

Abstract
The selective RNA-binding protein QKI is essential for myelination in the central nervous system (CNS). QKI belongs to the family of signal transduction activators of RNA (STARs), characteristic of binding RNA and signaling molecules, therefore is postulated to regulate RNA homeostasis in response to developmental signals. Here we report that QKI acts downstream of the Src family protein tyrosine kinases (Src-PTKs) during CNS myelination. QKI selectively interacted with the mRNA encoding the myelin basic protein (MBP). Such interaction stabilized MBP mRNA and was required for the rapid accumulation of MBP mRNA during active myelinogenesis. We found that the interaction between QKI and MBP mRNA was negatively regulated by Src-PTK-dependent phosphorylation of QKI. During early myelin development, tyrosine phosphorylation of QKI in the developing myelin drastically declined, presumably leading to enhanced interactions between QKI and MBP mRNA, which was associated with the rapid accumulation of MBP mRNA and accelerated myelin production. Therefore, developmental regulation of Src-PTK-dependent tyrosine phosphorylation of QKI suggests a novel mechanism for accelerating CNS myelinogenesis via regulating mRNA metabolism.
Keywords: mRNA metabolism/myelin development/RNA-binding protein/Src family protein tyrosine kinases/tyrosine phosphorylation
Introduction
In the central nervous system (CNS), accurate production of myelin structural proteins is essential for myelinogenesis, the process by which specialized plasma membrane extensions ensheath neuronal axons (Campagnoni and Macklin, 1988). Such a process is under tight regulation by developmental signals via undefined mechanisms. Deficiency of the RNA-binding protein QKI encoded by the qkI gene results in severe hypomyelination (Hogan and Greenfield, 1984; Campagnoni and Macklin, 1988; Hardy, 1998), suggesting that RNA homeostasis is a key regulatory step for myelinogenesis. In the quakingviable (qkv) mutant mice, a recessive genetic lesion affects a 5′ regulatory element of the qkI gene and impairs QKI expression specifically in myelin-producing cells of the homozygous qkv animals (Ebersole et al., 1996; Hardy et al., 1996). Consequently, a subclass of myelin structural protein mRNAs display post- transcriptional abnormalities, including destabilization, mislocalization and misregulated splicing (Li et al., 2000; Larocque et al., 2002; Wu et al., 2002).
Several protein isoforms of QKI are derived from differential usage of the 3′ coding exon via alternative splicing of the qkI transcript (Ebersole et al., 1996; Kondo et al., 1999). All the major QKI isoforms share an extended hnRNP K-homology (KH) RNA-binding domain at the N-terminus, followed by a few proline-rich putative Src-homology 3 (SH3)-binding motifs and a cluster of tyrosine residues at the C-terminus (Vernet and Artzt, 1997). These features implicate dual interactions of QKI with signaling molecules and its target mRNAs, which has led to an intriguing hypothesis that QKI may mediate developmental signals to regulate the cellular fate of myelin structural protein mRNAs and myelinogenesis. Indeed, QKI has been shown to selectively interact with the 3′ untranslated region (UTR) of the mRNA encoding the myelin basic protein (MBP) (Li et al., 2000; Zhang and Feng, 2001; Larocque et al., 2002), an essential structural component of CNS myelin (Inoue et al., 1981; Roach et al., 1985). Several alternatively spliced MBP isoforms are expressed specifically in myelin-producing cells, all harboring the same 3′-UTR, and the quantity of MBP production is under tight control during development (Campagnoni and Macklin, 1988). QKI deficiency in the qkv/qkv mutant results in destabilization of the MBP mRNA and a marked reduction of MBP expression during early myelin development (Li et al., 2000), suggesting that MBP mRNA is a target for QKI in CNS myelination. However, how QKI regulates MBP mRNA homeostasis in response to developmental signals remains unknown.
In this study, we found abundant QKI expression in the normal brain during the developmental window for the most active myelinogenesis, which was required for the rapid accumulation of the MBP mRNA. Interactions between QKI and the MBP 3′-UTR stabilized a reporter mRNA in transfected cells, and QKI deficiency resulted in destabilization and delayed accumulation of the MBP mRNA during CNS myelin development. However, in contrast to the rapid increase of the MBP mRNA, QKI expression was maintained at a steady level, raising the question of whether developmental signals may enhance the interactions between QKI and the MBP mRNA, which in turn leads to elevated MBP expression and accelerated myelinogenesis. Indeed, we found that Src family protein tyrosine kinases (Src-PTKs) phosphorylate QKI at the C-terminal tyrosine cluster and modulate the ability of QKI to bind MBP mRNA. Furthermore, Src-PTK-dependent tyrosine phosphorylation of QKI was vigorously regulated during active myelin production, providing a mechanism to enhance interactions between QKI and the MBP mRNA. These results suggest that QKI may act downstream of Src-PTKs to control CNS myelinogenesis via regulating MBP mRNA metabolism.
Results
QKI stabilizes MBP mRNA and is required for the rapid accumulation of MBP mRNA during active myelinogenesis
Three major QKI protein isoforms (QKI-5, 6 and 7, Figure 1A) exist in the myelin-producing cells of the CNS (Hardy et al., 1996). We developed a polyclonal antibody, which detected all three QKI protein isoforms in the whole tissue lysate derived from the normal brain (Figure 1B). Furthermore, QKI-6 and -7 were the predominant cytoplasmic isoforms detected in the postnuclear fraction of brain homogenate in which the nuclear isoform QKI-5 was largely diminished. Consistent with the previous report that QKI is negligible in brain neurons (Hardy et al., 1996), the neuronal cell line CAD1 (Qi et al., 1997) expressed very low levels of QKI (Figure 1B, right panel). Using this antibody, we examined the developmental expression of QKI in the brain stem of the qkv/qkv hypomyelination mutant mice in parallel with that in the non-phenotypic littermate controls (wt/qkv), and the result was superimposed with the expression profile of MBP mRNA (Figure 1C).

Fig. 1. QKI is required for the rapid accumulation of MBP mRNA during accelerated myelinogenesis. (A) Schematic representation of QKI isoforms (QKI-5, -6 and -7), each with a distinct C-terminus due to alternative splicing of the qkI primary transcript. QUA1 and QUA2 together with the single KH domain form the RNA-binding domain of QKI; the proline-rich PXXP motifs and the tyrosine cluster at the C-terminus are illustrated. (B) Detection of QKI protein isoforms in the brain. The left panel indicates detection of all three QKI isoforms in the whole tissue lysate derived from wild-type P10 brain stem (BS). The right panel shows detection of QKI-6 and -7 in the post-nuclear extracts derived from brain stem and CAD1 cells. The blot was re-probed by the antibody against the housekeeping protein eIF5α as a loading control. (C) Diminished QKI expression during accelerated myelinogenesis results in delayed MBP mRNA accumulation. The top panel illustrates the MBP mRNA expression profile in qkv/qkv and wt/qkv littermate controls. RPA was performed using total RNA derived from brain stems at P12, P20, P2M and P4M, and the MBP mRNA level was estimated by normalizing the PhosphorImager reading of MBP to that of GAPDH. The bottom panel shows the developmental profile of QKI protein expression in qkv/qkv and wt/qkv littermate controls. Brain stem extracts were prepared at indicated ages and subjected to SDS–PAGE immunoblot using anti-QKI antibody. The housekeeping protein LDH was used as a loading control. The level of QKI in each lane was estimated by normalization of total QKI signals to the LDH signal using NIH imaging software and indicated at the bottom.
In mice, the most active CNS myelinogenesis occurs in the first postnatal month. The non-phenotypic wt/qkv animals display normal myelin development, mimicking that in the wild type (Campagnoni and Macklin, 1988). During this period, accelerated expression of MBP mRNA proceeds along with myelin development, reaching its peak at postnatal day 20 (P20, Figure 1C). High level expression of QKI was detected in the wt/qkv brain and marked reduction of QKI was observed in the qkv/qkv brain during the most active period of myelinogenesis (P12–P20 in Figure 1C). The reduction of QKI in the qkv/qkv brain was specific for myelin-producing cells because QKI expression in non-myelinating glia is not affected (Hardy et al., 1996; our unpublished data). At later developmental stages, QKI gradually declined and was maintained at a low level in the non-phenotypic controls, accompanied with a concordant reduction of MBP mRNA (P2M and P4M in Figure 1C). In the qkv/qkv mutant brain, impaired QKI expression resulted in a failure in the rapid accumulation of MBP mRNA between P12 and P20 (Figure 1C). Instead, a slow accumulation occurred, resulting in a delayed peak by the end of the second postnatal month. At this age, the level of MBP mRNA detected in the qkv/qkv brain was comparable to that in the non-phenotypic littermates (Figure 1C). However, the delayed MBP mRNA accumulation, as a result of misregulation in response to developmental signals due to the lack of QKI, can not rescue the qkv hypomyelination phenotype. Therefore, the timing of MBP accumulation during early myelinogenesis is essential for CNS myelin development, and QKI appears to be an important factor required for such a process.
Our previous studies indicated that the failure of the rapid MBP mRNA accumulation during early myelin development in the qkv/qkv brain results from MBP mRNA destabilization (Li et al., 2000). To test whether QKI is directly involved in controlling the stability of MBP mRNA, we fused the QKI-binding MBP 3′-UTR to the 3′ end of the green fluorescence protein (GFP) that harbors negligible QKI-binding activity (Figure 2A). The MBP 3′-UTR conveyed strong QKI-binding to the GFP reporter mRNA (Figure 2A). When co-expressed with the cytoplasmic isoform QKI-6 in HEK293T host cells that lack endogenous QKI-6, the steady-state level of the GFP–MBP reporter mRNA was elevated ∼2.4-fold compared with control cells lacking QKI-6 (Figure 2B). In contrast, QKI-6 expression did not cause an increase of the GFP mRNA. Furthermore, the decay of the GFP–MBP reporter mRNA in QKI-expressing cells was significantly reduced (Figure 2C). This result supports our hypothesis that interactions between QKI and the MBP 3′-UTR leads to stabilization of the MBP mRNA.

Fig. 2. QKI stabilizes mRNA in a MBP 3′-UTR-dependent manner. (A) The MBP 3′-UTR conveys QKI-binding to the GFP reporter mRNA. In vitro RNA binding was performed using 35S-labeled QKI and biotin-labeled mRNA as described in Materials and methods. Parallel triplicates of RNA-binding reactions were performed and the average binding of the triplicate was calculated in each experiment. Four independent experiments were carried out (n = 4). The binding by GFP–MBP mRNA was normalized to that by the GFP mRNA in each paired reaction, and the standard deviation is indicated on top of the column. ** indicates P < 0.01 using paired t-test. (B) Co-expression of QKI results in elevated expression of the GFP–MBP reporter mRNA. GFP and GFP–MBP cell lines with or without stable expression of Flag-QKI-6 (QK+ and QK– respectively) were generated. The top panel shows a representative RPA gel that illustrates the steady state level of the reporter mRNA and the housekeeping β-actin mRNA. The quantitative analysis of the reporter mRNA (normalized to β-actin) from five independent experiments (n = 5) is illustrated in the bottom panel. * indicates P < 0.05 by paired t-test. The inset shows immunoblot analysis of Flag-QKI-6 and GFP expression in these cells. (C) QKI expression attenuates the decay of the GFP–MBP reporter mRNA. Cells were treated with actinomycin D (10 µg/ml) and RPA was carried out to determine the level of the GFP–MBP reporter mRNA normalized to that of the 18S rRNA at indicated time points. The level of reporter mRNA at 0 h was set as 100% and the decay curves were derived from five independent experiments (n = 5). Standard error is indicated for each data point. * indicates P < 0.05 by standard t-test when comparing results derived from QK+ and QK– cells.
We next examined the level of qkI gene expression during the developmental window when the level of MBP mRNA is vigorously elevated. In contrast to the MBP mRNA, expression of qkI mRNAs as well as the QKI proteins was maintained at a stable level (Figures 3 and 1C). Therefore, the QKI-dependent stabilization of MBP mRNA during active myelinogenesis is unlikely due to the elevated molecular ratio of QKI to MBP mRNA. This result raises a question of whether the RNA-binding activity of QKI is increased in response to developmental signals, perhaps by post-translational modification of QKI, which in turn leads to the rapid accumulation of MBP mRNA during accelerated myelin production.

Fig. 3. Expression of QKI and MBP mRNA during active myelinogenesis. The QKI mRNA isoforms were detected by RPA using an antisense riboprobe corresponding to the 3′-coding region of QKI-7 as described in Materials and methods. The full-length probe was protected by QKI-7; the rest of the QKI mRNA isoforms, mainly QKI-5 and QKI-6, protect a shorter fragment of the riboprobe that was evaluated in the same RPA gel. The steady state level of QKI-7, -5 and -6, as well as that of the MBP mRNA in wild-type brain stem was evaluated by RPA at postnatal day 7, 12, 16, 20 and 28. The top panel shows a representative RPA gel for detecting QKI mRNA isoforms and the housekeeping GAPDH mRNA. The PhosphorImager reading of QKI mRNAs is normalized to that of GAPDH, plotted against age and superimposed with the RPA result of MBP mRNA obtained from the same animals in the bottom panel.
Src-PTKs negatively regulate the RNA-binding activity of QKI
The putative SH3-binding motifs in the QKI C-terminus lead us to hypothesize that the RNA-binding activity of QKI is regulated by Src-PTK-mediated tyrosine phosphorylation, especially considering the fact that Src-PTKs modulate the activity of several RNA-binding proteins (Pype et al., 1994; Taylor and Shalloway, 1994; Wang et al., 1995; Ostareck-Lederer et al., 2002). We purified His-tagged recombinant QKI isoforms (His-QKI-5, 6 and 7) and incubated them with Src-PTKs in the presence of [γ-32P]ATP. As shown in Figure 4A, His-QKI isoforms were phosphorylated by members of the Src-PTK family, including Src and Fyn. We also incubated in vitro translated [35S]QKI-7 with Src (Figure 4B), which resulted in retarded migration of [35S]QKI-7 on SDS–PAGE, a common feature observed for many other phospho-proteins. To further test whether QKI can be tyrosine phosphorylated by Src-PTKs in living cells, we co-expressed Flag-tagged QKI isoforms with Src-PTKs in HEK293T cells that harbor low levels of endogenous Src-PTK activity. When co-expressed with Src-Y527F (SrcA in Figure 4C), a mutant Src that harbors constitutive kinase activity (Fresno Vara et al., 2001), all QKI isoforms were tyrosine phosphorylated as demonstrated by immunoprecipitation–immunoblot analysis using the phosphotyrosine-specific antibody PY20 (Figure 4C). In contrast, Src-K295R (SrcI), a mutant Src that lacks kinase activity (Kamps and Sefton, 1986), did not cause phosphorylation of QKI in parallel experiments.

Fig. 4. QKI isoforms are phosphorylated by Src-PTKs. (A) Phos phorylation of QKI by Src and Fyn in vitro. Src-PTK-dependent [γ-32P]ATP labeling of His-QKI isoforms and [32P]Src due to autophosphorylation are indicated on the left. (B) Retarded migration of Src-treated [35S]QKI-7 on SDS–PAGE (arrows on the right). The lower molecular weight [35S]QKI-7 band is an N-terminal truncated product derived from internal initiated translation. (C) Tyrosine phosphorylation of Flag-QKI isoforms in HEK293T cells. SrcA (Src-Y527F), constitutively active kinase; SrcI (Src-K295R), inactive kinase; IP, immunoprecipitation; IB, immunoblot; Anti-PTyr, phosphotyrosine-specific antibody PY20.
We next examined whether tyrosine phosphorylation modulates the ability of QKI for interaction with MBP mRNA. As shown in Figure 5A, Src treatment significantly attenuated the interaction between QKI and the MBP mRNA, despite the fact that the experimental conditions only resulted in tyrosine phosphorylation of <50% of input QKI (Figure 4B). The activity for binding MBP mRNA by all three QKI isoforms was regulated to a similar degree by Src treatment (Figure 5B). This offers a mechanism to control the RNA-binding activity of QKI via Src-PTKs whose activity is subjected to regulation by a variety of developmental and extracellular signals during CNS development (Kramer et al., 1999; Nakazawa et al., 2001). To address the specificity of the influence of Src-PTKs on QKI–RNA interaction, we also examined the effect of Src on the RNA-binding activity of the fragile X mental retardation protein (FMRP). FMRP has been shown to bind the MBP 3′-UTR in vitro (Li et al., 2001), and can be phosphorylated by Src-PTKs in vitro (data not shown). In contrast to QKI, Src treatment did not affect the activity of FMRP in binding MBP mRNA (Figure 5C).

Fig. 5. Tyrosine phosphorylation of QKI by Src-PTKs negatively regulates the interactions between QKI and the MBP mRNA. (A) Src treatment attenuates the interactions between QKI-7 and the MBP mRNA. RNA binding and Src treatment of QKI-7 are described in Materials and methods. [35S]QKI-7 input (ng) is plotted against RNA-bound [35S]QKI-7 determined by scintillation counting (c.p.m.). (B) Src treatment negatively regulates the activity of all three QKI isofroms (Q5, Q6 and Q7) for binding MBP mRNA. Ten nanograms of 35S-labeled protein estimated by comparison with known amounts of His-QKI on SDS–PAGE immunoblot was used in each reaction. For each isoform, the MBP mRNA-bound QKI from mock-treated reactions was defined as 100%, and MBP mRNA-bound QKI from Src-treated reactions was normalized to that of mock-treated parallel reaction. The lower panel shows a representative SDS–PAGE image of the RNA-bound QKI isoforms in Src- and mock-treated reactions. The captured QKI in each reaction accounts for ∼10% of the input after the stringent washes. (C) Src treatment specifically regulates the activity of QKI in binding MBP mRNA. Ten nanograms of 35S-labeled QKI-7 or FMRP was used in each reaction. The number of experiments performed (n) is indicated. Standard error is indicated for each column; *, P < 0.05, paired t-test.
The C-terminal tyrosine cluster of QKI mediates phosphorylation and regulation of RNA-binding by Src-PTKs
To identify the tyrosine residue(s) that mediates Src-dependent phosphorylation of QKI and regulation of RNA-binding activity, we performed mutagenesis experiments on QKI cDNA. Our initial approach indicated that C-terminal truncations of QKI completely abolished Src-dependent phosphorylation (data not shown). In the deleted QKI C-terminus, five tyrosines form a cluster (Y285, 288, 290, 292 and 303, marked Ya–Ye respectively in the top panel of Figure 6A for simplicity), which is conserved from fruit flies to humans (Vernet and Artzt, 1997). Site-directed mutagenesis, in which each of these tyrosines was replaced by phenylalanine, resulted in a reduction of Src-dependent phosphorylation of His-QKI (Figure 6A, the substituted Y was indicated for each mutant). Nonetheless, Src-dependent phosphorylation was completely abolished only when all five tyrosines were replaced (QKI Ya–e). When co-expressed with constitutively active Src-PTKs (Src-Y527F or Fyn-Y528F) in HEK293T cells, wild-type QKI was efficiently phosphorylated, whereas the mutant QKI Ya–e failed to be phosphorylated by Src-PTKs (Figure 6B). Apparently, the bona fide RNA-binding activity of QKI was not affec ted by mutations of the C-terminal tyrosine cluster (Figure 6C). However, the ability to respond to Src-PTK-dependent regulation of RNA-binding was markedly attenuated for QKI Yb–d and completely abrogated for QKI Ya–e (Figure 6C). These results demonstrated that the C-terminal tyrosine cluster of QKI mediates phosphorylation and regulation of RNA-binding by Src-PTKs.

Fig. 6. Identification of tyrosines that mediate Src-dependent regulation of QKI–RNA interaction. (A) The C-terminal tyrosine cluster mediates Src-dependent tyrosine phosphorylation of QKI (QKI–PTyr). Top panel, amino acids of QKI-7 (A281–V310) harboring the tyrosine cluster. Y285, 288, 290, 292 and 303 are marked Ya, Yb, Yc, Yd and Ye for simplicity. Middle panel, Src-dependent [γ-32P]ATP labeling of His-QKI-7. Phenylalanine substitutions at various Ys were obtained by site-directed mutagenesis and marked on top of each lane. Bottom panel, the quantity of His-QKI in each phosphorylation reaction was evaluated by immunoblot using anti-QKI antibody. (B) Mutation of the C-terminal tyrosine cluster blocks QKI–PTyr in HEK293T cells. The expression of Flag-QKI-7 and HA-tagged kinases in lysate (load), and Flag-QKI–PTyr in the immunoprecipitate (IP) was detected by immunoblot (IB). (C) Mutation of the C-terminal tyrosine cluster abrogates Src-dependent regulation of QKI–RNA interaction. RNA binding was performed as described in Figure 5. Standard error was indicated for each column; *, P < 0.05, paired t-test.
In order to ascertain that tyrosine phosphorylation of QKI indeed occurs in the brain, we performed immunoprecipitation analysis from the postnuclear fraction of brain homogenate using the anti-phosphotyrosine antibody and the anti-QKI antibody, respectively. The immunoprecipitated QKI (mainly QKI-6 and QKI-7) from brain cytoplasmic lysate contained phosphotyrosine (Figure 7A, upper panel). In the reciprocal experiment, we detected QKI in the pool of phosphotyrosine-containing proteins immunoprecipitated from the brain (Figure 7A, bottom panel). As expected, no signal was detected from immunoprecipitates derived from the CAD1 cells that express negligible levels of QKI (Figure 1B).

Fig. 7. The C-terminal tyrosine cluster is the target for Src-PTK-dependent tyrosine phosphorylation of QKI in the myelin compartment. (A) Detection of QKI–PTyr by immunoprecipitation (IP) from the post-nuclear extracts derived from P12 brain stem (BS) and CAD1. (+) and (–) indicate the presence or absence of the corresponding primary antibody in IP. (B) The C-terminal tyrosine cluster of QKI mediates QKI phosphorylation by kinases in isolated myelin. [γ-32P]ATP labeling assay was carried out using wild-type or mutant His-QKI-7 in the presence of Src, isolated myelin (myl) or whole cytoplasmic extract (cyt) derived from normal brain stem as described in Materials and methods. (C) Tyrosine phosphorylation of QKI in the isolated myelin is mediated by the PPXP motif. The top panel shows amino acids in the C-terminus of QKI-7, containing a single PPXP motif (marked by a bold underline) and a few PXXP proline-rich motifs (underlined). In vitro phosphorylation was carried out in isolated myelin using wild-type His-QKI-7, mutant His-QKI-7 Ya and the mutant His-QKI-7 DSH3 in which the prolines in the single PPXP motif were replaced by alanines. A representative SDS–PAGE illustrating 32P-labeled QKI was shown on the left. The PhosphorImager reading of wild-type His-QKI-7 was defined as 100%, and the PhosphorImager reading of each mutant was normalized to that of wild type in parallel experiments. Statistic analysis was carried out for three independent experiments (n = 3) and shown on the right. *, P < 0.05; ***, P < 0.001 compared with wild-type His-QKI by standard t-test. (D) Inhibition of QKI–PTyr in the developing myelin by Src-PTK inhibitor. Concentrations for each inhibitor are indicated on top of the corresponding lanes.
We next asked whether the C-terminal tyrosines of QKI are phosphorylated in the myelin compartment of brain, where MBP mRNA is deposited for localized translation (Colman et al., 1982; Ainger et al., 1997). A well-established procedure was employed to isolate myelin membrane from mouse brain stem (Kramer et al., 1999). His-QKI-7 was incubated with the isolated myelin under an optimized condition for Src-PTKs in the presence of [γ-32P]ATP in order to evaluate the activity of Src-PTKs in the myelin compartment. As shown in Figure 7B, wild-type QKI was labeled strongly whereas QKI Ya–e completely failed to be phosphorylated. This result demonstrated that phosphorylation of QKI in the myelin compartment is mediated by the C-terminal tyrosine cluster, which appears to control the RNA-binding activity of QKI. In contrast, both wild-type QKI and the mutant QKI Ya–e were phosphorylated by kinases in the whole cytoplasmic extract derived from the brain stem (Figure 7B). This result suggests that the C-terminal tyrosine cluster may specifically mediate tyrosine phosphorylation of QKI in myelin compartment and/or in myelin producing cells.
Using the same assay, we showed that mutation of the single PPXP putative SH3-binding motif in the C-terminus of QKI abrogated ∼70% of tyrosine phosphorylation of QKI in the isolated myelin (Figure 7C). Furthermore, tyrosine phosphorylation of QKI in the developing myelin was inhibited in a dose-dependent manner by pyrazolopyrimidine pp2 (Figure 7D), a specific inhibitor of Src-PTKs (Sanna et al., 2000). In contrast, QKI phosphorylation was not inhibited by pp3, a tyrosine kinase inhibitor that does not affect Src-PTKs (Sanna et al., 2000). Taken together, these results indicated that Src-PTKs were responsible for tyrosine phosphorylation of QKI in the myelin compartment, most likely via SH3-mediated interaction with QKI.
Declined tyrosine phosphorylation of QKI during active myelinogenesis associates with rapid MBP mRNA accumulation
The functional consequence of Src-PTK-dependent tyrosine phosphorylation of QKI on MBP mRNA homeo stasis depends on how such modification is regulated during myelin development. We examined Src-PTK-dependent tyrosine phosphorylation of QKI in the isolated myelin during active myelination. As shown in Figure 8, the level of tyrosine phosphorylation of QKI by Src-PTKs in the developing myelin was highest in the first postnatal week (P7). During the vigorous accumulation of MBP mRNA between P7 and P20 (Figure 3), tyrosine phosphorylation of QKI in the developing myelin drastically declined. By the end of the fourth postnatal week (P28), tyrosine phosphorylation of QKI was reduced ∼90%. Since Src-PTK-dependent phosphorylation reduced QKI’s activity in binding MBP mRNA (Figures 5 and 6), down-regulating tyrosine phosphorylation of QKI observed in the above experiment is expected to enhance the interaction between QKI and the MBP mRNA and increase the stability of MBP mRNA. Indeed, the decline of QKI tyrosine phosphorylation was associated with the vigorous accumulation of the MBP mRNA (Figure 3) as well as elevated expression of MBP protein (Figure 8). Therefore, the developmentally controlled down-regulation of QKI tyrosine phosphorylation suggested a novel mechanism to accelerate myelinogenesis via facilitation of the interaction between QKI and the MBP mRNA during this critical window of myelin development.
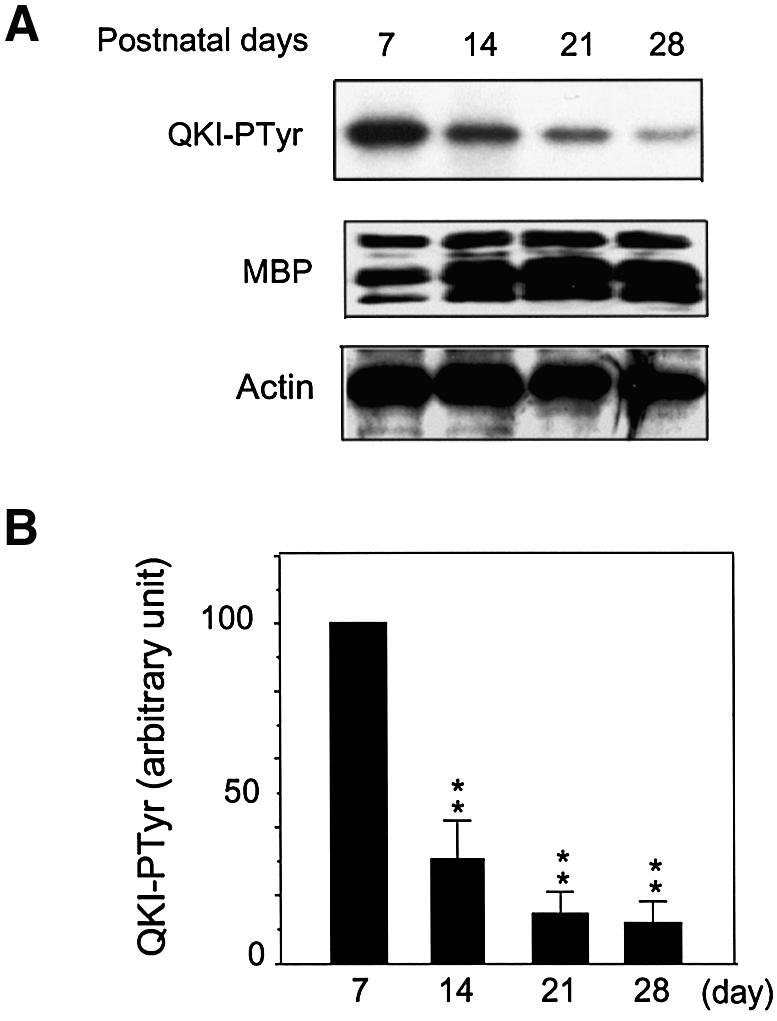
Fig. 8. Down-regulation of QKI–PTyr during the most active phase of myelinogenesis associates with MBP accumulation. (A) QKI–PTyr in the developing myelin and MBP expression during myelin development. His-QKI-7 was subjected to phosphorylation by Src-PTKs in brain stem myelin at indicated ages in the presence of [γ-32P]ATP. The top panel shows an image of SDS–PAGE indicating the down-regulation of [32P]QKI–Ptyr. The middle panel shows the concordant increase of MBP detected by immunoblot in isolated myelin during development. The multiple bands of MBP represent major MBP protein isoforms derived from alternative splicing of the MBP mRNA. The bottom panel shows β-actin in the re-probed immunoblot as a loading control. (B) Quantitative analysis of [32P]QKI–PTyr in the developing myelin by PhosphorImager analysis (n = 4). The standard deviation was indicated. P < 0.001 by one way ANOVA; **, P < 0.01 when compared with QKI–PTyr in the isolated myelin derived from P7 brain stem.
Discussion
The above results suggest that the selective RNA-binding protein QKI is a downstream target of Src-PTKs. QKI belongs to the fast growing protein family designated as signal transduction activator of RNA (STAR). STAR members harbor a single KH domain and several proline-rich SH3-binding motifs, featuring dual interaction with mRNA and signaling molecules (Vernet and Artzt, 1997). Other STAR members include the Src-associated protein at mitosis (Sam68) (Taylor et al., 1995), the Caenorhabditis elegans tumor suppressor GLD1 (Jones and Schedl, 1995), the mammalian splicing factor SF1 (Wang et al., 1999) and the Drosophila protein T-STAR/ETOILE that controls spermatogenesis (Venables et al., 1999). The function of STARs has been postulated to mediate developmental and/or extracellular signals to control the cellular fate of mRNA (Vernet and Artzt, 1997). The QKI-mediated MBP mRNA stabilization (Figures 1 and 2), the modulation of the RNA-binding activity of QKI by Src-PTKs (Figures 5 and 6) and the developmental regulation of QKI tyrosine phosphorylation (Figure 8) support the hypothesis that QKI is a pivotal intermediate for developmental signals to control CNS myelination via regulating mRNA homeostasis.
The C-terminal tyrosine cluster in QKI is responsible for QKI phosphorylation by Src-PTKs in vitro, in HEK293T cells and in the myelin compartment (Figures 6 and 7). Furthermore, phosphorylation of QKI on these tyrosine residues regulates QKI–mRNA interaction (Figures 5 and 6). There are five additional tyrosines in all QKI isoforms, located at the N-terminus and the KH domain respectively. All the tyrosines in QKI are conserved from Xenopus to humans (Vernet and Artzt, 1997; Zorn et al., 1997). However, the N-terminal tyrosines are not targets for Src-PTKs, but are phosphorylated by other kinases perhaps in other cell types, as suggested by the phosphorylation of the mutant QKI Ya–e in the complete brain cytoplasmic extract (Figure 7B). Selective kinase inhibitors and mutation of the PPXP motif of QKI (Figure 7) indicated that the kinase(s) responsible for phosphorylating QKI in the myelin compartment belongs to the family of Src-PTKs, which most likely interacts with QKI via the SH3 domain. It is worth pointing out that SH3-mediated phosphorylation by Src-PTKs appears to be a common mechanism for regulating RNA-binding activity of STARs (Taylor et al., 1995; Wang et al., 1995) as well as that of other RNA-binding proteins including hnRNPK (Ostareck-Lederer et al., 2002). In all the reported studies, Src-PTK-dependent phosphorylation negatively regulates the RNA-binding activity. Nonetheless, our studies characterized such regulation in the developmental context for the first time, providing a comprehensive model of how STARs may function as positive regulators in mRNA homeostasis via developmentally controlled down-regulation of tyrosine phosphorylation.
The activity of Src-PTKs is subjected to regulation by a variety of developmental and extracellular signals (Umemori et al., 1994; Lu et al., 1998; Yu and Salter, 1999; Nakazawa et al., 2001). During CNS development, Src-PTK-dependent phosphorylation of the N-methyl-d-aspartate (NMDA) receptor is up-regulated and further induced by long term potentiation (Lu et al., 1998; Nakazawa et al., 2001). In addition, the expression of the Src-PTK member Fyn is elevated during early differentiation of oligodendrocytes (Osterhout et al., 1999), which appears to play important roles in regulating the morphological development of oligodendrocytes (Osterhout et al., 1999; Klein et al., 2002). Interestingly, Fyn activity gradually declined in the developing myelin during the first three postnatal weeks (Umemori et al., 1994; Kramer et al., 1999), which is consistent with the developmentally controlled down-regulation of tyrosine phosphorylation of QKI (Figure 8). The remarkable regulation of QKI tyrosine phosphorylation in the myelin compartment during the defined developmental window suggests that QKI is a critical factor that mediates developmental signals to control myelinogenesis. Whether all Src-PTK members are involved in regulating tyrosine phosphorylation of QKI in developing myelin needs to be further delineated by future studies.
The high level of QKI expression before P20 and the declined expression of QKI in later stages of normal myelin development (Figure 1C) suggest that QKI may be mostly required during the first few postnatal weeks where accelerated myelinogenesis occurs. Sufficient expression of QKI is essential for the developmentally regulated accumulation of MBP mRNA before it reaches its peak at P20. The severe reduction of QKI in qkv/qkv brain results in a failure of MBP mRNA accumulation at P20 (Figure 1C) due to decreased stability of the MBP mRNA (Li et al., 2000), presumably as a result of insufficient protection from nuclease attack. In contrast, no abnormality at the level of MBP transcription or translation was observed in the qkv/qkv brain (Li et al., 2000). In addition, QKI-5 was reported to regulate mRNA splicing (Wu et al., 2002). Although tyrosine phosphorylation of QKI-5 may regulate its influence on splicing which in turn may affect mRNA stability, QKI-6-mediated mRNA stabilization (Figure 2) can occur independently of QKI-5, because the HEK293T host cells employed in our study harbor negligible levels of QKI-5 (data not shown). Besides the stabilization of MBP mRNA by QKI, How, the Drosophila homolog of QKI, stabilizes its target mRNA during tendon cell differentiation (Nabel-Rosen et al., 2002). These observations suggest that QKI-mediated mRNA stabilization is employed by many developmental systems. The down-regulation of tyrosine phosphorylation of QKI during active myelinogenesis is expected to enhance the interaction between QKI and the MBP mRNA, which provides a novel mechanism for accelerating the accumulation of MBP mRNA via mRNA stabilization.
The functional significance of regulating QKI–RNA interaction by tyrosine phosphorylation is not limited to MBP mRNA metabolism. In fact, QKI isoforms may play different roles at various steps of post-transcriptional regulation, including splicing, subcellular localization and translation of mRNA (Barbarese, 1991; Saccomanno et al., 1999; Li et al., 2000; Wu et al., 2002). For example, QKI-6 was reported to inhibit translation of its bound reporter mRNA in vitro (Saccomanno et al., 1999). In addition, QKI-5 can cause nuclear retention of the MBP mRNA and hence inhibits MBP production (Larocque et al., 2002). During accelerated myelinogenesis, the abundant expression of QKI and the decline in tyrosine phosphorylation of QKI most likely facilitates QKI–MBP mRNA interaction, and hence is expected to strengthen the influence by QKI. Therefore, in contrast to the role of QKI on MBP mRNA stabilization, the potential inhibitory effects of QKI by nuclear retention and translation suppression of MBP may not contribute to the vigorous elevation of MBP for the peak of myelination. However, Src-PTK-dependent tyrosine phosphorylation of QKI may regulate these inhibitory effects by QKI at other developmental stages. Besides myelin-producing cells, QKI and its homologues are broadly expressed in various tissues (Kondo et al., 1999). The function of QKI has been implicated in cardiovascular development (Zaffran et al., 1997; Noveroske et al., 2002) as well as in apoptosis and tumorigenesis (Jones and Schedl, 1995; Pilotte et al., 2001; Li et al., 2002). Therefore, the novel mechanism of Src-PTK-dependent regulation of QKI–RNA interaction in CNS myelinogenesis may be broadly employed in other systems for regulating the cellular fate of mRNA in response to various developmental and extracellular signals.
Materials and methods
Animals and RNA–protein isolation
The qkv colony was purchased from Jackson Laboratory. qkv/wt and qkv/qkv littermates were produced as described previously (Li et al., 2000). For total RNA preparation, brain stem was dissected at various ages, immediately followed by RNA extraction using Trizol™ (Life Technology). The quantity of RNA from each sample was determined by OD260 absorption and further confirmed by ethidium bromide-stained agarose gel electrophoresis. Cytoplasmic extract from brain tissues was prepared as described previously (Li et al., 2000). For whole tissue lysate preparation, brain stems were minced and sonicated in 1 × Laemmli buffer. The quantity of total protein in each sample was estimated by Bradford assay (Bio-Rad). Equal amounts of protein (∼10 µg) or RNA (∼5 µg) from each sample was used for further analysis.
Recombinant QKI protein production
His-QKI-5 and His-QKI-6 in PET28a (Novagen) were generously provided by Dr Artzt. QKI-7 coding region (Li et al., 2000) was PCR- amplified and cloned into PET28a (Novagen) to fuse with the His tag, or into PGEX4 vector to fuse with glutathione S-transferase (GST) (Pharmacia). Mutagenesis for replacing the C-terminal tyrosines by phenylalanines as well as the prolines by alanines in the SH3-binding motif was carried out using the QuikChange kit following manufacturer’s protocols (Stratagene). The constructs were sequenced to confirm 100% identity in comparison to published sequences before production of recombinant His-QKI and GST–QKI respectively following manufacturer’s instructions (Qiagen). Recombinant proteins were subjected to SDS–PAGE to determine the purity and quantity, and were eluted from SDS–PAGE before being used as immunogen.
Cell culture and transgene
The MBP 3′-UTR (provided by Dr Campagnoini, UCLA) was subcloned into the NotI site downstream of the open reading frame of GFP–N3 (Clontech) to obtain the construct of GFP–MBP. GFP–N3 and GFP–MBP were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogene), and stable cell lines expressing each of the transgene were established by G418 selection. Flag-tagged QKI-6 expression was introduced into these host cell lines by the Zeocin resistant retroviral vector Zeo96 and stable cell lines were established by Zeocin selection. QKI expression was confirmed by immunoblot analysis using anti-Flag M2 antibody (Sigma). To measure the decay of the GFP–MBP reporter mRNA, cells were treated with actinomycin D (CalBiochem) at 10 µg/ml to block transcription. At indicated time points, RNase protection assay (RPA) was performed and the level of reporter mRNA was measured by PhosphorImager quantification and normalized to the RPA signal for the 18S rRNA (Ambion).
Immunoblot analysis and antibodies
Anti-Flag M2 antibody and anti-lactate dehydrogenase (LDH) antibody were purchased from Sigma, anti-eIF5α antibody was purchased from Santa Cruz, the monoclonal anti-MBP antibody that recognizes all major MBP isoforms was purchased from Chemicon (MAB386) and PY20 antibody was purchased from Upstate. To generate anti-QKI antibody, recombinant His-QKI-7 was used to immunize rabbit and guinea pig (Cocalico Inc.). The serum was subjected to IgG purification followed by affinity purification using GST–QKI-7. The specificity of anti-QKI antibodies was defined by immunoprecipitation of [35S]QKI isoforms generated from in vitro translation (results will be published elsewhere).
RNase protection assay
The RPA for MBP and GAPDH transcripts was performed as described previously (Li et al., 2000). For QKI RPA, a 351 bp cDNA fragment containing the 3′-end of QKI-7 cDNA was PCR amplified and cloned into TA-Cloning Vector (Invitrogen). The cDNA was sequenced to identify orientation and to confirm 100% identity as published. Antisense riboprobe was generated by in vitro transcription in the presence of T7 polymerase (Stratagene) and [32P]UTP (Amersham) using linearized plasmid. The GFP RPA cDNA fragment was obtained by PCR using primers GGAGTACAACTACAACAGC and GAGCTCGAGATCTGAGTCC. This fragment was cloned into pDrive vector (Qiagen) and 32P-labeled antisense RPA probe was generated from in vitro transcription as described above. Hybridization and RPA were performed according to procedures published previously (Li et al., 2000).
RNA-binding assay
RNA-binding assay was performed according to the previously published procedure (Ashley et al., 1993). [35S]methionine-labeled QKI-7 and FMRP were generated by TNT reaction containing T7 RNA polymerase (Promega). Biotinylated RNA encoding for the 14 kDa isoform of MBP (the major isoform in the brain), GFP and the GFP–MBP reporter were synthesized by in vitro transcription (Stratagene) using protocols published previously (Li et al., 2000). For Src treatment of in vitro translated [35S]QKI, 10 µl of translation reaction was incubated with 1 unit of Src in 50 µl of 1 × kinase buffer (Upstate). Mock reactions were carried out in the same buffer without Src. For RNA binding, 120 ng of the biotinylated RNA was incubated with [35S]QKI-7 or [35S]FMRP in the presence of 1 µg of yeast t-RNA as indicated in the corresponding figures. Reactions in which Src- or mock-treated [35S]QKI were incubated with no RNA was set as background control. Biotinylated RNA was captured by Streptavidin–magnetic beads (Dynal). After three washes, the co-captured [35S]QKI-7 was eluted by 1 × Laemmli buffer followed by scintillation counting, or SDS–PAGE analysis to visualize [35S]QKI-7 by a PhosphorImager.
Myelin isolation and kinase assay
Myelin membranes were isolated by two rounds of discontinuous sucrose gradients with two rounds of hypotonic shock using the procedure described previously (Umemori et al., 1994). The isolated myelin was re-suspended in 1 × kinase buffer (Upstate) containing 0.25 mM NaVO3, 1 mM PMSF and 0.3% IGPCA630 (Sigma). Total protein from isolated myelin was quantified by Bradford assay (Bio-Rad). Purified Src and Fyn were purchased from Upstate. In vitro kinase assay was performed under conditions following the manufacturer’s instructions. For purified kinases, 100 ng of His-QKI was incubated with 0.5 unit of kinase at 30°C for 30 min before SDS–PAGE analysis. For kinase reactions using isolated myelin, 500 ng of His-QKI-7 (wt or Ya–e mutant) was bound to Magnetic Ni beads (Qiagene). After three washes, the bound QKI was incubated with myelin lysate containing 3 µg protein in the presence of 10 µCi [γ-32P]ATP in 50 µl 1 × kinase buffer as described above followed by SDS–PAGE analysis. The phosphorylation level of QKI in each reaction was determined by PhosphorImager analysis.
Acknowledgments
Acknowledgements
We are grateful to Drs A.Campagnoni, X.Xu, K.Artzt and T.J.Murphy for cDNA constructs of MBP, Src-PTKs and QKI, as well as for the retroviral vector Zeo96. We thank S.T.Warren, R.Dingledine and D.Reines for valuable discussion in the process of preparation of this manuscript. The work is supported by NMSS grant RG3296 and NIH grant NS 39551 to Y.F.
References
- Ainger K., Avossa,D., Diana,A.S., Barry,C., Barbarese,E. and Carson,J.H. (1997) Transport and localization elements in myelin basic protein mRNA. J. Cell Biol., 138, 1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley C.T. Jr, Wilkinson,K.D., Reines,D. and Warren,S.T. (1993) FMR1 protein: conserved RNP family domains and selective RNA binding. Science, 262, 563–566. [DOI] [PubMed] [Google Scholar]
- Barbarese E. (1991) Spatial distribution of myelin basic protein mRNA and polypeptide in quaking oligodendrocytes in culture. J. Neurosci. Res., 29, 271–281. [DOI] [PubMed] [Google Scholar]
- Campagnoni A.T. and Macklin,W.B. (1988) Cellular and molecular aspects of myelin protein gene expression. Mol. Neurobiol., 2, 41–89. [DOI] [PubMed] [Google Scholar]
- Colman D.R., Kreibich,G., Frey,A.B. and Sabatini,D.D. (1982) Synthesis and incorporation of myelin polypeptides into CNS myelin. J. Cell Biol., 95, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole T.A., Chen,Q., Justice,M.J. and Artzt,K. (1996) The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat. Genet., 12, 260–265. [DOI] [PubMed] [Google Scholar]
- Fresno Vara J.A., Caceres,M.A., Silva,A. and Martin-Perez,J. (2001) Src family kinases are required for prolactin induction of cell proliferation. Mol. Biol. Cell, 12, 2171–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy R.J. (1998) Molecular defects in the dysmyelinating mutant quaking. J. Neurosci. Res., 51, 417–422. [DOI] [PubMed] [Google Scholar]
- Hardy R.J., Loushin,C.L., Friedrich,V.L.,Jr, Chen,Q., Ebersole,T.A., Lazzarini,R.A. and Artzt,K. (1996) Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J. Neurosci., 16, 7941–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan E.L. and Greenfield,S. (1984) Animal Models of Genetic Disorders of Myelin. Plenum Press, New York, NY.
- Inoue Y., Nakamura,R., Mikoshiba,K. and Tsukada,Y. (1981) Fine structure of the central myelin sheath in the myelin deficient mutant Shiverer mouse, with special reference to the pattern of myelin formation by oligodendroglia. Brain Res., 219, 85–94. [DOI] [PubMed] [Google Scholar]
- Jones A.R. and Schedl,T. (1995) Mutations in gld-1, a female germ cell-specific tumor suppressor gene in Caenorhabditis elegans, affect a conserved domain also found in Src-associated protein Sam68. Genes Dev., 9, 1491–1504. [DOI] [PubMed] [Google Scholar]
- Kamps M.P. and Sefton,B.M. (1986) Neither arginine nor histidine can carry out the function of lysine-295 in the ATP-binding site of p60src. Mol. Cell. Biol., 6, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C., Kramer,E.M., Cardine,A.M., Schraven,B., Brandt,R. and Trotter,J. (2002) Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J. Neurosci., 22, 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T., Furuta,T., Mitsunaga,K., Ebersole,T.A., Shichiri,M., Wu,J., Artzt,K., Yamamura,K. and Abe,K. (1999) Genomic organization and expression analysis of the mouse qkI locus. Mamm. Genome 10, 662–669. [DOI] [PubMed] [Google Scholar]
- Kramer E.M., Klein,C., Koch,T., Boytinck,M. and Trotter,J. (1999) Compartmentation of Fyn kinase with glycosylphosphatidylinositol-anchored molecules in oligodendrocytes facilitates kinase activation during myelination. J. Biol. Chem., 274, 29042–29049. [DOI] [PubMed] [Google Scholar]
- Larocque D. et al. (2002) Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron, 36, 815–829. [DOI] [PubMed] [Google Scholar]
- Li Z., Zhang,Y., Li,D. and Feng,Y. (2000) Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J. Neurosci., 20, 4944–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Zhang,Y., Ku,L., Wilkinson,K.D., Warren,S.T. and Feng,Y. (2001) The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res., 29, 2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.Z., Kondo,T., Murata,T., Ebersole,T.A., Nishi,T., Tada,K., Ushio,Y., Yamamura,K. and Abe,K. (2002) Expression of Hqk encoding a KH RNA binding protein is altered in human glioma. Jpn J. Cancer Res., 93, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.M., Roder,J.C., Davidow,J. and Salter,M.W. (1998) Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science, 279, 1363–1367. [DOI] [PubMed] [Google Scholar]
- Nabel-Rosen H., Volohonsky,G., Reuveny,A., Zaidel-Bar,R. and Volk,T. (2002) Two isoforms of the Drosophila RNA binding protein, How, act in opposing directions to regulate tendon cell differentiation. Dev. Cell, 2, 183–193. [DOI] [PubMed] [Google Scholar]
- Nakazawa T., Komai,S., Tezuka,T., Hisatsune,C., Umemori,H., Semba,K., Mishina,M., Manabe,T. and Yamamoto,T. (2001) Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem., 276, 693–699. [DOI] [PubMed] [Google Scholar]
- Noveroske J.K., Lai,L., Gaussin,V., Northrop,J.L., Nakamura,H., Hirschi,K.K. and Justice,M.J. (2002) Quaking is essential for blood vessel development. Genesis, 32, 218–230. [DOI] [PubMed] [Google Scholar]
- Ostareck-Lederer A., Ostareck,D.H., Cans,C., Neubauer,G., Bomsztyk,K., Superti-Furga,G. and Hentze,M.W. (2002) c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol. Cell. Biol., 22, 4535–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterhout D.J., Wolven,A., Wolf,R.M., Resh,M.D. and Chao,M.V. (1999) Morphological differentiation of oligodendrocytes requires activation of Fyn tyrosine kinase. J. Cell Biol., 145, 1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte J., Larocque,D. and Richard,S. (2001) Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev., 15, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pype S., Slegers,H., Moens,L., Merlevede,W. and Goris,J. (1994) Tyrosine phosphorylation of a M(r) 38 000 A/B-type hnRNP protein selectively modulates its RNA binding. J. Biol. Chem., 269, 31457–31465. [PubMed] [Google Scholar]
- Qi Y., Wang,J.K., McMillian,M. and Chikaraishi,D.M. (1997) Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci., 17, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach A., Takahashi,N., Pravtcheva,D., Ruddle,F. and Hood,L. (1985) Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell, 42, 149–155. [DOI] [PubMed] [Google Scholar]
- Saccomanno L., Loushin,C., Jan,E., Punkay,E., Artzt,K.B. and Goodwin,E.B. (1999) The STAR protein QKI-6 is a translational repressor. Proc. Natl Acad. Sci. USA, 96, 12605–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna P.P., Berton,F., Cammalleri,M., Tallent,M.K., Siggins,G.R., Bloom,F.E. and Francesconi,W. (2000) A role for Src kinase in spontaneous epileptiform activity in the CA3 region of the hippocampus. Proc. Natl Acad. Sci. USA, 97, 8653–8657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S.J. and Shalloway,D. (1994) An RNA-binding protein associated with src through its SH2 and SH3 domains in mitosis. Nature, 368, 867–871. [DOI] [PubMed] [Google Scholar]
- Taylor S.J., Anafi,M., Pawson,T. and Shalloway,D. (1995) Functional interaction between c-Src and its mitotic target, Sam 68. J. Biol. Chem., 270, 10120–10124. [DOI] [PubMed] [Google Scholar]
- Umemori H., Sata,S., Yagi,T., Aizawa,S. and Yamamoto,T. (1994) Initial events of myelination involve Fyn tyrosine kinase signalling. Nature, 367, 572–576. [DOI] [PubMed] [Google Scholar]
- Venables J.P., Vernet,C., Chew,S.L., Elliott,D.J., Cowmeadow,R.B., Wu,J., Cooke,H.J., Artzt,K. and Eperon,I.C. (1999) T-STAR/ETOILE: a novel relative of SAM68 that interacts with an RNA-binding protein implicated in spermatogenesis. Hum. Mol. Genet., 8, 959–969. [DOI] [PubMed] [Google Scholar]
- Vernet C. and Artzt,K. (1997) STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet., 13, 479–484. [DOI] [PubMed] [Google Scholar]
- Wang L.L., Richard,S. and Shaw,A.S. (1995) P62 association with RNA is regulated by tyrosine phosphorylation. J. Biol. Chem., 270, 2010–2013. [DOI] [PubMed] [Google Scholar]
- Wang X., Bruderer,S., Rafi,Z., Xue,J., Milburn,P.J., Kramer,A. and Robinson,P.J. (1999) Phosphorylation of splicing factor SF1 on Ser20 by cGMP-dependent protein kinase regulates spliceosome assembly. EMBO J., 18, 4549–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.I., Reed,R.B., Grabowski,P.J. and Artzt,K. (2002) Function of quaking in myelination: regulation of alternative splicing. Proc. Natl Acad. Sci. USA, 99, 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.M. and Salter,M.W. (1999) Src, a molecular switch governing gain control of synaptic transmission mediated by N-methyl-d-aspartate receptors. Proc. Natl Acad. Sci. USA, 96, 7697–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffran S., Astier,M., Gratecos,D. and Semeriva,M. (1997) The held out wings (how) Drosophila gene encodes a putative RNA-binding protein involved in the control of muscular and cardiac activity. Development, 124, 2087–2098. [DOI] [PubMed] [Google Scholar]
- Zhang Y and Feng,Y (2001) Distinct molecular mechanisms lead to diminished myelin basic protein and 2′,3′-cyclic nucleotide 3′-phosphodiesterase in qk(v) dysmyelination. J. Neurochem., 77, 165–172. [DOI] [PubMed] [Google Scholar]
- Zorn A.M., Grow,M., Patterson,K.D., Ebersole,T.A., Chen,Q., Artzt,K. and Krieg,P.A. (1997) Remarkable sequence conservation of transcripts encoding amphibian and mammalian homologues of quaking, a KH domain RNA-binding protein. Gene, 188, 199–206. [DOI] [PubMed] [Google Scholar]