

Abstract
Hen egg-white lysozyme dissolved in glycerol containing 1% water was studied by using CD and amide proton exchange monitored by two-dimensional 1H NMR. The far- and near-UV CD spectra of the protein showed that the secondary and tertiary structures of lysozyme in glycerol were similar to those in water. Thermal melting of lysozyme in glycerol followed by CD spectral changes indicated unfolding of the tertiary structure with a Tm of 76.0 ± 0.2°C and no appreciable loss of the secondary structure up to 85°C. This is in contrast to the coincident denaturation of both tertiary and secondary structures with Tm values of 74.8 ± 0.4°C and 74.3 ± 0.7°C, respectively, under analogous conditions in water. Quenched amide proton exchange experiments revealed a greater structural protection of amide protons in glycerol than in water for a majority of the slowly exchanging protons. The results point to a highly ordered, native-like structure of lysozyme in glycerol, with the stability exceeding that in water.
Is water truly a unique solvent for biochemical processes or can proteins dissolve and conduct their normal biological functions in some nonaqueous solvents as well? The realization that proteins, if lyophilized from an aqueous solution at a pH remote from their pI, can readily dissolve in numerous neat organic solvents (1, 2) has allowed a systematic investigation of this question. For example, the solubility of the well studied and representative hydrophilic protein hen egg-white lysozyme (pI ≈11) (3), lyophilized from pH 6, exceeds 10 mg/ml in more than a dozen nonaqueous solvents without any extra additives (1). In one such solvent, 99% glycerol, fully reduced and unfolded lysozyme can correctly reoxidize and refold with an efficiency comparable with that in aqueous solution (4). Another protein, the protease subtilisin Carlsberg, in this solvent exhibits a catalytic activity similar to that in water (5). The key issue in rationalizing these striking findings, as well as in answering the question posed above, is that of protein structure and conformational flexibility in glycerol.
Although structural properties of proteins in water/glycerol mixtures have been examined (6), essentially no work has been done in neat (or nearly neat) glycerol. And yet, it is becoming clear that the protein structure in organic solvents cannot be deduced by extrapolation from aqueous/organic mixtures—the native conformation may be retained both in water and in a neat solvent but not in mixtures thereof (7). Therefore, in the present study we have used two independent experimental methodologies, hydrogen isotope exchange followed by two-dimensional NMR spectroscopy and far- and near-UV CD spectroscopy, to assess conformational properties of hen egg-white lysozyme dissolved in glycerol.§ The results obtained reveal that the structure of lysozyme in glycerol is highly ordered, is more thermostable than in water, and has local structural fluctuations discernibly distinct from those in aqueous solution.
MATERIALS AND METHODS
Materials.
Hen egg-white lysozyme (EC 3.2.1.17), thrice crystallized, dialyzed, and lyophilized, was purchased from Sigma. Poly-dl-alanine (PDLA), also from Sigma, was purified on Sephadex G-15 and lyophilized from D2O (Cambridge Stable Isotopes, Cambridge, MA). Glycerol (99.8% pure) was purchased from Mallinckrodt. All other chemicals used in this work were obtained from commercial suppliers and were of analytical grade or purer.
Preparation of Lysozyme Samples.
Lysozyme was dialyzed and lyophilized from pH 3.8 (1) and stored in a desiccator at −18°C. Amide protons of lysozyme were totally exchanged for deuterons in D2O (pD 3.4) at 82°C (8). Protein concentrations were determined spectrophotometrically at 280 nm, using the extinction coefficient of 2.63 (mg protein)−1⋅ml⋅cm−1 (9).
CD Analysis.
Lysozyme was dissolved in water at pH 3.8, diluted 1:100 either into the same aqueous solvent or into glycerol, and mixed vigorously. Final protein concentrations were 0.15 and 0.25 mg/ml for far- and near-UV CD experiments, respectively. CD spectra were measured in a Jasco (Easton, MD) 710 spectropolarimeter at 25°C. Far-UV measurements were performed in 0.1-cm (water) and 0.05-cm (glycerol) circular cuvettes, and near-UV measurements in 1.0-cm cells. Spectra (four accumulations for each sample) were acquired at a scan speed of 2 nm/min with response time of 8 sec. Spectra of water (pH 3.8) and glycerol containing 1% water (pH 3.8) were measured under the same conditions and used for baseline corrections. Noise reduction was achieved by Fourier transformation processing using software provided by Jasco. Ellipticity is reported as per residue molar ellipticity,[Θ] (deg⋅cm2⋅dmol−1).
Secondary structure determination from the far-UV CD spectra was performed by using the lincom program (10) with spectral basis sets derived by Sreerama and Woody (11). The quality of the spectral fitting was slightly better for water (the resulting SD = 0.74) than for glycerol (SD = 0.81).
For thermal melting experiments, lysozyme was dissolved in H2O (pH 3.8) at 12 mg/ml, filtered through a 0.22 μm Millipore filter, and diluted 1:100 into either water (pH 3.8) or glycerol. Heat-induced spectral changes were followed at 222 and 290 nm. Temperature scans performed by adjustment either manually (at approximately 30°C/hr) or automatically (at heating rates of 20°C/hr and 50°C/hr for water and 20°C/h for glycerol) superimposed, indicating the equilibrium character of the transition under these conditions. The spectra of lysozyme before and after melting agreed within 90%, confirming reversibility of the protein denaturation under these conditions. Melting temperatures, Tm, were determined as described (12) by using the two-state approximation. The measured Tm values for different heating rates were averaged.
Amide Proton Exchange of PDLA.
PDLA was used as a model for the amide proton exchange of an unfolded polypeptide (13). Deuterated PDLA was dissolved in D2O at 20 mg/ml and then diluted 1:100 into either H2O or glycerol to initiate the exchange. The experiments were performed for PDLA dissolved at pD 2.6, 3.4, and 4.6, which corresponds to pH values of 3.0, 3.8, and 5.0, respectively (14). Proton exchange at 30°C was monitored by measuring the absorbance at 220 nm and found to obey first-order kinetics. Exchange constants, kint (intrinsic), were determined for five samples at each pH value and then averaged. Log kint was found to correlate linearly with the pD of the initial PDLA solution for both solvents. Rate constants for other pH values were determined by using linear extrapolation.
Amide proton exchange of lysozyme in glycerol was monitored by using a modification of the quenched flow technique (15, 16). Deuterated lysozyme was dissolved in D2O at 500 mg/ml (pD 3.4) or 400 mg/ml (pD 7.1). Amide proton exchange was initiated by a 1:100 dilution into regular (nondeuterated) glycerol. After vigorous mixing and incubation at 30°C for certain time periods, the exchange was quenched by a 1:10 dilution into H2O at pH 3.8 and 0°C [under these conditions, the amide proton exchange is very slow (17)]. The resultant lysozyme solution subsequently was processed at 4°C by first concentrating 1:10 by using a Miniplate device (Millipore) and then diafiltered against 3 vol of H2O (pH 3.8) to obtain a final concentration of glycerol below 0.5%. The lysozyme sample then was concentrated in Centriplus tubes (Millipore), diluted 1:10 into D2O (pD 3.4), and concentrated in Centriplus and Centricon tubes (Millipore) to approximately 90 mg/ml.
The degree of exchange of amide deuterons for protons was determined by 1H NMR using purged correlation spectroscopy (PCOSY) experiments (18) performed with a JEOL GX 400 NMR spectrometer. Spectra were measured at 35°C with water presaturation, a 1.3-sec repetition rate, 64 scans per each t2 transient, and 512 t1 points. Peaks were assigned according to ref. 8. The absolute intensities of all four components of the anti-phase crosspeaks between amide and α-protons were measured and averaged. Crosspeaks between α- and β-protons were used as an internal reference for quantification and comparison of different spectra.
Analysis of the Amide Proton Exchange.
Amide proton exchange in a protein may be slowed primarily because of the presence of intramolecular hydrogen bonds formed on folding. The protection factor, Pf, of a given amide proton is the ratio of the intrinsic exchange constant, kint, to the observed exchange constant, kobs (19). The latter rate constant for each individual proton is determined from the first-order kinetic analysis of the corresponding NMR signal intensity as a function of the exchange time. The intrinsic exchange constant corresponds to the exchange measured in the absence of the secondary and tertiary structures and usually is calculated from model-compound studies (13). The ratio of the protection factors of an amide proton in glycerol (Pfg) to that in water (Pfw) represents the relative stabilization between the two solvents and is termed here the stability factor, Sf. The latter can be determined from the experimental data according to the equation:
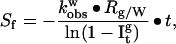 |
1 |
where kobsw is the exchange constant of this amide proton in water taken from ref. 20, Rg/w is the ratio of kint in glycerol to that in water for the model compound PDLA; and Itg is the average peak intensity of the crosspeaks of a particular amide proton in the PCOSY spectrum after the protein has been incubated in glycerol for time t, normalized as outlined in the previous section.
This exchange/quench methodology allowed us to follow the exchange of 33 of the slowest exchanging amide protons in lysozyme (17, 20). The exchange experiments were performed in duplicates, and the resultant percentages of the exchange for a given proton varied <10%. The data were obtained in this manner for Leu-8, Ala-11, Asn-27, Asn-39, Ala-42, Asn-65, Cys-76, Leu-83, Val-92, Lys-97, and Val-99, after 24 hr, and for Ala-9, Lys-13, Phe-34, Arg-61, Trp-63, and Cys-64, after 4 weeks, of incubation in glycerol. For the remaining 16 protons, the exchange was so slow that no peaks were detected in PCOSY spectra even after 4 weeks. For these protons, the upper limits of the peak intensities could be estimated from the baseline noise of the PCOSY spectra and used to provide lower limit estimates for the stability factors. The lower limits of the Sf values for Ala-10, Trp-28, Val-29, Ala-32, Lys-33, Asp-52, Gln-57, Ile-58, and Ser-60 were found to exceed unity, indicating the structural protection in glycerol higher than in water. For the remaining protons (Met-12, Cys-30, Ala-31, Tyr-53, Ala-95, Lys-96, and Ile-98), it was not possible to determine whether structural protection is above or below that in water.
RESULTS AND DISCUSSION
We recently discovered that unfolded/reduced hen egg-white lysozyme could correctly refold/reoxidize even in 99% glycerol (4). To rationalize this striking observation, i.e., correct protein folding in essentially a nonaqueous medium, in the present study we examined the structure and rigidity of lysozyme dissolved in glycerol by using two independent biophysical methods—CD and modified quenched-flow amide proton exchange (15, 16) monitored by two-dimensional 1H NMR.
CD analysis of lysozyme dissolved in water and in glycerol was carried out in the far- and near-UV regions. The former (shorter wavelengths) reflects the secondary structure, whereas the latter (longer wavelengths) arises from the tertiary structure of the protein (21, 22).
Fig. 1A depicts the far-UV CD spectra of lysozyme dissolved in water at pH 3.8 and then diluted 1:100 either with the same aqueous solution (curve a) or with neat glycerol (curve b). One can see that the two spectra, although not identical, are very similar. Both display strong negative bands in the range from 200 to 260 nm and signal intensity at 208 nm greater than at 222 nm, which is characteristic of an α + β protein (23). The contents of the secondary structural elements in the lysozyme molecule both in water and in glycerol were determined (Table 1) by fitting these spectra to published basis sets (11). The results obtained in water were essentially the same as those reported by others using either CD (24) or Fourier-transform IR spectroscopies (25). The α-helix and β-turn contents of lysozyme were found to be virtually the same in glycerol as in water, whereas the β-sheet content is greater.
Figure 1.

Far-UV (A) and near-UV (B) CD spectra of lysozyme in water at pH 3.8 (a) and in glycerol (b). The lyophilized protein was dissolved in water at pH 3.8 at 15 and 25 mg/ml for far- and near-UV measurements, respectively, and then diluted 1:100 with either water (pH 3.8) or glycerol. Spectra of water (pH 3.8) and glycerol containing 1% water (pH 3.8) were measured at 25°C under the same conditions and subtracted for baseline corrections.
Table 1.
The secondary structure of lysozyme dissolved in water and in glycerol as determined from the far-UV CD spectra
| Component | Secondary structure content, %
|
|
|---|---|---|
| Water | Glycerol | |
| α-Helix | 30 | 30 |
| β-Sheet | 13 | 21 |
| β-Turn | 27 | 25 |
| Unordered | 30 | 24 |
CD spectra were measured at 25°C. Lyophilized lysozyme was dissolved in water (pH 3.8) at 15 mg/ml, and then diluted 1:100 either by the same aqueous solution or by glycerol. The secondary structure contents were determined as outlined in Materials and Methods.
Fig. 1B shows the near-UV CD spectra of the aqueous and glycerol solutions of lysozyme. The spectrum in water (curve a) is dominated by a positive peak intensity in the 280–300 nm range and is identical to those reported (21, 26). The spectrum in glycerol (curve b) is similar to that in water, although the peaks are broadened and slightly shifted. The presence of a positive peak intensity in this region reflects significant tertiary structure of the protein (22). It is worth mentioning that such signals are not observed for lysozyme dissolved in other, evidently denaturing, organic solvents, including ethylene glycol, methanol, dimethyl sulfoxide, formamide, and dimethylformamide (27), where the protein is devoid of the tertiary structure. The similarity of the aqueous and glycerol spectra suggests a similarity in the environments of the aromatic amino acid side chains and, in turn, in the tertiary structures.
Next, we addressed the structural stability of lysozyme dissolved in water and in glycerol. This was achieved by following its thermal denaturation both at 222 nm (secondary structure) and at 290 nm (tertiary structure). In water, the secondary structure of lysozyme undergoes a cooperative thermal unfolding transition with a Tm of 74.3 ± 0.7°C (Fig. 2A, curve a), whereas in glycerol there is no transition in the temperature range from 25°C to 85°C (Fig. 2A, curve b). In contrast, the melting of the tertiary structure of lysozyme occurred both in water and in glycerol, and the Tm values were similar, 74.8 ± 0.4°C (Fig. 2B, curve a) and 76.0 ± 0.2°C (Fig. 2B, curve b), respectively. [Note that the Tm value determined herein for the temperature unfolding of lysozyme in water is the same as reported by others (20)].
Figure 2.

Temperature melting of the secondary (A) and tertiary (B) structure of lysozyme in water at pH 3.8 (a) and in glycerol (b). The lyophilized protein was dissolved in water (pH 3.8) at 12 mg/ml, filtered, and diluted 1:100 with either water (pH 3.8) or glycerol. Unfolding of the secondary and tertiary structures of lysozyme was followed by measuring the changes in the mean residue ellipticity. The heating rate was 20°C/hr. Mean residue ellipticities of water at pH 3.8 and of glycerol containing 1% water (pH 3.8) were examined separately as a function of temperature and subtracted for baseline corrections. The Tm values of lysozyme’s unfolding were determined as described in ref. 12.
In water, thermal unfolding of the secondary and tertiary structures of lysozyme occurs concurrently (curves a in Fig. 2), in agreement with the previous observations (e.g., ref. 28), and suggests no significant accumulation of intermediates. In glycerol, the situation is markedly different. Although lysozyme dissolved in glycerol loses its tertiary structure in a cooperative event at nearly the same melting temperature as in water, the secondary structure in the resultant intermediate state is still highly populated. Such conformational states that are devoid of the tertiary structure but contain extensive secondary structure may be viewed as molten globule-like states (29).
To gain further, more detailed insights into the structural stability of lysozyme dissolved in glycerol, we used an additional methodology, amide proton exchange, that has been used extensively to study protein stability and structural fluctuations in water (19, 30). The experimental design involved a quenched technique similar to the quenched flow methods used to study folding intermediates (31–33). In our experiments, lysozyme with amide protons totally exchanged for deuterons was dissolved in D2O, diluted 1:100 into nondeuterated glycerol, allowed to exchange for a certain time period, and quenched by dilution into H2O at a pH (3.8) and temperature (0°C) where the exchange is minimal (17, 20, 34); the samples were concentrated, and the extent of the exchange in glycerol was assessed by using two-dimensional 1H NMR.
The initial exchange experiments were carried out for lysozyme predissolved in D2O at pD 3.4 (which corresponds to pH 3.8, i.e., the same value as that used in the CD experiments described above). However, no measurable exchange was detected even after a 6-week incubation; incubation for significantly longer periods resulted in lysozyme degradation. These data clearly indicate that the amide protons are trapped by the secondary and/or tertiary structure of lysozyme in glycerol to a far greater extent than in the nonstructured PDLA control, which fully exchanged in a matter of minutes. This observation, although qualitative, unequivocally demonstrates that lysozyme dissolved in glycerol is highly ordered.
To accelerate the amide proton exchange reaction, the pD value of the lysozyme solution in D2O before dilution with glycerol was raised from 3.4 to 7.1. Such a change in aqueous solution results in some 100-fold increase in the exchange rates (17, 20, 34). A similar increase may be expected in glycerol because of the phenomenon of protein “pH memory” (4, 35) and was, in fact, observed. Lysozyme was incubated in glycerol for both 24 hr and 4 weeks. Fig. 3 displays the stability factors, Sf, of those slowly exchanging amide protons of lysozyme which were determined quantitatively from two-dimensional 1H NMR spectra (see Materials and Methods). These stability factors vary more than 300-fold—from 0.15 to 48. Four of the amide protons (Leu-8, Leu-83, Lys-97, and Val-99) have Sf values less than unity, indicating that the structure of lysozyme in glycerol is more amenable to the exchange at these positions than in water. However, the great majority of the measured stability factors exceed unity, indicating that the structure at the corresponding amino acid residues is less open to the exchange than in water.
Figure 3.

Stability factors, Sf, (for glycerol vs. water) of the slowly exchanging amide protons belonging to different amino acid residues of lysozyme (see Materials and Methods for details).
The observed variations of the stability factors reveal that changes in the protection of amide protons against the exchange on transition from water to glycerol depend on the location of the specific amide proton in the protein globule. Because the protons monitored in the present study exchange in water through local unfolding (17, 20, 34), the detected wide range of stability factors likely reflects significant changes in the pattern of local unfolding of lysozyme in glycerol compared with that in water.
All of the slowest exchanging amide protons of lysozyme can be divided into three classes: those where the measured Sf is below one, those where the measured or estimated Sf is above one, and those for which the Sf values could not be reliably classified (see Materials and Methods). These classes are depicted on a ribbon diagram of lysozyme in Fig. 4. One can see that three of the four amide protons that are less structurally protected in glycerol than in water (Leu-8, Lys-97, and Val-99) are located at the ends of α-helices (Fig. 4, red spheres), which, therefore, are concluded to be more open to the exchange in glycerol than in water, presumably because of greater structural fluctuations. (The last of the four amide protons from the first class, Leu-83, is located in the 310 helix.) Green spheres in Fig. 4 illustrate that lysozyme exhibits increased structural protection from exchange for most of its slowly exchanging amide protons in glycerol as compared with water. This is true for those amide protons involved in hydrogen bonds in α-helices (Ala-9, Ala-10, Ala-11, Lys-13, Trp-28, Val-29, Ala-32, Phe-34, and Val-92) and the β-sheet (Ala-42, Asp-52, Gln-57, Ile-58, and Ser-60). This also is true for the residues from the loop region (Arg-61, Trp-63, Cys-64, Asn-65, and Cys-76), including Cys-64, which is not involved in hydrogen bonds (36). In fact, the largest stability factors are exhibited by Arg-61 and Trp-63, located in the loop region, suggesting that this area is greatly stabilized in glycerol compared with water. It is interesting to note that Trp-63 lies at the active site hinge region of the protein between the α- and β-domains and is not buried in the hydrophobic core.
Figure 4.

A ribbon diagram of the three-dimensional structure of lysozyme. Spheres are drawn at the positions of the appropriate amide nitrogens and color-coded by the stability factor class (see text): Sf measured to be <1 (red spheres), Sf measured or estimated to be >1 (green spheres), and Sf that could not be reliably estimated from our data (gray spheres). This figure was prepared with molmol (45) using the Protein Data Bank file 1hew. The NH2 and COOH groups mark the amino and carboxyl termini, respectively, of the protein.
Thus the bulk of our data indicates that in glycerol, compared with water, the structural equilibria of lysozyme are shifted toward closed forms, thereby protecting the majority of the amide protons from the exchange. The simplest explanation for this is that the folded structure of lysozyme is stabilized in glycerol more than in water. The increase in the stability factors would then arise from a general increase in the closed-form population. This mechanism, however, seems incompatible with the CD-monitored thermal unfolding of the tertiary structure of lysozyme, which occurs at nearly the same temperature as in water (Fig. 2B). This observation suggests that the increase in Sf in glycerol does not arise from a general increase in structural stability, although a lack of information on the apparent changes in the heat capacity in glycerol does not allow the complete exclusion of this possibility.
Another possibility is that the unfolding mechanism of lysozyme changes in glycerol. The thermal denaturation data (Fig. 2) reveal that in glycerol lysozyme unfolds via a three-state mechanism involving an intermediate with extensive secondary structure populated at high temperature. Both the observed thermal stability of the tertiary structure of lysozyme in glycerol and the increase in the Sf values (Figs. 3 and 4) are consistent with the presence of an intermediate conformational state that shifts the overall equilibrium away from the unfolded forms. Thus, even with the stability of the tertiary structure unchanged, the product of its unfolding is an intermediate still protected against the amide proton exchange. This mechanism also is supported by the following observations: (i) the thermal unfolding intermediate detected by CD herein has properties similar to those observed in low-pH and other destabilized forms of the protein (37, 38), as well as in its folding intermediates (21); and (ii) folding/unfolding intermediates have been detected by the amide proton exchange in other proteins (39, 40). Furthermore, the largest amide proton protection factors in water for the molten globule form of apomyoglobin, 200 (41), in the early folding intermediates of lysozyme, 200 (42), and of RNase A, 1000 (43), and in a peptide model for early folding intermediates, 260 (44), suggest that these sorts of intermediate forms have sufficient structural stability to account for the increase in the amide protons protection that we have observed.
In summary, the CD data suggest that the secondary and tertiary structures of lysozyme in glycerol resemble those in water. Thermal melting studies indicate that (i) the tertiary structure of lysozyme in glycerol unfolds at almost the same temperature as in water, and (ii) the result of this transition, in contrast to the water situation, is a form that contains high levels of the secondary structure. The amide proton exchange data reveal that for most of the amide protons examined, on transition from water to glycerol the equilibrium between the open (i.e., capable of the amide proton exchange) and closed (protected from the exchange) forms shifts toward the latter.
The data obtained here support the notion that just as water is not a unique medium for protein folding (4), it is likewise not unique for the maintenance of highly ordered and stable native-like protein structures. Despite some differences, the basic structure and stability of lysozyme in water and in glycerol are similar. This similarity does not extend to other, including structurally related, organic solvents, such as ethylene glycol (27), although a wider range of solvents will have to be examined. Also, it needs to be established whether other proteins will behave in glycerol the same way as lysozyme.
Acknowledgments
We are indebted to Prof. O. B. Ptitsyn for insightful discussion of the lysozyme unfolding process and to Drs. D. Schaak and O. Gursky for helpful comments during this study. This work was financially supported by the U.S. Department of Energy, the Biotechnology Process Engineering Center at the Massachusetts Institute of Technology, and the Rowland Institute for Science.
ABBREVIATIONS
- PCOSY
purged correlation spectroscopy
- PDLA
poly-d,l-alanine
Footnotes
Glycerol herein always contained 1% (vol/vol) of water to allow direct correlations with our previous lysozyme folding data (4). For brevity, this 99% solvent is referred to as glycerol.
References
- 1.Chin J T, Wheeler S L, Klibanov A M. Biotechnol Bioeng. 1994;44:140–145. doi: 10.1002/bit.260440120. [DOI] [PubMed] [Google Scholar]
- 2.Bromberg L E, Klibanov A M. Proc Natl Acad Sci USA. 1995;92:1262–1266. doi: 10.1073/pnas.92.5.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jolles P, editor. Lysozyme: Model Enzyme in Biochemistry and Biology. Basel: Birkhauser; 1996. [Google Scholar]
- 4.Rariy R V, Klibanov A M. Proc Natl Acad Sci USA. 1997;94:13520–13523. doi: 10.1073/pnas.94.25.13520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu K, Griebenow K, Klibanov A M. Biotechnol Bioeng. 1997;56:485–491. doi: 10.1002/(SICI)1097-0290(19971205)56:5<485::AID-BIT2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 6.Lapanje S. Physicochemical Aspects of Protein Denaturation. New York: Wiley; 1978. pp. 143–156. [Google Scholar]
- 7.Griebenow K, Klibanov A M. J Am Chem Soc. 1996;118:11695–11700. [Google Scholar]
- 8.Redfield C, Dobson C M. Biochemistry. 1988;27:122–135. doi: 10.1021/bi00401a020. [DOI] [PubMed] [Google Scholar]
- 9.Hamaguchi K, Kurono A. J Biochem. 1963;54:111–122. [PubMed] [Google Scholar]
- 10.Perczel A, Park K, Fasman G D. Anal Biochem. 1992;203:83–93. doi: 10.1016/0003-2697(92)90046-a. [DOI] [PubMed] [Google Scholar]
- 11.Sreerama N, Woody R W. Anal Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- 12.Pace C N, Shirley B A, Thomson J A. In: Protein Structure: A Practical Approach. Creighton T E, editor. Oxford: IRL; 1989. pp. 311–330. [Google Scholar]
- 13.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasoe P K, Long F A. J Phys Chem. 1960;64:188–190. [Google Scholar]
- 15.Paterson Y, Englander W S, Roder H. Science. 1990;249:755–759. doi: 10.1126/science.1697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y Z, Paterson Y, Roder H. Protein Sci. 1995;4:804–814. doi: 10.1002/pro.5560040420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedersen T G, Thomsen N K, Andersen K V, Madsen J C, Poulsen F M. J Mol Biol. 1993;230:651–660. doi: 10.1006/jmbi.1993.1176. [DOI] [PubMed] [Google Scholar]
- 18.Marion D, Bax A. J Magn Reson. 1988;80:528–533. [Google Scholar]
- 19.Hvidt A, Nielson S O. Adv Protein Chem. 1966;21:287–386. doi: 10.1016/s0065-3233(08)60129-1. [DOI] [PubMed] [Google Scholar]
- 20.Radford S E, Buck M, Topping K D, Evans P A. Proteins Struct Funct Genet. 1992;14:237–248. doi: 10.1002/prot.340140210. [DOI] [PubMed] [Google Scholar]
- 21.Kuwajima K, Hiraoka Y, Ikeguchi M, Sugai S. Biochemistry. 1985;24:874–881. doi: 10.1021/bi00325a010. [DOI] [PubMed] [Google Scholar]
- 22.Kelly S M, Price N C. Biochim Biophys Acta. 1997;1338:161–185. doi: 10.1016/s0167-4838(96)00190-2. [DOI] [PubMed] [Google Scholar]
- 23.Venyaminov S Yu, Yang J T. In: Circular Dichroism and the Conformational Analysis of Biomolecules. Fasman G D, editor. New York: Plenum; 1996. pp. 69–104. [Google Scholar]
- 24.Chang C T, Wu C S C, Yang J T. Anal Biochem. 1978;91:13–31. doi: 10.1016/0003-2697(78)90812-6. [DOI] [PubMed] [Google Scholar]
- 25.Costantino H R, Griebenow K, Mishra P, Langer R, Klibanov A M. Biochim Biophys Acta. 1995;1253:69–74. doi: 10.1016/0167-4838(95)00156-o. [DOI] [PubMed] [Google Scholar]
- 26.Buck M, Radford S E, Dobson C M. Biochemistry. 1993;32:669–678. doi: 10.1021/bi00053a036. [DOI] [PubMed] [Google Scholar]
- 27.Knubovets, T., Osterhout, J. J. & Klibanov, A. M. (1999) Biotechnol. Bioeng.63, in press. [PubMed]
- 28.Dobson C M, Evans P A. Biochemistry. 1984;23:4267–4270. [Google Scholar]
- 29.Ptitsyn O B. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- 30.Clarke J, Itzhaki L S. Curr Opin Struct Biol. 1998;8:112–118. doi: 10.1016/s0959-440x(98)80018-3. [DOI] [PubMed] [Google Scholar]
- 31.Loftus D, Gbenle G O, Kim P S, Baldwin R L. Biochemistry. 1986;25:1428–1436. doi: 10.1021/bi00354a036. [DOI] [PubMed] [Google Scholar]
- 32.Radford S E, Dobson C M, Evans P A. Nature (London) 1992;358:302–307. doi: 10.1038/358302a0. [DOI] [PubMed] [Google Scholar]
- 33.Jennings P A, Wright P E. Science. 1993;262:892–896. doi: 10.1126/science.8235610. [DOI] [PubMed] [Google Scholar]
- 34.Delepierre M, Dobson C M, Karplus M, Poulsen F, States D J, Wedin R E. J Mol Biol. 1987;197:111–130. doi: 10.1016/0022-2836(87)90613-9. [DOI] [PubMed] [Google Scholar]
- 35.Klibanov A M. Nature (London) 1995;374:596. doi: 10.1038/374596a0. [DOI] [PubMed] [Google Scholar]
- 36.Miranker A, Radford S E, Karplus M, Dobson C M. Nature (London) 1991;349:633–636. doi: 10.1038/349633a0. [DOI] [PubMed] [Google Scholar]
- 37.Haezebrouck P, Joniau M, Van Dael H, Hooke S D, Woodruff N D, Dobson C M. J Mol Biol. 1995;246:382–387. doi: 10.1006/jmbi.1994.0093. [DOI] [PubMed] [Google Scholar]
- 38.Hamaguchi K, Sakai H. J Biochem. 1965;57:721–732. doi: 10.1093/oxfordjournals.jbchem.a128138. [DOI] [PubMed] [Google Scholar]
- 39.Chamberlain A K, Handel T M, Marqusee S. Nat Struct Biol. 1996;3:782–787. doi: 10.1038/nsb0996-782. [DOI] [PubMed] [Google Scholar]
- 40.Raschke T M, Marqusee S. Nat Struct Biol. 1997;4:298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 41.Hughson F M, Wright P E, Baldwin R L. Science. 1990;249:1544–1548. doi: 10.1126/science.2218495. [DOI] [PubMed] [Google Scholar]
- 42.Lu J R, Dahlquist F W. Biochemistry. 1992;31:4749–4756. doi: 10.1021/bi00135a002. [DOI] [PubMed] [Google Scholar]
- 43.Udgaonkar J B, Baldwin R L. Proc Natl Acad Sci USA. 1990;87:8197–8201. doi: 10.1073/pnas.87.21.8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fezoui, Y., Braswell, E. H., Xian, W. & Osterhout, J. J. (1999) Biochemistry 38, in press. [DOI] [PubMed]
- 45.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]