

Abstract
Pseudouridylation of snRNAs in vertebrates is guided by small nucleolar/Cajal body-specific RNAs (sno/scaRNAs). We developed an in vitro system using cell extracts and single site-radiolabeled U2 snRNAs to study pseudouridylation in Saccharomyces cerevisiae. Micrococcal nuclease-treated cell extracts are fully competent to catalyze U2 pseudouridylation, suggesting an RNA-independent process. A pseudouridylase activity for Ψ35 within yeast U2 is identified via a screen of an S.cerevisiae GST–ORF protein library. This activity is associated with YOR243c ORF, which has not previously been assigned function. When the GST–YOR243c protein is expressed in Escherichia coli, pseudouridylation activity is comparable to that expressed in S.cerevisiae, demonstrating that this protein (designated Pus7) alone can catalyze Ψ35 formation in U2. Both in vitro and in vivo analyses using wild-type and pus7-Δ strains show that Pus7 is indispensable for Ψ35 formation in U2. Using site-specific radiolabeled U2 and U2 fragments, we show that Pus7 activity is specific for Ψ35 and that the U2 stem– loop II region is essential for the pseudouridylation reaction. A BLAST search revealed Pus7 homologs in various organisms.
Keywords: GST–ORF protein library/pseudouridylation/Pus7/S.cerevisiae/U2 snRNA
Introduction
Virtually all cellular RNAs undergo post-transcriptional processing and modification. Pseudouridylation and 2′-O-methylation are the most common internal modifications, and are found in three major types of stable RNA including the spliceosomal small nuclear RNAs (snRNAs), ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs) (reviewed in Reddy and Busch, 1988; Maden, 1990; Bjork, 1995; Grosjean et al., 1995; Massenet et al., 1998). While the modifications in rRNA and tRNA have been studied extensively (for reviews, see Peculis, 1997; Smith and Steitz, 1997; Tollervey and Kiss, 1997; Weinstein and Steitz, 1999; Kiss, 2001; Filipowicz and Pogacic, 2002; Terns and Terns, 2002), relatively little is known about those that occur in spliceosomal snRNAs. Although it has been well documented that HeLa cell extracts are functionally active in catalyzing the formation of pseudouridine (Ψ) in all major spliceosomal snRNAs (U1, U2, U4, U5 and U6) (Patton, 1991, 1993a,b, 1994; Patton et al., 1994; Zerby and Patton, 1996, 1997), due at least in part to technical difficulties, the molecular mechanism of this process remains largely unclear.
All five spliceosomal snRNAs contain extensive modifications that are implicated in pre-mRNA splicing. It has long been known that these modified nucleotides are extremely well conserved (Massenet et al., 1998), consistent with findings that they are virtually all clustered in regions known to be important for splicing (Massenet et al., 1998; Yu et al., 1999). For instance, there are a number of pseudouridines (Ψ) present in the branch-site recognition region of vertebrate U2 snRNA (Massenet et al., 1998; Yu et al., 1999), and three of these are also present at equivalent positions in yeast U2 (Massenet et al., 1998; also see Figure 1A). Their high level of conservation as well as their location in critical regions within the spliceosomal snRNAs (e.g. U2 branch-site recognition region) strongly suggests that modified nucleotides are functionally important. Direct experimental evidence comes from our previous work demonstrating that the modified nucleotides of U2 snRNA are indeed required for snRNP biogenesis and pre-mRNA splicing in Xenopus oocytes (Yu et al., 1998). Furthermore, recent NMR structural studies from Greenbaum and coworkers showed that the base-pairing interaction between Ψ35 in yeast U2 snRNA and the nucleotide next to the pre-mRNA branch point adenosine induces a change in conformation of the U2-branch site helix (Newby and Greenbaum, 2001, 2002). This change causes the branch point adenosine to bulge out, a configuration known to be critical for the first chemical step of splicing.

Fig. 1. In vitro pseudouridylation of yeast U2 snRNA. (A) The sequence of the 5′ 123 nucleotides of S.cerevisiae U2 is shown. There are in total three pseudouridines in S.cerevisiae U2, indicated by three arrows (positions 35, 42 and 44). The gray box indicates the Sm binding site, and the underlined nucleotides (GΨAGUA) are involved in base-pairing interaction with the branch site of pre-mRNA. (B) Yeast U2 snRNA containing a single 32P radiolabel 5′ of U35 (lane 1), U38 (lane 2), U42 (lane 3) or U44 (lane 4) was incubated at 30°C for 1 h in cell extracts prepared from the wild-type S.cerevisiae strain. RNA was recovered, treated with nuclease P1 and analyzed by TLC (see Materials and methods). (C) Yeast U2 containing a single 32P radiolabel 5′ of U35 was incubated at 30°C for 1 h with the yeast cell extracts (lane 1) or micrococcal nuclease-treated yeast cell extracts (lane 2). Recovered RNA was subjected to nuclease P1 digestion and TLC analysis. Radiolabeled uridylate (32pU) and pseudouridylate (32pΨ) are indicated.
A growing body of evidence suggests that the mechanism of spliceosomal snRNA modification in vertebrates is similar to that of rRNAs, which is facilitated by guide RNAs that contain either an H/ACA motif (for pseudouridylation) or a C/D box (for 2′-O-methylation) (Tycowski et al., 1998; Ganot et al., 1999; Huttenhofer et al., 2001; Jady and Kiss, 2001; Darzacq et al., 2002; Zhao et al., 2002). Each guide RNA contains a stretch of nucleotides complementary to its target substrate RNA. By forming a duplex with its target RNA, the guide RNA directs modification at a specific site (Peculis, 1997; Smith and Steitz, 1997; Tollervey and Kiss, 1997; Weinstein and Steitz, 1999; Kiss, 2001; Filipowicz and Pogacic, 2002; Terns and Terns, 2002). In several instances, the guide function of H/ACA and C/D motif RNAs for spliceosomal snRNA modification has been experimentally confirmed (Tycowski et al., 1998; Jady and Kiss, 2001; Zhao et al., 2002).
Interestingly, despite an intensive search, no specific guide RNAs have been identified for spliceosomal snRNA modifications in Saccharomyces cerevisiae. Recent work from Branlant’s group showed that the yeast Pus1 gene product, which was originally identified as a tRNA pseudouridine synthase, can also pseudouridylate yeast U2 snRNA at position 44 (Massenet et al., 1999), one of the three pseudouridines in the branch-recognition region (Figure 1A). Therefore, an important question arises as to whether spliceosomal snRNA modifications in S.cerevisiae are guided by RNAs that contain a C/D or H/ACA motif, as is the case for higher eukaryotes, or whether modifications are catalyzed by protein enzymes acting alone.
In our present work, we demonstrate that a previously unknown S.cerevisiae protein encoded by the YOR243c gene is responsible for yeast U2 pseudouridylation at position 35. Our results indicate that the internal modification of U2 (and perhaps other spliceosomal snRNAs) in S.cerevisiae is mechanistically distinct from that of higher eukaryotes.
Results
U2 pseudouridylation in S.cerevisiae is RNA independent
To study the mechanism of U2 pseudouridylation in S.cerevisiae, we established a cell-free system using yeast cell extracts that efficiently and faithfully catalyzes U2 pseudouridylation. Four synthetic yeast U2 snRNAs, each containing a single 32P label 5′ of uridine 35, 38, 42 or 44 (U35, U38, U42, U44) were separately incubated with the extract at 30°C for 1 h. Total RNA was recovered, digested with nuclease P1 and analyzed by thin-layer chromatography (TLC). The uridylates labeled at U35, U42 and U44, three positions that are naturally pseudouridylated (Figure 1A), were converted to pseudouridylates (Figure 1B, lanes 1, 3 and 4). However, no conversion was observed in the U2 that contained a single label at U38, a position that is not naturally modified in S.cerevisiae (Figure 1B, lane 2). Although the conversion efficiency at position 42 (lane 3) was low (∼20%), the conversion efficiency at positions 35 and 44 (lanes 1 and 4) was ∼100% under the conditions used (see Materials and methods).
To determine whether U2 pseudouridylation in S.cerevisiae is mediated by guide RNAs, the extracts were treated with micrococcal nuclease and subsequently tested for pseudouridylation activity. Northern analysis of RNAs recovered from micrococcal nuclease-treated extracts confirmed that C/D box and H/ACA motif RNAs that guide yeast rRNA modifications were degraded (data not shown). As shown in Figure 1C, micrococcal nuclease-treated extracts (lane 2) and untreated extracts (lane 1) were equally active in catalyzing U2 pseudouridylation. This result suggests that pseudouridylation of S.cerevisiae U2 snRNA may be an RNA-independent process.
A GST–ORF fusion containing YOR243c catalyzes U2 pseudouridylation at position 35
To identify the protein enzyme responsible for U2 pseudouridylation at position 35, we utilized a yeast GST–ORF genomic library consisting of 6144 strains (Martzen et al., 1999; Phizicky et al., 2002). Each of the strains expresses a unique GST–ORF fusion protein under control of the PCUP1 promoter. The expression library was grown in 64 pools of 96 strains each. Pools of purified GST–ORF fusion proteins were prepared (Martzen et al., 1999; Phizicky et al., 2002) and assayed for pseudouridylation activity with the U2 substrate containing a single 32P label 5′ of U35. Only pool 56 contained pseudouridylase activity (Figure 2A). Subpools of GST– ORF fusion proteins were then prepared from pool 56 [containing 96 strains in 12 columns and eight rows (A–H)] and screened for pseudouridylation activity (see Materials and methods). Only subpools ‘column 10’ and ‘row B’ showed pseudouridylation activity (Figure 2B, top and middle panels). Strain 10B expresses a GST–ORF of the gene YOR243c, which has not been assigned function according to the current Saccharomyces Genome Database (Cherry et al., 2002). The GST–YOR243c fusion protein was purified from strain 10B and tested to confirm that the Ψ35-specific activity coincided with GST–YOR243c (Figure 2B, bottom panel).

Fig. 2. Screening of a S.cerevisiae GST–ORF fusion protein library. (A) Sixty-four purified pools of yeast GST–ORF proteins were each incubated with yeast U2 snRNA containing a single 32P radiolabel 5′ of U35. After the reaction, radiolabeled U2 was recovered, treated with nuclease P1 and analyzed by TLC. Among the 64 pools, only pool 56 showed pseudouridylase activity. As controls, the same U2 with a single radiolabel 5′ of U35 was digested with nuclease P1 prior to screening (lane –), or digested with nuclease P1 after incubation with the wild-type yeast cell extracts (lane +), and analyzed on the same TLC plate. (B) Twelve columns (each containing eight different strains) (top panel) and eight rows (each containing 12 individual strains) (middle panel) were further screened for pseudouridylase activity, as described in (A). The activity was contained in column 10 and row B, suggesting that strain 10B was the source strain. This conclusion was confirmed by subsequent screening of individual strains in column 10 (bottom panel).
To rule out the possibility that some other yeast factor(s) (protein or RNA) copurified with GST–YOR243c, GST– YOR243c was expressed in Escherichia coli, purified and assayed for pseudouridylation activity. As shown in Figure 3, the E.coli-expressed fusion protein catalyzed the formation of Ψ35 (lanes 5–8) at levels comparable to that of the yeast-expressed fusion protein (lanes 1–4). These results clearly demonstrate that YOR243c encodes a protein that catalyzes yeast U2 snRNA pseudouridylation at position 35, and demonstrate further that the activity occurs in a guide RNA-independent manner. Thus, the enzyme is a pseudouridine synthase, and was designated Pus7.

Fig. 3. Pseudouridylase activity of the GST–Pus7 (YOR243c) fusion protein expressed in either S.cerevisiae (lanes 1–4) or E.coli (lanes 5–8) was assayed in vitro and compared. The range of final concentrations of fusion protein (ng/ml) in the pseudouridylation reaction (see Materials and methods, and previous figure legends) is indicated above the lanes.
Pus7 is responsible for U2 snRNA pseudouridylation at position 35 in vitro
To assess whether Pus7 is the only S.cerevisiae protein responsible for Ψ35 formation in S.cerevisiae U2, and whether this pseudouridylation activity is specific for position 35, purified GST–Pus7 protein as well as cell extracts prepared from the pus7-Δ strain and the wild-type parental strain were incubated with a synthetic yeast U2 RNA containing a single 32P label 5′ of U35, U42 or U44. As shown in Figure 4, GST–Pus7 protein converted U35 to Ψ35 (lane 2) but was unable to catalyze pseudouridylation at positions U42 (lane 6) and U44 (lane 10). In contrast, extracts prepared from the pus7-Δ strain were capable of converting U42 and U44 to Ψ (lanes 8 and 12), whereas no conversion of U35 to Ψ was detected (lane 4). When wild-type parental extract was used, all three positions were pseudouridylated (lanes 3, 7 and 11), as occurs naturally (Massenet et al., 1998). Taken together, these results show that Pus7 is indeed the only protein required for U2 pseudouridylation at position 35 in vitro, and that the pseudouridylation activity of Pus7 is site specific for Ψ35 in S.cerevisiae U2 snRNA.

Fig. 4. Pus7 is indispensable for Ψ35 formation in yeast U2 in vitro and the pseudouridylase activity of Pus7 is specific for position 35. Three yeast U2 snRNAs, each containing a single 32P radiolabel 5′ of either U35 (lanes 1–4), U42 (lanes 5–8) or U44 (lanes 9–12), were incubated separately with purified GST–Pus7 (lanes 2, 6 and 10), or incubated in the extracts prepared from the yeast wild-type strain (lanes 3, 7 and 11) or the pus7-Δ strain (lanes 4, 8 and 12). After the reaction, radiolabeled U2 was recovered and subjected to nuclease P1 digestion and TLC analysis (see Materials and methods, and previous figure legends). Lanes 1, 5 and 9 are controls in which the single radiolabeled U2 was digested with nuclease P1 before the pseudouridylation reaction.
Pus7 is responsible for Ψ35 formation in S.cerevisiae U2 in vivo
To check whether Pus7 is also responsible for Ψ35 formation in vivo, pseudouridylation of endogenous U2 snRNA from the parental and pus7-Δ strains was also analyzed, using the widely used method involving N-cyclohexyl-N′-(2-morpholinoethyl)-carbodiimid metho-p-toluolsulfonate (CMC) modification followed by primer extension (Bakin and Ofengand, 1993). It is well established that pseudouridines in RNAs are specifically modified by CMC, which in turn causes primer-extension ‘stops’ at positions one nucleotide before the pseudouridines (Bakin and Ofengand, 1993; Massenet et al., 1999). Total cellular RNA was prepared, modified by CMC and primer-extended with a radiolabeled DNA oligonucleotide complementary to yeast U2 snRNA. When total RNA prepared from the wild-type strain was used, primer-extension revealed three stops at positions one nucleotide before Ψ35, Ψ42 and Ψ44 (Figure 5, lane 2). In contrast, primer-extension of total RNA prepared from pus7-Δ strain showed only two stops at positions one nucleotide before Ψ42 and Ψ44 (Figure 5, lane 4). No stop was observed one nucleotide before position 35 (lane 4), indicating that pseudouridylation at this position was completely abolished. These data indicate that YOR243c is the only S.cerevisiae gene that encodes a protein enzyme (Pus7) responsible for U2 pseudouridylation at position 35.
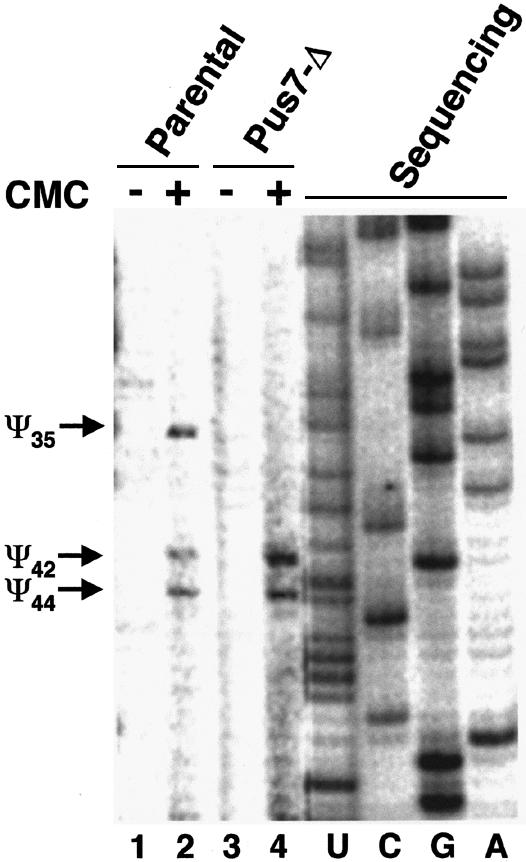
Fig. 5. Pus7 is responsible for yeast U2 pseudouridylation at position 35 in vivo. Total yeast RNA isolated from the wild-type strain (lanes 1 and 2) or the pus7-Δ strain (lanes 3 and 4) was used for CMC modification (lanes 2 and 4) followed by U2 primer-extension analysis (see Materials and methods). Lanes 1 and 3 are mock-treated controls where CMC was omitted. Primer-extension ‘stops’ caused by pseudouridine-specific CMC modification are indicated on the left (Ψ35, Ψ42 and Ψ44). A U2 primer-extension sequencing ladder (lanes U, C, G and A) was electrophoresed in parallel to facilitate pseudouridine mapping.
Stem–loops IIa and IIb of U2 are required for Pus7 pseudouridylation activity
To understand how U2 snRNA is recognized by Pus7, pseudouridylation experiments were performed using truncated U2 snRNAs. As shown in Figure 6, sequence was successively deleted either from the 5′ end or the 3′ end of yeast U2 snRNA, approaching the target pseudouridylation nucleotide, U35. A mutant U2 substrate lacking the 5′-most 21 nucleotides was efficiently pseudouridylated at position 35 (lane 1). Even when the 5′ 27 or 32 nucleotides were removed, conversion of U35 to Ψ35 still occurred, albeit with lower efficiency (lanes 2 and 3). These results suggest that a large majority of the yeast U2 snRNA nucleotides that lie 5′ to U35 are dispensable for Pus7-catalyzed pseudouridylation. When the entire 3′ sequence downstream of stem–loops IIa and IIb (starting from position 104) was deleted, the remainder of U2 was efficiently pseudouridylated (Figure 6, lane 7). However, when deletion was extended to include stem–loops IIa and IIb, conversion of U35 to Ψ35 was abrogated (lanes 4–6). These results suggest that the primary sequence or the secondary structure of stem–loops IIa and IIb of U2 constitutes a Pus7 recognition motif necessary for the conversion of U35 to Ψ35. Since stem–loops IIa and IIb are distant from the target uridine, the critical RNA sequence recognized by S.cerevisiae Pus7 is clearly distinct from the sequence immediately flanking the target uridine, which is usually recognized by H/ACA motif RNPs that catalyze pseudouridylation of spliceosomal snRNAs in higher eukaryotes.

Fig. 6. U2 stem–loop II region is essential for Pus7 function. Yeast U2 snRNA containing a single 32P radiolabel 5′ of U35 was site-specifically cleaved. The RNA fragment containing the radiolabel was recovered and subjected to a pseudouridylation assay with purified GST–Pus7 (see Materials and methods, and previous figure legends). RNAs cleaved between positions 21 and 22, 27 and 28, or 32 and 33, begin with nucleotide 22 (lane 1), 28 (lane 2) or 33 (lane 3), respectively. RNAs cleaved between positions 37 and 38, 39 and 40, or 103 and 104, begin with nucleotide 1 and end with nucleotide 37 (lane 4), 39 (lane 5), 47 (lane 6) or 103 (lane 7), respectively. A schematic of yeast U2 is shown, and the approximate cleavage positions are indicated by arrows. Stem–loop I and stem–loop IIa,b are indicated. In lanes 1–7, equivalent molar amounts of all substrates and GST–Pus7 were used in the assays. Lanes 8 and 9 are controls in which the intact single radiolabeled U2 was digested with nuclease P1 before (–) or after (+) pseudouridylation.
Identification of Pus7 homologs in several species
To understand the relationship between Pus7 and the other pseudouridine synthases, we compared the amino acid sequence of Pus7 with that of other known protein pseudouridylases, namely those of the TruA, TruB, RluA and RsuA families (Gustafsson et al., 1996; Koonin, 1996). Interestingly, the comparison revealed no significant sequence homology (data not shown). Thus, Pus7 may belong to a different pseudouridylase family whose other members have yet to be identified.
A BLAST search of all available databases revealed Pus7 homologs in several different organisms, including Schizosaccharomyces pombe (gene name: SPBCIA4.09), Caenorhabditis elegans (B0024.11), Drosophila melanogaster (CG6745) and humans (KIAA1987) (Figure 7). Remarkably, there is 40% amino acid identity between yeast Pus7 and human KIAA1987 (Figure 7), suggesting that Pus7 might be evolutionarily conserved. Thus, in higher eukaryotes such as human where pseudouridylation is catalyzed by H/ACA RNPs, the Pus7 homolog may serve as a back-up that ensures pseudouridylation of U2 snRNA at the position equivalent to U35 (see Discussion).

Fig. 7. Pus7 homologous proteins in various species. BLAST search identified Pus7 (S.cer.) homologs in a number of different organisms, including S.pombe (S.pom.) (SPBC1A4.09), C.elegans (C.el.) (B0024.11), D.melanogaster (Dros.) (CG6745) and human (H.sap.) (KIAA1897). A comparison of the amino acid sequences of these proteins is shown. The conserved residues (including the identical and similar amino acids) are highlighted in black.
Discussion
Using the yeast GST–ORF fusion protein library, we identified and characterized a pseudouridylase (Pus7) responsible for pseudouridylation at position 35 of S.cerevisiae U2. Pus7 is encoded by YOR243c ORF, which has not previously been assigned function according to the current Saccharomyces Genome Database (Cherry et al., 2002). Here, we showed that the GST–Pus7 fusion protein expressed either in yeast or in E.coli is highly active in catalyzing the formation of Ψ35 in yeast U2. This activity is completely abolished in a pus7-Δ strain. These results suggest that Pus7 is necessary and sufficient for yeast U2 pseudouridylation at position 35. We also showed that Pus7 pseudouridylation activity is specific for Ψ35 and dependent on the stem–loop II region within U2 snRNA.
Pseudouridylation in S.cerevisiae U2 snRNA differs from that in higher eukaryotes
It is widely believed that eukaryotic spliceosomal snRNA modifications are catalyzed by sno/scaRNPs (small nucleolar/small Cajal body-specific ribonucleoproteins), as are the modifications of rRNAs (Yu et al., 1998; Kiss, 2001). This view was bolstered by the recent discovery of a large number of modification-specific guide RNAs (C/D box and H/ACA motif RNAs) in higher eukaryotic cells (Tycowski et al., 1998; Ganot et al., 1999; Huttenhofer et al., 2001; Jady and Kiss, 2001; Darzacq et al., 2002; Zhao et al., 2002). Surprisingly, however, extensive searches in S.cerevisiae have failed to identify any guide RNAs for spliceosomal snRNA modifications, although a near-complete set was identified for rRNA modifications (Lowe and Eddy, 1999, 2000; Samarsky and Fournier, 2000). Consistent with these results, the Branlant group (Massenet et al., 1999) reported that Pus1p, a tRNA pseudouridylase, can also catalyze yeast U2 snRNA pseudouridylation at position 44, one of the three pseudouridines identified in S.cerevisiae U2. This raises the interesting hypothesis that the modification of yeast spliceosomal snRNAs may follow a protein-only mechanism that differs completely from that in higher eukaryotes.
We present three independent lines of evidence which suggest that S.cerevisiae U2 pseudouridylation (and perhaps the other spliceosomal snRNAs) is indeed catalyzed by an RNA-independent mechanism. First, micrococcal nuclease-treated yeast cell extracts retain pseudouridylation activity specific for U35, U42 and U44 in U2 snRNA (Figure 1C; data not shown). Secondly, pseudouridylation catalyzed by Pus7 requires a U2 RNA sequence (or structure, i.e. stem–loop II; Figure 6) that is distant from the site of catalysis, unlike the RNA-guided mechanism in higher eukaryotes, which utilizes the RNA sequence immediately flanking the target site (Kiss, 2001). Thirdly, the GST–Pus7 fusion protein purified from either S.cerevisiae or E.coli efficiently and accurately catalyzes U2 pseudouridylation at position 35 (Figure 3). Currently, we are carrying out similar experiments using different yeast spliceosomal snRNAs to test the possibility that the internal modifications of all yeast spliceosomal snRNAs are catalyzed by RNA-independent mechanisms.
Why does S.cerevisiae U2 contain far fewer pseudouridines than vertebrate U2?
A comparison of vertebrate and S.cerevisiae U2 snRNAs reveals a >4-fold difference in the number of pseudouridines: 13 in vertebrates versus only three in S.cerevisiae (Reddy and Busch, 1988; Massenet et al., 1998; Yu et al., 1998). In contrast, a huge number of pseudouridines are found in rRNAs in both species (∼50 in S.cerevisiae and ∼100 in human) (Maden, 1990). How did this scenario arise? We suggest that the answer lies in the differential mechanisms by which pseudouridylation occurs in these species.
Internal modifications within higher eukaryotic spliceosomal snRNAs are catalyzed by sno/scaRNPs within which the RNAs serve as guides that direct a single pseudouridylase (presumably shared by all sno/scaRNPs) to act on the RNA substrate at specific sites. Thus, substrate specificity is governed completely by a relatively short guide sequence within each guide RNA. In contrast, we and others (Massenet et al., 1999) have shown that S.cerevisiae U2 pseudouridylation is catalyzed by protein only, and thus substrate specificity is governed by the amino acid sequence of the protein enzymes. It has been postulated that duplication of guide RNA genes as well as mutations in the relatively short guide sequences during evolution could have given rise to similar yet new guide RNAs that directed pseudouridylation at novel sites (Lafontaine and Tollervey, 1998). Such processes would most likely occur much less readily in protein-based mechanisms where a new protein enzyme must evolve for each new site (Lafontaine and Tollervey, 1998). Consistent with these suppositions is the fact that there are only three protein enzymes that catalyze three independent pseudouridylation reactions in S.cerevisiae U2 snRNA, whereas a number of similar H/ACA guide RNAs (only the guide sequences are different) evolved in higher eukaryotes to direct U2 pseudouridylation at 13 specific sites. It is possible that H/ACA snoRNPs for spliceosomal snRNA pseudouridylation either evolved in S.cerevisiae but were subsequently lost from the genome (e.g. via chromosomal deletion) or simply never evolved. Clearly, yeast and vertebrate lineages underwent substantially different selective pressures throughout evolution, so it is not surprising that the independent evolution of specialized biochemical functions such as pseudouridylation proceeded by radically different means.
Pus7 and its homologs in higher eukaryotes
Using BLAST search, we identified Pus7 homologs in several different species, including higher eukaryotes such as humans (there is 40% sequence identity between yeast Pus7 and its human homolog; Figure 7). Why do higher eukaryotes carry Pus7 homologs despite the fact that U2 pseudouridylation, including Ψ34 (equivalent to Ψ35 in yeast U2) (Darzacq et al., 2002; Zhao et al., 2002), is catalyzed by H/ACA motif sno/scaRNPs? Perhaps the Pus7 homologs act on different substrates from those of H/ACA motif sno/scaRNPs––the Pus7 homologs may catalyze pseudouridylation of yet-to-be identified RNAs. This possibility is reasonable since, at present, it is not possible to predict substrate specificity based on homologous sequences given that nothing is known about Pus7 domain structure. Alternatively, the possibility that Pus7 homologs pseudouridylate their respective U2 snRNA at a position equivalent to U35 in S.cerevisiae suggests that there are redundant site-specific activities in higher eukaryotes that ensure U2 pseudouridylation. Consistent with this argument, we have experimental evidence from Xenopus oocytes that there are redundant activities for Ψ34 formation in U2 (equivalent to Ψ35 in yeast U2) (Zhao et al., 2002).
Redundant activities for the formation of Ψ35 in U2 also imply that this pseudouridine may be critical for U2 function, and our recent experimental results support this idea. We showed that the pseudouridines in the Xenopus U2 branch site recognition region, including Ψ34, are required for pre-mRNA splicing in oocytes (X.Zhao and Y.-T.Yu, unpublished data). Although viable in rich medium, the S.cerevisiae pus7-Δ strain is growth- disadvantaged under 1 M NaCl or when grown in competition with the wild-type strain (X.Ma and Y.-T.Yu, unpublished data). Furthermore, recent NMR studies from Greenbaum’s group strongly suggest the importance of this pseudouridine (Newby and Greenbaum, 2001, 2002). Together, these results make a reasonable case for the need for redundant pseudouridylase activities (and therefore Pus7 homologs) in higher eukaryotes.
Finally, given that the number of guide RNAs identified to date is still far smaller than the number of modified nucleotides identified within spliceosomal snRNAs, it is possible that not all modifications of vertebrate spliceosomal snRNAs are guided by sno/scaRNAs. Those that are not guided by sno/scaRNAs would be catalyzed by classical pseudouridine synthases (a protein-only mechanism).
Materials and methods
Construction of site-specific radiolabeled U2 snRNA
Yeast U2 snRNA was generated by in vitro transcription using pT7U2Δ107 plasmid as template (a gift from D.McPheeters). The transcription reaction contained 1.2 mM each of ATP, CTP, UTP and GTP (New England Biolabs), 40 mM Tris–HCl pH 7.5, 6 mM MgCl2, 2 mM spermidine, 5 mM DTT, 0.1 mg/ml AflII-linearized pT7U2Δ107 and 4 U/µl T7 RNA polymerase. The synthesized U2 was purified on a denaturing polyacrylamide gel and site-specifically cleaved by RNase H (a gift from R.Crouch) in the presence of specific 2′-O-methyl RNA–DNA chimeric oligonucleotides, as described previously (Yu, 2000). Four oligonucleotides, UmAmdCdAdCdTUmGmAmUmCmUmUmAmGmCmCm, AmUmAmdCdTdAdCAmCmUmUmGmAmUmCmUmUmAmGm, AmCmAmdGdAdTdACmUmAmCmAmCmUmUmGmAmUm and GmAmAmdCdAdGdAUmAmCmUmAmCmAmCmUmUmGm, were used for cleavage of the phosphodiester bond between G34 and U35, between G37 and U38, between C41 and U42, and between G43 and U44, respectively. The cleaved half-RNAs were gel-purified, and the 3′ half was further subjected to a dephosphorylation reaction containing 1× dephosphorylation buffer (Roche), 10–20 pmol of 3′ half U2 and 1 U of alkaline phosphatase (AP) (Roche). The reaction was carried out at 50°C for 1 h. After extraction with phenol/chloroform/isoamyl alcohol mixture (1:1:0.01) (PCA) and ethanol precipitation, the 3′ half RNA was rephosphorylated at its 5′ end in a 10 µl reaction containing 1× phosphorylation buffer (Amersham), 10–20 pmol of 3′ half U2, ∼150 µCi of [γ-32P]ATP (6000 Ci/mmol, Du Pont/NEN) and ∼20 U of T4 polynucleotide kinase (Amersham). The radiolabeled 3′ half RNA was then religated with the 5′ half U2 in the presence of a bridging DNA oligonucleotide (complementary to nucleotides 20–64 of yeast U2) and T4 DNA ligase, as described (Yu, 2000).
To create 5′ or 3′ truncated U2 RNAs (Figure 6), U2 RNA with a single radiolabel 5′ of U35 was subjected to site-specific RNase H cleavage directed by 2′-O-methyl RNA–DNA chimeras (UmAmGmdCdCdAdAAmAmGmGmCmCmGmAmGmAmAmGm, for the cleavage between positions 21 and 22; UmGmAmdTdCdTdTAmGmCmCmAmAmAmAmGmGmCmCm, for the cleavage between positions 27 and 28; AmCmAmdCdTdTdGAmUmCmUmUmAmGmCmCmAmAmAmAm, for the cleavage between positions 32 and 33; AmUmAmdAdGdAdACmAmGmAmUmAmCmUmAmCmAmCmUmUmGmAm, for the cleavage between positions 47 and 48), or was digested by DNA enzymes (TGATAAGAACAGATAGGCTAGCTACAACGATACACTTGATCTTA for the cleavage between positions 37 and 38; ACTGATAAGAACAGAGGCTAGCTACAACGAACTACACTTGATCT for the cleavage between positions 39 and 40; AATGTGTATTGTAAGGCTAGCTACAACGAAAATTAAAAGGTAATG for the cleavage between positions 103 and 104) (Santoro and Joyce, 1997). Following purification by denaturing gel electrophoresis, the U2 fragments containing the single 32P radiolabel were excised, recovered and used in the experiments described in Figure 6.
Preparation of yeast cell extracts and micrococcal nuclease-treated extracts
The homozygous diploid pus7-Δ strain and its wild-type parental strain (BY4743) were purchased from Research Genetics. Yeast cell extracts were prepared essentially as described (Martzen et al., 1999; Phizicky et al., 2002). In brief, wild-type and deletion strains were grown at 30°C in 500 ml yeast extract-peptone-dextrose (YPD) medium to an OD600nm of 2–4. Cells were then harvested at 4°C by centrifugation (Sorvall SLA-3000 rotor, 3000 r.p.m. for 10 min). Pelleted cells were resuspended in 20 ml of lysis buffer [50 mM Tris–HCl pH 7.5, 1 mM EDTA, 4 mM MgCl2, 5 mM DTT, 10% (v/v) glycerol, 1 M NaCl, 1 mM each leupeptin and pepstatin] and lysed at 4°C by beating with glass beads (0.45–0.5 mm) on a mini bead beater (Biospec). Cell extracts were subsequently centrifuged at 4°C to remove insoluble debris (Sorvall SS-34 rotor, 17 000 g for 20 min). The supernatant was then dialyzed overnight against 1 l of storage buffer [20 mM Tris–HCl pH 7.5, 2 mM EDTA, 4 mM MgCl2, 1 mM DTT, 55 mM NaCl, 50% (v/v) glycerol]. The extracts contained 30–35 mg/ml protein and were frozen at –75°C in small aliquots.
To prepare micrococcal nuclease-treated extracts, an aliquot of 200 µl of the wild-type yeast cell extract was mixed with 10 µl of 44 mM CaCl2 and 10 µl of 30 U/µl micrococcal nuclease. The reaction was carried out at 30°C for 20 min, and was subsequently stopped by the addition of 10 µl of 92 mM EGTA. Micrococcal nuclease-treated extracts contained only short RNA species (<34 nt) as judged by 3′ end labeling with 32pCp and T4 RNA ligase (England and Uhlenbeck, 1978) or by 5′ end labeling using AP-catalyzed dephosphorylation followed by rephosphorylation with [γ-32P]ATP and polynucleotide kinase (see above).
Purification of GST–ORF fusion proteins from S.cerevisiae
Pools of yeast GST–ORF fusion proteins and the GST–Pus7 (YOR243c) fusion protein were all purified from 250 ml cultures, exactly as described previously (Martzen et al., 1999; Phizicky et al., 2002). Purified proteins (∼150–350 µg/ml) were ∼50–70% pure according to SDS–PAGE and silver staining, and stored at –20°C in storage buffer containing 20 mM Tris–HCl pH 7.5, 2 mM EDTA, 4 mM MgCl2, 1 mM DTT, 55 mM NaCl and 50% (v/v) glycerol.
Expression and purification of GST–Pus7 fusion protein from E.coli
Using a pair of DNA oligonucleotides (the 5′ primer, GATGTCTGACTCCTCAGAAGCC, and the 3′ primer, TTAGATATTCTCCTTAACATC), YOR243c gene was amplified via PCR from the original plasmid (purified from strain 10B) that contains YOR243c ORF (pYEX4T-1) (Martzen et al., 1999). The PCR product was inserted in frame into the SmaI site of pGEX-2T vector, which contains a GST tag upstream of the SmaI site. The sequence of the insert was verified by DNA sequencing. This recombinant plasmid containing YOR243c was then transformed into E.coli BL21 (DE3) cells and grown in 250 ml LB medium [with ampicillin (100 µg/ml)] at 37°C to an OD600nm of ∼0.8. IPTG (1 mM) was then added to the culture and the cells were allowed to grow at 20°C for another 12 h. Cells were collected by centrifugation, resuspended in lysis buffer (buffer A) [40 mM sodium phosphate pH 7.6, 20 mM NaCl, 5 mM β-mercaptoethanol, 5% (v/v) glycerol] plus 1 mM each leupeptin and pepstatin, and disrupted by sonication. The cell homogenate was centrifuged at 10 000 g for 15 min to remove insoluble debris. The supernatant was then diluted with an equal volume of buffer B containing 40 mM sodium phosphate pH 7.6, 5 mM β-mercaptoethanol, 5% (v/v) glycerol, and was subsequently applied to a glutathione–Sepharose 4B column (1 ml) (Amersham). After a thorough wash with buffer A, the GST– Pus7 fusion protein was eluted from the column with buffer A containing 25 mM glutathione. The resultant GST–Pus7 solution was dialyzed at 4°C against 1 l of storage buffer [20 mM Tris–HCl pH 7.5, 2 mM EDTA, 4 mM MgCl2, 1 mM DTT, 55 mM NaCl, 50% (v/v) glycerol]. Purified GST–Pus7 protein was of >90% purity (analyzed by SDS–PAGE and Coomassie Blue staining) and stored at –20°C in small aliquots.
Pseudouridylation assay
Single 32P-radiolabeled yeast U2 snRNA was mixed with 2 µl yeast cell extracts in a final volume of 20 µl containing 100 mM Tris–HCl pH 8.0, 100 mM ammonium acetate, 5 mM MgCl2, 2 mM DTT, 0.1 mM EDTA and 10 000 c.p.m. (<0.001 fmol) single radiolabeled U2. Reactions were carried out at 30°C for 1 h. Radiolabeled U2 was then re-purified by denaturing gel electrophoresis, recovered and subjected to nuclease P1 digestion and TLC analysis, as described previously (Patton, 1991; Yu et al., 1998). For screening of the GST–ORF fusion protein library, yeast cell extracts were substituted with ∼150–350 ng of the pool of purified GST–ORF fusion proteins or ∼1–5 ng of single GST–Pus7 fusion protein expressed in and purified from S.cerevisiae or E.coli.
To analyze pseudouridylation of endogenous U2 from wild-type and pus7-Δ strains, CMC modification followed by U2 primer-extension was employed (Bakin and Ofengand, 1993). Briefly, total cellular RNA (∼10 µg) isolated from corresponding yeast strains was treated with CMC at 37°C for 20 min in a 30 µl reaction mixture (∼10 µg RNA sample, 0.17 M CMC, 50 mM Bicine pH 8.3, 4 mM EDTA and 7 M urea). The modified RNA was then incubated with 50 mM bicarbonate buffer (pH 10.4) at 37°C for 2 h. Recovered RNA was subjected to primer-extension analysis, as described previously (Yu et al., 1998). The primer was a 5′ 32P-radiolabeled DNA oligonucleotide complementary to nucleotides 104–126 of S.cerevisiae U2. Primer-extension stops caused by pseudouridine-specific CMC modification were visualized by autoradiography.
Acknowledgments
Acknowledgements
We are indebted to E.M.Phizicky and E.J.Grayhack for providing the S.cerevisiae GST–ORF fusion protein library and for much advice on protein purification procedures. We thank D.McPheeters for S.cerevisiae U2 constructs. We also thank E.M.Phizicky and T.Taylor for critical reading of the manuscript. This work was supported by grant GM62937 from the National Institutes of Health.
References
- Bakin A. and Ofengand,J. (1993) Four newly located pseudouridylate residues in Escherichia coli 23S ribosomal RNA are all at the peptidyltransferase center: analysis by the application of a new sequencing technique. Biochemistry, 32, 9754–9762. [DOI] [PubMed] [Google Scholar]
- Bjork G.R. (1995) Biosynthesis and function of modified nucleosides in tRNA. In Soll,D. and RajBhandary,U.L. (eds), tRNA: Structure, Biosynthesis and Function. ASM Press, Washington, DC, pp. 165–205.
- Cherry J.M. et al. (2002) Saccharomyces Genome Database, http://genome-www.stanford.edu/Saccharomyces/
- Darzacq X., Jady,B.E., Verheggen,C., Kiss,A.M., Bertrand,E. and Kiss,T. (2002) Cajal body-specific small nuclear RNAs: a novel class of 2′-O-methylation and pseudouridylation guide RNAs. EMBO J., 21, 2746–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England T.E. and Uhlenbeck,O.C. (1978) Enzymatic oligoribonucleotide synthesis with T4 RNA ligase. Biochemistry, 17, 2069–2076. [DOI] [PubMed] [Google Scholar]
- Filipowicz W. and Pogacic,V. (2002) Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol., 14, 319–327. [DOI] [PubMed] [Google Scholar]
- Ganot P., Jady,B.E., Bortolin,M.L., Darzacq,X. and Kiss,T. (1999) Nucleolar factors direct the 2′-O-ribose methylation and pseudouridylation of U6 spliceosomal RNA. Mol. Cell. Biol., 19, 6906–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H., Sprinzl,M. and Steinberg,S. (1995) Posttranscriptionally modified nucleosides in transfer RNA: their locations and frequencies. Biochimie, 77, 139–141. [DOI] [PubMed] [Google Scholar]
- Gustafsson C., Reid,R., Greene,P.J. and Santi,D.V. (1996) Identification of new RNA modifying enzymes by interactive genome search using known modifying enzymes as probes. Nucleic Acids Res., 24, 3756–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhofer A., Kiefmann,M., Meier-Ewert,S., O’Brien,J., Lehrach,H., Bachellerie,J.P. and Brosius,J. (2001) RNomics: an experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. EMBO J., 20, 2943–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jady B.E. and Kiss,T. (2001) A small nucleolar guide RNA functions both in 2′-O-ribose methylation and pseudouridylation of the U5 spliceosomal RNA. EMBO J., 20, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T. (2001) Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J., 20, 3617–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E.V. (1996) Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res., 24, 2411–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine D.L.J. and Tollervey,D. (1998) Birth of the snoRNPs: the evolution of the modification-guide snoRNAs. Trends Biochem. Sci., 23, 383–388. [DOI] [PubMed] [Google Scholar]
- Lowe T.M. and Eddy,S.R. (1999) A computational screen for methylation guide snoRNAs in yeast. Science, 283, 1168–1171. [DOI] [PubMed] [Google Scholar]
- Lowe T.M. and Eddy,S.R. (2000) S. cerevisiae methyl guide snoRNAs (snoRNA Database). http://rna.wustl.edu/snoRNAdb/Sc/Sc-snos-bysno.html
- Maden B.E.H. (1990) The numerous modified nucleotides in eukaryotic ribosomal RNA. Prog. Nucleic Acid Res. Mol. Biol., 39, 241–303. [DOI] [PubMed] [Google Scholar]
- Martzen M.R., McCraith,S.M., Spinelli,S.L., Torres,F.M., Fields,S., Grayhack,E.J. and Phizicky,E.M. (1999) A biochemical genomics approach for identifying genes by the activity of their products. Science, 286, 1153–1155. [DOI] [PubMed] [Google Scholar]
- Massenet S., Mougin,A. and Branlant,C. (1998) Posttranscriptional modifications in the U small nuclear RNAs. In Grosjean,H. and Benne,R. (eds), The Modification and Editing of RNA. ASM Press, Washington, DC, pp. 201–228.
- Massenet S., Motorin,Y., Lafontaine,D.L.J., Hurt,E.C., Grosjean,H. and Branlant,C. (1999) Pseudouridine mapping in the Saccharomyces cerevisiae spliceosomal U small nuclear RNAs (snRNAs) reveals that pseudouridine synthase Pus1p exhibits a dual substrate specificity for U2 snRNA and tRNA. Mol. Cell. Biol., 19, 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby M.I. and Greenbaum,N.L. (2001) A conserved pseudouridine modification in eukaryotic U2 snRNA induces a change in branch-site architecture. RNA, 7, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby M.I. and Greenbaum,N.L. (2002) Sculpting of the spliceosomal branch site recognition motif by a conserved pseudouridine. Nat. Struct. Biol., 9, 958–965. [DOI] [PubMed] [Google Scholar]
- Patton J.R. (1991) Pseudouridine modification of U5 RNA in ribonucleo protein particles assembled in vitro. Mol. Cell. Biol., 11, 5998–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J.R. (1993a) Multiple pseudouridine synthase activities for small nuclear RNAs. Biochem. J., 290, 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J.R. (1993b) Ribonucleoprotein particle assembly and modification of U2 small nuclear RNA containing 5-fluorouridine. Biochemistry, 32, 8939–8944. [DOI] [PubMed] [Google Scholar]
- Patton J.R. (1994) Formation of pseudouridine in U5 small nuclear RNA. Biochemistry, 33, 10423–10427. [DOI] [PubMed] [Google Scholar]
- Patton J.R., Jacobson,M.R. and Pederson,T. (1994) Pseudouridine formation in U2 small nuclear RNA. Proc. Natl Acad. Sci. USA, 91, 3324–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peculis B.A. (1997) RNA processing-pocket guides to ribosomal RNA. Curr. Biol., 7, 480–482. [DOI] [PubMed] [Google Scholar]
- Phizicky E.M. et al. (2002) Biochemical genomics approach to map activities to genes. Methods Enzymol., 350, 546–559. [DOI] [PubMed] [Google Scholar]
- Reddy R and Busch,H. (1988) Small nuclear RNAs: RNA sequences, structure and modifications. In Birnstiel,M.L. (ed.), Structure and Function of Major and Minor Small Nuclear Ribonucleoprotein Particles. Springer-Verlag, Berlin, Germany, pp. 1–37.
- Samarsky D. and Fournier,M.J. (2000) Small nucleolar RNAs (snoRNAs) from the yeast Saccharomyces cerevisiae. http://www.bio.umass.edu/biochem/rna-sequence/Yeast_snoRNA_Database/snoRNA_DataBase.html
- Santoro S.W. and Joyce,G.F. (1997) A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C.M. and Steitz,J.A. (1997) Sno storm in the nucleolus: new roles for myriad small RNPs. Cell, 89, 669–672. [DOI] [PubMed] [Google Scholar]
- Terns M.P. and Terns,R.M. (2002) Small nucleolar RNAs: versatile trans-acting molecules of ancient evolutionary origin. Gene Expr., 10, 17–39. [PMC free article] [PubMed] [Google Scholar]
- Tollervey D. and Kiss,T. (1997) Function and synthesis of small nucleolar RNAs. Curr. Opin. Cell Biol., 9, 337–342. [DOI] [PubMed] [Google Scholar]
- Tycowski K.T., You,Z.H., Graham,P.J. and Steitz,J.A. (1998) Modification of U6 spliceosomal RNA is guided by other small RNAs. Mol. Cell, 2, 629–638. [DOI] [PubMed] [Google Scholar]
- Weinstein L.B. and Steitz,J.A. (1999) Guided tours: from precursor snoRNA to functional snoRNP. Curr. Opin. Cell Biol., 11, 378–384. [DOI] [PubMed] [Google Scholar]
- Yu Y.-T. (2000) Site-specific 4-thiouridine incorporation into RNA molecules. Methods Enzymol., 318, 71–88. [DOI] [PubMed] [Google Scholar]
- Yu Y.-T., Shu,M.-D. and Steitz,J.A. (1998) Modifications of U2 snRNA are required for snRNP assembly and pre-mRNA splicing. EMBO J., 17, 5783–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.-T., Scharl,E.C., Smith,C.M. and Steitz,J.A. (1999) The growing world of small nuclear ribonucleoprotein particles. In Gesteland,R.F., Cech,T.R. and Atkins,J.F. (eds), The RNA World, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 487–524.
- Zerby D.B. and Patton,J.R. (1996) Metabolism of pre-messenger RNA splicing cofactors: modification of U6 RNA is dependent on its interaction with U4 RNA. Nucleic Acids Res., 24, 3583–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerby D.B. and Patton,J.R. (1997) Modification of human U4 RNA requires U6 RNA and multiple pseudouridine synthases. Nucleic Acids Res., 25, 4808–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. Li,Z.-H., Terns,R.M., Terns,M.P. and Yu,Y.-T. (2002) An H/ACA guide RNA directs U2 pseudouridylation at two different sites in the branch point recognition region in Xenopus oocytes. RNA, 8, 1515–1525. [PMC free article] [PubMed] [Google Scholar]