Chem. 2002;277:1941–1948. doi: 10.1074/jbc.M109718200. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Yoon M, Gonzalez FJ. Antagonism of the actions of peroxisome proliferator-activated receptor-alpha by bile acids. J Biol Chem. 2001;276:47154–47162. doi: 10.1074/jbc.M107000200. [DOI] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- Stein B, Yang MX. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Mol Cell Biol. 1995;15:4971–4979. doi: 10.1128/mcb.15.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivat-Hannah V, Bourguet W, Gottardis M, Gronemeyer H. Separation of retinoid X receptor homo- and heterodimerization functions. Mol Cell Biol. 2003;23:7678–7688. doi: 10.1128/MCB.23.21.7678-7688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Shao YY, Ballock RT. Peroxisome proliferator activated receptor-gamma (PPARgamma) represses thyroid hormone signaling in growth plate chondrocytes. Bone. 2005;37:305–312. doi: 10.1016/j.bone.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, Drouin J, Karin M. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein–protein interaction. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Ide T, Shimano H, Yahagi N, Amemiya-Kudo M, Mat-suzaka T, Yatoh S, Kitamine T, Okazaki H, Tamura Y, Sekiya M, Takahashi A, Hasty AH, Sato R, Sone H, Osuga J, Ishibashi S, Yamada N. Cross-talk between peroxisome proliferator-activated receptor (PPAR) alpha and liver X receptor (LXR) in nutritional regulation of fatty acid metabolism. I PPARs suppress sterol regulatory element binding protein-1c promoter through inhibition of LXR signaling. Mol Endocrinol. 2003;17:1240–1254. doi: 10.1210/me.2002-0190. [DOI] [PubMed] [Google Scholar]