

Abstract
β-Lactamases confer bacterial resistance to β-lactam antibiotics, such as penicillins. The characteristic class C β-lactamase AmpC catalyzes the reaction with several key residues including Ser64, Tyr150, and Lys67. Here, we describe a 1.07 Å X-ray crystallographic structure of AmpC β-lactamase in complex with a boronic acid deacylation transition-state analogue. The high quality of the electron density map allows the determination of many proton positions. The proton on the Tyr150 hydroxyl group is clearly visible and is donated to the boronic oxygen mimicking the deacylation water. Meanwhile, Lys67 hydrogen bonds with Ser64Oγ, Asn152Oδ1, and the backbone oxygen of Ala220. This suggests that this residue is positively charged and has relinquished the hydrogen bond with Tyr150 observed in acyl—enzyme complex structures. Together with previous biochemical and NMR studies, these observations indicate that Tyr150 is protonated throughout the reaction coordinate, disfavoring mechanisms that involve a stable tyrosinate as the general base for deacylation. Rather, the hydroxyl of Tyr150 appears to be well positioned to electrostatically stabilize the negative charge buildup in the tetrahedral high-energy intermediate. This structure, in itself, appears consistent with a mechanism involving either Tyr150 acting as a transient catalytic base in conjunction with a neutral Lys67 or the lactam nitrogen as the general base. Whereas mutagenesis studies suggest that Lys67 may be replaced by an arginine, disfavoring the conjugate base mechanism, distinguishing between these two hypotheses may ultimately depend on direct determination of the pKa of Lys67 along the reaction coordinate.
Introduction
β-Lactamases are the major cause of bacterial resistance to the β-lactam family of antibiotics, such as penicillins and cephalosphorins.1,2Among the four classes of β-lactamases, classes A and C are the most common clinically and have been the subjects of extensive biochemical and structural studies.1,3 Class C β-lactamases, represented by AmpC from Escherichia coli, are widespread in Gram-negative hospital pathogens and constitute a major medical concern.4-6These enzymes have been the subject of intense study.7-9
β-Lactamases catalyze the hydrolysis of the critical β-lactam ring in penicillins, cephalosporins, and related β-lactams. The reaction proceeds through a precovalent first-encounter complex, two high-energy intermediates, a relatively low-energy covalent intermediate, and a product complex (Figure 1).7,10,11After initial binding of the substrate, the catalytic serine attacks the lactam carbonyl carbon to form a tetrahedral acylation high-energy intermediate (Figure 1). This intermediate collapses, opening the four-member β-lactam ring to form a covalent acyl-enzyme complex. The covalent intermediate is then attacked by an activated water molecule resulting in the tetrahedral deacylation high-energy intermediate. The covalent bond between the serine Oγ atom and the lactam carbonyl carbon is subsequently broken, releasing the hydrolyzed, inactivated product (Figure 1).
Figure 1.

Reaction coordinate for serine β-lactamases. The reaction proceeds through a pre-covalent first encounter complex (left), a tetrahedral high-energy acylation intermediate, a low-energy acyl—enzyme complex, a tetrahedral high-energy deacylation intermediate following the attack of the catalytic water, and finally the hydrolyzed product, which is subsequently released from the enzyme complex.
For AmpC β-lactamase, several key catalytic residues have been identified.12-18In addition to Ser64, which directly attacks the β-lactam ring, Lys67, Tyr150, Asn152, Lys315, and the main chain of Ala318 play important roles in the reaction pathway. Substitution of these residues typically results in dramatic decreases in enzymatic activities. Thus, the Lys67Glu substitution abolished catalytic activity, though, intriguingly, the Lys67Arg substitution retained significant activity. This suggests that a positive charge is important at this position but not a lysine in particular, a point to which we will return.12,13 Substituting Tyr150 with a serine or phenylalanine residue decreased the reaction rate by as much as 10 000-fold.17 Lys315His and Lys315Gln substitutions reduced activities dramatically for both the acylation and deacylation steps.14 Replacing the nucleophilic serine by an alanine or glycine also reduced activity by 4 orders of magnitude, as expected.19
Whereas much has been learned about the AmpC mechanism, some of the specific roles of these residues remain unclear despite crystallographic snapshots of AmpC at almost every step along the reaction coordinate.4,6,10,11,20-23Both Tyr150 and Lys67 are within hydrogen-bonding distance to Ser64, suggesting that they may be involved in its deprotonation or protonation in the acylation and deacylation steps, respectively.20,24Tyr150 appears to hydrogen bond with Lys67, Lys315, and the deacylation water in many steps along the reaction coordinate. Thus, many investigators have proposed that Tyr150 is the catalytic base for deacylation, accepting a proton from the deacylating water as it attacks the acyl-enzyme intermediate.7,17 Recent calculations have also suggested a conjugate-base hypothesis involving Tyr150 and Lys67.25Others have proposed mechanisms that involve substrate groups themselves, particularly the lactam ring nitrogen, as possible catalytic bases, i.e., via substrate-assisted catalysis.21,26The differences among these mechanisms often center on the protonation state of Tyr150 and whether it functions as a donor or acceptor in a particular hydrogen bond. The phenolate anion of Tyr150 would favor its role as the general base to deprotonate the catalytic water during the deacylation process. Whereas small-molecule studies suggested that a Tyr150-like phenolic group was deprotonated at pH 7,27NMR experiments on the class C β-lactamase from Citrobacter freundii GN346 demonstrated that Tyr150 retained its Oη proton up to pH 11, at least for the apo-enzyme.28The pKa of this residue in complex with the substrate, however, is not known. For hypotheses involving Tyr150 as a conjugate base together with Lys67 or Lys315, the tyrosine can be in a protonated, neutral form while acting as a hydrogen-bond donor in its interaction with the respective neutral lysine. A neutral Tyr150 may also be able to activate Ser64 or the catalytic water and thus play an activating role that does not involve general base catalysis. The protonation state of Tyr150 and the residue and substrate groups with which it interacts in the rate-limiting deacylation high-energy intermediate are therefore crucial to understanding the mechanism of this class of antibiotic-resistance enzyme.
Here, we report the X-ray crystal structure of AmpC N289A in complex with a boronic acid deacylation transition-state analogue determined at 1.07 Å resolution. The tetrahedral geometry adopted by the boronic acid, the putative concentration of its negative charge on the boronic acid oxygens, and the β-lactam side-chain functionality displayed by this inhibitor (see below) make this molecule a good analogue of the tetrahedral intermediate for deacylation. The ultrahigh resolution data allowed us to determine multiple main-chain and side-chain conformations and to visualize the positions of many protons, including those on polar residues. In this structure, Tyr150 is indeed protonated and donates a proton to the oxygen representing the catalytic water in the transition-state analogue. Moreover, Tyr150 has lost the hydrogen bond with Lys67 that it maintains in the acyl-enzyme complex.21,29Lys67, meanwhile, appears to hydrogen bond with three acceptor atoms including Ser64Oγ. The implications of these observations for the various possible mechanisms of deacylation by class C β-lactamases will be considered.
Materials and Methods
Protein Purification. An AmpC mutant enzyme, N289A, was used in these studies. Asn289 is a nonconserved residue and is not involved in substrate binding or catalysis. N289A is effectively isofunctional with wild-type AmpC but diffracts to higher resolution than the WT enzyme.30,31N289A AmpC was expressed and purified with an m-aminophenylboronic acid affinity column and concentrated using Centricon spin concentrators as described previously.31
Crystal Growth and Data Collection. Cocrystals of AmpC N289A in complex with compound 1 (see Table 2) were grown using microseeding with hanging drops over 1.7 M potassium phosphate buffer at pH 8.7. The initial drops contained 3.8 mg/mL protein and 588 μM compound. Compound 1 was added to the drop in 1.2% DMSO and 1 M potassium phosphate (pH 8.7). Crystals appeared within 5-7 days at 23°C. Before data collection, the crystals were transferred to a cryoprotectant solution of 23% sucrose, 1.7 M potassium phosphate (pH 8.7), and flash-frozen in liquid nitrogen after 30 s. Reflections were measured on the DND-CAT beam line (5IDB) of the Advanced Photon Source at Argonne National Laboratory at 100 K using a Mar-CCD detector. Reflections were processed using the HKL software package.32
Table 2.
Compound 1 Structure and Key Distances in the Active Sitea
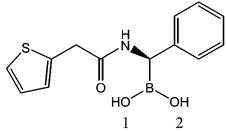 | |||
|---|---|---|---|
| atom pair | distance (Å) | atom pair | distance (Å) |
| Tyr150Oη—Lys67Nζ | 3.25 | Lys315Nζ—Thr316O | 2.86 |
| Tyr150Oη—Ser64Oγ | 3.00 | Lys67 Nζ—Ser64Oγ | 2.74 |
| Tyr150Oη—O2 | 2.63 | Lys67 Nζ—Asn152Oδ1 | 2.77 |
| Lys315Nζ—Tyr150Oη | 2.97 | Lys67 Nζ—Ala220O | 2.88 |
| Lys315Nζ—HOH130 | 2.75 | ||
Distances in monomer 1 are shown. Those in monomer 2 are similar.
Structure Determination and Refinement. A lower-resolution (1.39 Å, PDB ID 1PI4) structure of the same complex was used as the initial model, and refinement was carried out in SHELXL-97.31,33The model was refined using anisotropic displacement parameters (ADPs) with standard DELU (rigid-bond restraint), SIMU (restraint for spatially adjacent atoms), and ISOR (isotropic restraint for individual atoms). These values were later adjusted using the program PARVATI.34The occupancies and anistropic B factors for alternative conformations were also refined in the program. Riding hydrogen atoms were added in the last few rounds of refinement. Well-ordered hydrogen atoms on certain polar residues were also modeled when they were identified by positive peaks at a minimum of 1.5σ level in the hydrogen-omitted Fo-Fc map. We note that whereas a 1.5σ level feature is considered borderline for a second row element, we consider it significant for a hydrogen.35The hydrogen atom positions were fixed during refinement. The figures of protein structures were generated by Pymol (Delano Scientific). The structure has been deposited in the RCSB Protein Data Bank with ID code 2FFY.
Results
Structural Details at Ultrahigh Resolution. The complex crystallized in the C2 space group with a unit cell similar to previously determined AmpC structures. The final structure had an R-value of 13.4% and an Rfree of 16.4% (Table 1). There are two monomers of AmpC N289A in each asymmetric unit; their backbone conformations are nearly identical except for residues 275-294. This is a flexible region that adopts several different conformations or is partially disordered in monomer 1 of previous AmpC apo and complex structures.10,31,36Two conformations were modeled in monomer 1 for these residues in our structure; a few residues were missing in electron density for conformation 2. Conformation 1 is not observed in the wild type AmpC structures and may result from the Asn289→Ala substitution that places Ala289 in a pocket slightly too small to accommodate the original Asn residue. This allows Asp288 to hydrogen bond with Arg148 and enables other residues, such as Ile291, to make crystal packing interactions. Despite its partial occupancy, this conformation may contribute to the better diffraction quality of N289A crystals compared with that of wild-type AmpC. The same flexible region is ordered in the single conformation observed in the second monomer. The rmsd between the backbone atoms of the two monomers is 0.914 Å overall and 0.329 Å without residues 275-294. Monomer 1 is slightly less ordered than monomer 2, with a non-hydrogen-atom average B factor of 17.3 Å2 for the former and 15.5 Å2 for the latter.
Table 1.
Crystallographic Statistics for N289A AmpC/Compound 1
| resolution (Å) | 15.00-1.07 (1.11-1.07)a |
| completeness (%) | 94.7 (85.7) |
| Rmerge (%)b | 5.5 (47.7) |
| 12.07 (2.34) | |
| unique reflections | 325 964 (30 545) |
| space group | C2 |
| unit cell | 118.840, 76.063, 97.898 (Å) 90.00, 115.85, 90.00 (deg) |
| number of protein residues | 716 |
| number of residues with | 126c |
| double conformations | |
| number of ions | 1 HPO42-, 2 K + |
| number of water molecules | 1114 |
| Rwork (%)d | 13.4 (36.6) |
| Rfree (%) | 16.4 |
| rms deviation | |
| bond lengths | 0.013 Å |
| angle distances | 0.029 Å |
| average isotropic B factor (Å2) | |
| protein residues | 16.4 |
| solvent | 30.2 |
| transition-state analog | 16.6 |
| Ramanchandran plote | |
| most favored region (%) | 92.0 |
| additionally allowed (%) | 8.0 |
| generously allowed (%) | 0.0 |
Alternate conformations were modeled for a total of 69 residues for monomer 1 and 57 residues for monomer 2 (126 residues in total). An interesting example of these is the multiple conformations observed for Ile243 and Ile252 (Figure 2a). The two isoleucines are in direct contact with one another, with Ile243 near the protein surface and Ile252 completely buried. Conformation 1 of Ile243 makes favorable van der Waals interactions with conformation 1 of Ile252; the same is true for the second conformations of both residues. However, conformation 2 of Ile252 would sterically clash with conformation 1 of Ile243. Simultaneous adoption of conformation 1 of Ile252 and conformation 2 of Ile243, on the other hand, would create a gap between these two residues and expose more solvent-accessible surface area on Ile243. These observations suggest a coordinated conformation switch for the two residues, with conformation 1 of both residues likely to be the more favorable state. Whether or not this conformational switch has implications for catalysis is unclear, but we find it an interesting example of motion within the tightly packed core of the enzyme, observed at ultrahigh resolution. We note that no alternate conformations are observed for the catalytic residues at this resolution.
Figure 2.
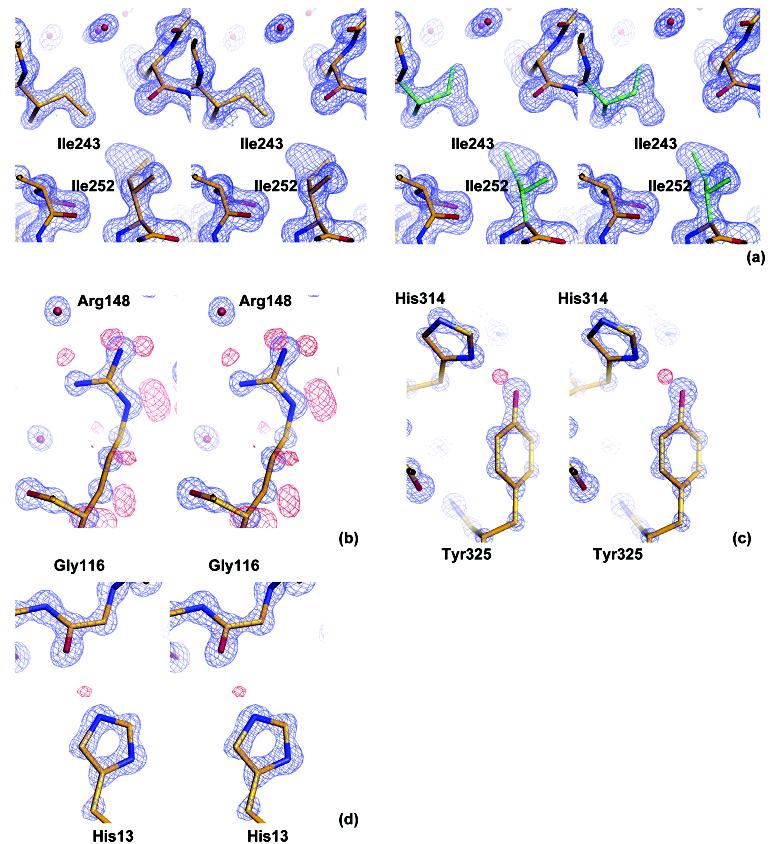
Stereoviews of characteristic structural details revealed at ultrahigh resolution. The 2Fo—Fc electron density map is displayed in blue and positive peaks in the hydrogen-omitted Fo—Fc electron density map are shown in red. (a) Double conformations of Ile243 and Ile252, showing correlated conformational changes in the protein core. Conformation 1 is shown in gold on the left, and conformation 2 is shown in green on the right. The 2Fo—Fc electron density map is shown at a 1.5σlevel. (b) Side-chain hydrogen atoms of Arg148, showing a 2Fo—Fc electron density map at a 3.0σlevel and positive peaks in a hydrogen-omitted Fo—Fc electron density map at a 2.0σlevel. (c) Tyr325, with the 2.5σpositive peak corresponding to a proton donated from Tyr325 to His314. A 2Fo—Fc electron density map is displayed at a 3.0σlevel. (d) His13, showing a possible proton, contoured at 1.5σ, on the N∊2 atom. This atom interacts with Gly116O in a crystal packing interface. The 2Fo—Fc electron density map is displayed at a 3.0σlevel.
The ultrahigh resolution data also allowed us to accurately determine the positions of individual atoms and to distinguish C, N and O atoms from the electron density volumes. This helped us correct the orientations of some side chain end groups, such as histidine and asparagines that had been incorrectly modeled in the lower resolution structures of AmpC.31For example, the conformation of His108gwas corrected by flipping the imidazole ring by approximately 180°. The atoms of the five member ring occupy the same positions in the two conformations, which could not be distinguished at medium resolution. At 1.07 Å, the difference between nitrogen and carbon atoms is clear in the electron density, allowing us to unambiguously determine the exact orientation of the imidazole ring. The subtle difference between the two carboxylate oxygen atoms in glutamate and aspartate also becomes discernible. For Asp217, the lengths of Cγ-Oδ1 and Cγ-Oδ2 are 1.27 and 1.24 Å respectively (the bond length rmsd for the whole model is 0.013 Å in refinement), with more electron density at the latter, suggesting double bond character for Oδ2 and single bond character for Oδ1.
Many hydrogen atoms of the backbone and ordered side chains are apparent in the positive peaks in the hydrogen-omitted Fo-Fc map at 1.5σ or higher levels (Figure 2). Not only do we observe the ‘riding’ hydrogen atoms, whose coordinates can be easily determined by the heavy atom positions, but also the positions of hydroxyl group protons on 21 out of a total of 112 SER/THR/TYR residues in the two monomers. The phenol hydroxyl proton is discernible for 11 out of 30 tyrosine residues (Figure 2). Overall, this structure should provide an unusually good template for future inhibitor discovery against this drug target.
Hydrogen-Bonding Network in the Active Site. The most exciting single feature in this ultrahigh-resolution structure is the hydrogen atom on the putative base for deacylation, the Oη atom of Tyr150. A positive peak in the hydrogen-omitted Fo-Fc electron density map at 1.5σclearly identifies a proton on the Tyr150 Oη atom; this peak is apparent up to a sigma level of 2.5, suggesting the proton is well-ordered. The proton is donated to the boronic acid O2 atom, with an oxygen-to-oxygen distance of 2.63 Å and an Oη—H—O2 angle of 147°(Figure 3a). This observation determines both the protonation state and the hydrogen-bonding partner of this catalytic residue. In addition to the boronic acid O2 atom, there are three other atoms that have been suggested to hydrogen bond with the tyrosine Oη atom: the Ser64 Oγ, Lys67 Nζ and Lys315 Nζ. However, the Ser64 Oγ can only be a hydrogen-bond acceptor in this complex and the position of the tyrosyl Oη proton suggests that this proton is not shared with Ser64. The distance between Ser64 Oγ and Tyr150 Oη is 3.00 Å, longer than the distance of 2.63 Å between the boronic acid O2 atom and Tyr150 Oη (Figure 3a, Table 2). More importantly, the angle formed by Ser64 Cβ, Ser64 Oγ, and Tyr150 Oη is 77.3°, compared with an optimal value of approximately 109° for a possible hydrogen bond. Taken together, these observations suggest that there is no hydrogen bond between Tyr150 and the catalytic serine in the deacylation high-energy intermediate.
Figure 3.
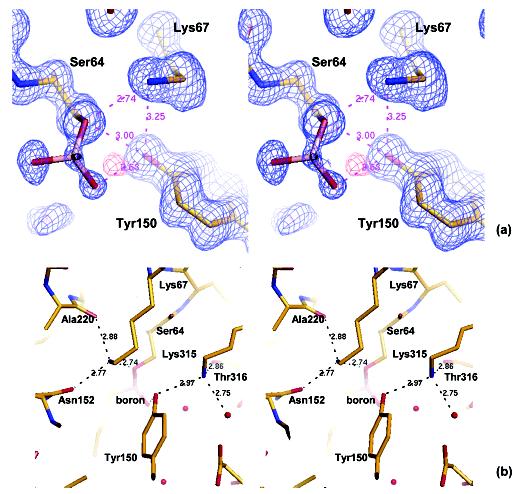
Hydrogen bonding network in the active site. (a) Stereoview of Tyr150, Lys67, Ser64, and the boronic acid group, showing distances between key residues. A 2Fo—Fc electron density map is displayed in blue at a 1σlevel, and the positive Fo—Fc peak in hydrogen-omitted electron density map is shown in red at a 1.5σcontour level. The positive peak corresponds to the hydrogen atom on Tyr150 Oη. Only the boronic group of the transition-state analogue is shown, with the rest of the molecule projecting out of the plane of the paper. (b) Stereoview of the active site residues showing key interactions in which Tyr150, Lys67, and Lys315 are involved. Each lysine interacts with three hydrogen bond acceptors. Lys67 Nζhydrogen bonds with Ser64 Oγ, Asn152 Oδ1, and the backbone O atom of Ala220. Lys315 Nζhydrogen bonds with Tyr150 Oη, the backbone O atom of Thr316, and a water molecule.
Lys67 Nζ has been proposed to hydrogen bond with Tyr150 Oη at various steps along the reaction coordinate of AmpC, and has been observed to do so in multiple crystal structures.7,21,29In the deacylation transition state, analogous to this structure, it has been suggested that Lys67 can cooperate with Tyr150, either stabilizing a tyrosinate base or accepting a proton from the hydrolytic water by way of the tyrosine.24,25In either scenario, Lys67 might be expected to hydrogen bond with Tyr150. In the transition-state analogue complex, however, the distance between the Lys67 Nζ and Tyr150 Oη is 3.25 Å and the angle formed by Lys67 Cε, Lys67 Nζ and Tyr150 Oη is 79.1°, suggesting that a hydrogen bond is not present and that the two atoms are merely in van der Waals contact (at 1.07 Å resolution, the difference between 3.25 Å and a characteristic hydrogen-bond distance of 2.8 Å is significant). Instead, Lys67 appears to hydrogen bond with three other residues in our structure: Ser64 Oγ, Asn152 Oδ1, and the backbone O atom of Ala220 (Figure 3b). Although the protonation state of Lys67 is not directly observed in this structure, determined at pH 8.7, it can only be the proton donor in all three possible hydrogen bonds, and thus it appears to be positively charged, bearing its full proton complement.
The last residue that Tyr150 can hydrogen bond with is Lys315. The Lys315 Nζ is also positioned to make three hydrogen bonds: Tyr150 Oη, the backbone O atom of Thr316, and a water molecule (Figure 3b). Lys315 is believed to be positively charged and it is likely that it functions as the donor in all three hydrogen bonds as well.25At a dihedral angle of 46.3°, the hydrogen bond between Lys315 and Tyr150 is more in plane with the phenol ring, compared with Lys67 whose Nζ is out of the plane by 82.2°. Thus, Tyr150 seems to form two hydrogen bonds in the active site, donating a hydrogen to the boronic acid O2 atom, which represents the position of the attacking deacylating water, and accepting a hydrogen from the Lys315 Nζ atom.
Discussion
Whether Tyr150 exists as an anionic phenolate or a protonated phenol in the active site of AmpC β-lactamase is actively debated.7,24,25A phenolate state of the tyrosine would support a general base hypothesis in which Tyr150 deprotonates the catalytic water in the deacylation step and, subsequently, the newly protonated Tyr150 could then act as a general acid to protonate the Oγ atom of Ser64, either directly or indirectly through Lys67. This first model is disfavored, though not ruled out, by the ultrahigh-resolution structure described here and by previous NMR studies. In a second model, the tyrosine acts as conjugate general base together with Lys67 (Figure 4a).25This hypothesis is more consistent with this structure and previous studies, though we do not fully favor it either. Rather, together with previous biochemical studies, our ultrahigh-resolution structure favors a third model where a phenol form of Tyr150 stabilizes the tetrahedral deacylation transition state in conjunction with the lactam nitrogen of the substrate (Figure 4b).
Figure 4.
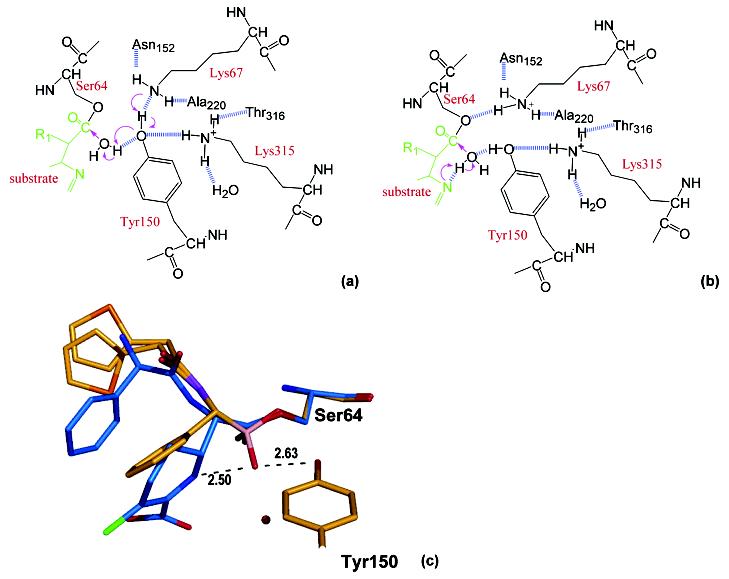
Conjugate base versus substrate-activated catalysis hypotheses. (a) Schematic diagram of the conjugate base hypothesis for deacylation. Arrows indicate the electron-transfer direction, and the dashed lines show the hydrogen bonds observed in the transition-state analogue structure except the one between Lys67 and Tyr150, which is not observed but drawn because it is essential for the conjugate base hypothesis. (b) Schematic diagram of the substrate-activated catalysis. (c) The deacylation transition-state analogue structure (in gold, with the end group modeled in two conformations) overlaid with the acyl—enzyme complex with loracarbef (in blue, PDB ID 1FCN, 2.35 Å resolution). The catalytic water from the acyl—enzyme structure is shown as a red sphere; its distance to the boronic O2 oxygen of the transition-state analogue is 3.17 Å. Also shown are the distances between the boronic group O2 atom, which mimics the deacylation water, Tyr150 Oηand the ring nitrogen of loracarbef.
Protonation State of Tyr150. The presence of a proton on the Tyr150 Oγ atom in our complex structure is consistent with recent NMR studies on apo wild type and mutant class C β-lactamases.28Ishiguro and colleagues found that the pKa of Tyr150 is above pH 11 in the wild type C. freundii GN346 β-lactamase and found that this residue was protonated up to pH 10.4 in the K67C mutant. Compared with the pKa value of 10.3 for a free tyrosine, this suggests that the burial of Tyr150 causes an upshift of its pKa, and that Lys67 has at most a small effect on this shift. These observations indicate that the phenolate state is unstable for Tyr150 inside the AmpC active site and that the catalytic tyrosine exists in its neutral form, at least in the apo enzyme. Our X-ray crystallographic complex structure determined at a basic pH value, together with the NMR studies, suggests that Tyr150 is in fact protonated throughout the reaction coordinate. This structure thus offers no harbor to hypotheses favoring an anionic tyrosinate base, although it does not completely rule them out.7,24,25It is also unlikely that a neutral Tyr150 acts as the general acid to protonate Ser64. Whereas the tyrosine Oη is nominally within a longish hydrogen-bond distance with the Ser Oγ at 3.00 Å, the angle between Ser64 Cβ, Ser64 Oγ, and Tyr150 Oη is far from optimal at 77.3°, disfavoring a hydrogen bond between them. Instead, Tyr150 interacts with the catalytic water, i.e., the O2 atom in our transition-state analogue, and the Oγ atom of Ser64 accepts a hydrogen bond from Lys67.
Recent quantum mechanical/molecular mechanics calculations have led to a coordinate base hypothesis involving Lys67 and Tyr150.25Rather than invoking a stably anionic Tyr150, this mechanism involves a transient transfer of a proton from the attacking water through the Tyr150 Oη atom and onto a neutral Lys67 Nζ, making the latter cationic. At the transition state, this model predicts that Tyr150 would accept a proton from the hydrolytic water and that Lys67 would hydrogen bond with Tyr150, from which it would in turn accept this proton. We observe the opposites-that Tyr150 is protonated, that Lys67 is protonated, and that the Lys67-Tyr150 hydrogen bond, observed in the acyl-enzyme complex, has broken.
An explanation that might resolve these discrepancies is if the ultrahigh resolution structure represents the state after the proton has been transferred from the hydrolytic water to Tyr150 and then onto Lys67. An attractive aspect of this proposal is that it would couple progress along the reaction coordinate to motion in the active site. Thus, the breaking of the Lys67-Tyr150 hydrogen bond, observed in acyl-enzyme class C β-lactamase structures,21,29could be evidence that the lysine, upon protonation, moves both physically and in the sense of the reaction coordinate toward the activation of Ser64, which is necessary for the final step in deacylation of the enzyme. The charged form of Lys67 would thus be a candidate for the general acid activating Ser64, leading to product release and regeneration of the neutral form of Lys67. This neutral form would then be in place to act as a catalytic base, ultimately to the Tyr150 in deacylation but possibly also for acylation of Ser64 by a β-lactam substrate, as has been proposed by others.10,26,38
This coordinate base hypothesis, though consistent with the ultrahigh-resolution structure on its own, is disfavored by previous biochemical experiments. Perhaps the strongest evidence against this hypothesis is the effect of the substitution Lys67→Arg, explored by Frère and colleagues.13The catalytic rate constant is diminished only by 2- to 3-fold for this mutant enzyme, which is hard to reconcile with a role for Lys67 as a catalytic base. From a structural standpoint, whereas the high-resolution structure may represent the post-proton transfer state, the boronic acid in the structure was designed to mimic the transition-state structure for deacylation preceding the water deprotonation. If the enzyme is pre-organized to recognize this state above all others, one might expect that this would be the state captured by crystallography. Admittedly, this is not a definitive argument. Finally, NMR evidence on apo mutant and wild-type class C β-lactamases suggests that Lys67 does not significantly perturb the pKa of Tyr150, and correspondingly this might suggest that there is no strong proton-coupling between the two residues.28This too, of course, is not definitive. Overall, whereas we do not favor the Lys67-Tyr150 coordinate base hypothesis, our structure does not disprove it. More direct experiments, such as exploring the pKa of Lys67 throughout the reaction coordinate, may ultimately be necessary to completely resolve this issue.
Substrate-Activated Catalysis. If neither an anionic Tyr150 nor a coordinate mechanism involving a neutral Lys67-Tyr150 pair is responsible for deprotonating the catalytic water, what mechanism do we favor? Mobashery and colleagues proposed that the lactam nitrogen from the substrate can play such a role, and our subsequent medium-resolution crystal structures seemed consistent with this hypothesis.26The ultrahigh resolution structure described here remains consistent with this proposal.
When we superimpose the deacylation transition-state structure with an acyl-enzyme intermediate structure, the lactam ring nitrogen lies 2.5 Å away from the boronic acid O2 oxygen that mimics the deacylation water (Figure 4c).21The proton on the Tyr150 Oη is collinear with the position of the lactam nitrogen on the other side of the boronic O2 atom and 2.63 Å away from the O2 atom. The tyrosine proton is thus well placed to stabilize developing negative charge on the deacylating water, while the substrate nitrogen acts as a base, transiently accepting the proton from the other side. Although the pKa value of this lactam nitrogen has been a point of controversy,25model studies suggest that it might be between pH 5 and 6.39This would be consistent with its ability to accept a proton from the activated deacylating water. This mechanism is consistent with substrate modification studies by Mobashery, which suggested that analogues lacking the equivalent nitrogen were trapped in the acyl adduct, as well as inhibition and structural studies that suggested that β-lactams with displaced nitrogens acted as inhibitors.26,31We note that studies of alternative depsipeptides substrates are not easily reconciled with this hypothesis since these substrates lack the lactam nitrogen.40That said, analogous roles for the equivalent thiol in these alternative substrates cannot be excluded.
We conclude by returning to the key observation of this studys-the protonation state and hydrogen-bonding network of Tyr150 in class C β-lactamases. In complex with a deacylation transition-state analogue at basic pH, Tyr150 of AmpC not only is protonated but also donates its proton to a boronic acid oxygen representing the deacylating water as it attacks the acyl-enzyme intermediate. This structure is thus consistent with earlier apo-enzyme studies, suggesting that Tyr150 remains protonated in class C β-lactamases.28On the other hand, the structure is difficult to reconcile with this residue acting as an anionic catalytic base and is inconsistent with this residue acting as a catalytic acid to activate Ser64 as a leaving group in the final deacylation step. This structure remains consistent with the role of Tyr150 in coordinate activation of the deacylating water in cooperation with the lactam nitrogen of the substrate itself (substrate-assisted catalysis), although it does not rule out, in itself, the alternative Lys67-Tyr150 coordinate base mechanism. Distinguishing between these two may ultimately require direct study of the pKa of Lys67. Finally, AmpC is a primary resistance determinant to β-lactam antibiotics among Gram-negative pathogens. The 1.07 Å structure described here provides an unusually detailed template for inhibitor discovery against this target.
Acknowledgments
Acknowledgment. This work was supported by NIH Grant GM63813 (to B.K.S.). We thank John Irwin, Matt Jacobson, Johannes Herman, Shahriar Mobashery, and Sammy Meroueh for helpful discussions. We also thank Kerim Babaoglu, Veena Thomas, Johannes Herman, and Sarah Boyce for reading the manuscript.
References
- (1).Fisher JF, Meroueh SO, Mobashery S. Chem. Rev. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- (2).Davies J. Science. 1994;264:375–382. doi: 10.1126/science.8153624. [DOI] [PubMed] [Google Scholar]
- (3).Bush K, Jacoby GA, Medeiros AA. Antimicrob. Agents Chemother. 1995;39:1211–1233. doi: 10.1128/aac.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lobkovsky E, Moews PC, Liu H, Zhao H, Frère J-M, Knox JR. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11257–11261. doi: 10.1073/pnas.90.23.11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vakulenko SB, Golemi D, Geryk B, Suvorov M, Knox JR, Mobashery S, Lerner SA. Antimicrob. Agents Chemother. 2002;46:1966–1970. doi: 10.1128/AAC.46.6.1966-1970.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Crichlow GV, Kuzin AP, Nukaga M, Mayama K, Sawai T, Knox JR. Biochemistry. 1999;38:10256–10261. doi: 10.1021/bi9908787. [DOI] [PubMed] [Google Scholar]
- (7).Oefner C, D’Arcy A, Daly JJ, Gubernator K, Charnas RL, Heinze I, Hubschwerlen C, Winkler FK. Nature. 1990;343:284–288. doi: 10.1038/343284a0. [DOI] [PubMed] [Google Scholar]
- (8).Galleni M, Lamotte-Brasseur J, Raquet X, Dubus A, Monnaie D, Knox JR, Frère J-M. Biochem. Pharmacol. 1995;49:1171–1178. doi: 10.1016/0006-2952(94)00502-d. [DOI] [PubMed] [Google Scholar]
- (9).Maiti SN, Phillips OA, Micetich RG, Livermore DM. Curr. Med. Chem. 1998;5:441–456. [PubMed] [Google Scholar]
- (10).Beadle BM, Trehan I, Focia P, Shoichet BK. Structure. 2002;10:413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]
- (11).Curley K, Pratt RF. J. Am. Chem. Soc. 1997;119:1529–1538. [Google Scholar]
- (12).Tsukamoto K, Tachibana K, Yamazaki N, Ishii Y, Ujiie K, Nishida N, Sawai T. Eur. J. Biochem. 1990;188:15–22. doi: 10.1111/j.1432-1033.1990.tb15365.x. [DOI] [PubMed] [Google Scholar]
- (13).Monnaie D, Dubus A, Frère J-M. Biochem. J. 1994;302:1–4. doi: 10.1042/bj3020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Monnaie D, Dubus A, Cooke D, Marchand-Brynaert J, Normark S, Frère J-M. Biochemistry. 1994;33:5193–5201. doi: 10.1021/bi00183a024. [DOI] [PubMed] [Google Scholar]
- (15).Dubus A, Normark S, Kania M, Page MG. Biochemistry. 1994;33:8577–8586. doi: 10.1021/bi00194a024. [DOI] [PubMed] [Google Scholar]
- (16).Dubus A, Normark S, Kania M, Page MG. Biochemistry. 1995;34:7757–7764. doi: 10.1021/bi00023a023. [DOI] [PubMed] [Google Scholar]
- (17).Dubus A, Ledent P, Lamotte-Brasseur J, Frère J-M. Proteins. 1996;25:473–485. doi: 10.1002/prot.7. [DOI] [PubMed] [Google Scholar]
- (18).Murphy BP, Pratt RF. Biochem. J. 1988;256:669–672. doi: 10.1042/bj2560669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Beadle BM, Shoichet BK. J. Mol. Biol. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- (20).Lobkovsky E, Billings EM, Moews PC, Rahil J, Pratt RF, Knox JR. Biochemistry. 1994;33:6762–6772. doi: 10.1021/bi00188a004. [DOI] [PubMed] [Google Scholar]
- (21).Patera A, Blaszczak LC, Shoichet BK. J. Am. Chem. Soc. 2000;122:10504–10512. [Google Scholar]
- (22).Chen CC, Rahil J, Pratt RF, Herzberg O. J. Mol. Biol. 1993;234:165–178. doi: 10.1006/jmbi.1993.1571. [DOI] [PubMed] [Google Scholar]
- (23).Nukaga M, Abe T, Venkatesan AM, Mansour TS, Bonomo RA, Knox JR. Biochemistry. 2003;42:13152–13159. doi: 10.1021/bi034986b. [DOI] [PubMed] [Google Scholar]
- (24).Lamotte-Brasseur J, Dubus A, Wade RC. Proteins. 2000;40:23–28. doi: 10.1002/(sici)1097-0134(20000701)40:1<23::aid-prot40>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- (25).Gherman BF, Goldberg SD, Cornish VW, Friesner RA. J. Am. Chem. Soc. 2004;126:7652–7664. doi: 10.1021/ja036879a. [DOI] [PubMed] [Google Scholar]
- (26).Bulychev A, Massova I, Miyashita K, Mobashery S. J. Am. Chem. Soc. 1997;119:7619–7625. [Google Scholar]
- (27).Kato-Toma Y, Ishiguro M. Bioorg. Med. Chem. Lett. 2001;11:1161–1164. doi: 10.1016/s0960-894x(01)00168-8. [DOI] [PubMed] [Google Scholar]
- (28).Kato-Toma Y, Iwashita T, Masuda K, Oyama Y, Ishiguro M. Biochem. J. 2003;371:175–181. doi: 10.1042/BJ20021447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. Biochemistry. 2001;40:9207–9214. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- (30).Trepanier S, Knox JR, Clairoux N, Sanschagrin F, Levesque RC, Huletsky A. Antimicrob. Agents Chemother. 1999;43:543–548. doi: 10.1128/aac.43.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Roth TA, Minasov G, Morandi S, Prati F, Shoichet BK. Biochemistry. 2003;42:14483–14491. doi: 10.1021/bi035054a. [DOI] [PubMed] [Google Scholar]
- (32).Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- (33).Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- (34).Merritt EA. Acta Crystallogr. Sect. D: Biol. Crystallogr. 1999;55:1109–1117. doi: 10.1107/s0907444999003789. [DOI] [PubMed] [Google Scholar]
- (35).Minasov G, Wang X, Shoichet BK. J. Am. Chem. Soc. 2002;124:5333–5340. doi: 10.1021/ja0259640. [DOI] [PubMed] [Google Scholar]
- (36).Powers RA, Shoichet BK. J. Med. Chem. 2002;45:3222–3234. doi: 10.1021/jm020002p. [DOI] [PubMed] [Google Scholar]
- (37).Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- (38).Varetto L, De Meester F, Monnaie D, Marchand-Brynaert J, Dive G, Jacob F, Frère J-M. Biochem. J. 1991;278:801–807. doi: 10.1042/bj2780801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Proctor P, Gensmantel NP, Page MI. J. Chem. Soc., Perkin Trans. 2. 1982:1185–1192. [Google Scholar]
- (40).Xu Y, Soto G, Hirsch KR, Pratt RF. Biochemistry. 1996;35:3595–3603. doi: 10.1021/bi952106q. [DOI] [PubMed] [Google Scholar]