

Abstract
We report here that the E7 oncoprotein encoded by the oncogenic human papillomavirus (HPV) type 16 binds to the glycolytic enzyme type M2 pyruvate kinase (M2-PK). M2-PK occurs in a tetrameric form with a high affinity to its substrate phosphoenolpyruvate and a dimeric form with a low affinity to phosphoenolpyruvate, and the transition between both conformations regulates the glycolytic flux in tumor cells. The glycolytic intermediate fructose 1,6-bisphosphate induces the reassociation of the dimeric to the tetrameric form of M2-PK. The expression of E7 in an experimental cell line shifts the equilibrium to the dimeric state despite a significant increase in the fructose 1,6-bisphosphate levels. Investigations of HPV-16 E7 mutants and the nononcogenic HPV-11 subtype suggest that the interaction of HPV-16 E7 with M2-PK may be linked to the transforming potential of the viral oncoprotein.
Keywords: glycolysis, cell transformation, cancer, virus
Unicellular organisms have a variety of sensing mechanisms to adapt the cell proliferation rate to variations in their environmental nutrient supply. Several gene products, like the ras or cdc kinase proteins, which are involved in nutrient sensing in yeast (1, 2), are conserved during the evolution of multicellular organisms, and in mammals, these gene products often are altered in tumors. Despite our knowledge about the protein machinery regulating cell proliferation increasing tremendously over the recent years, we are still at the beginning to understand how nutrients contribute to proliferation control in multicellular organisms. There is, however, quite good evidence that phosphometabolites derived from both glycolysis (for recent review, see ref. 3) and the pentose phosphate pathway (ref. 4 and references therein) provide some of the signals linking metabolic conditions to cell proliferation. The glycolytic phosphometabolites, which are necessary for the biosynthesis of nucleic acids, phospholipids, and complex carbohydrates, are up-regulated in the G1 phase of the cell cycle (for recent review, see ref. 3), and constant high levels of phosphometabolites have been detected by 31P NMR spectroscopy in rapidly proliferating tumor cells (5).
The key enzymes regulating the glycolytic phosphometabolite pools and glycolytic flux rate are hexokinase, 6-phosphofructo 1-kinase, and especially pyruvate kinase (PK) (6, 7). The activity of PK, the key enzyme controlling the exit of the glycolytic pathway, determines the relative amount of glucose that is channeled into synthetic processes or used for glycolytic energy production (6, 8). Proliferating mammalian cells express the M2 type isoenzyme of PK (M2-PK) (9), and expression of M2-PK is cell cycle regulated in proliferating rat thymocytes (10). M2-PK occurs in an active tetrameric and a less active dimeric form (8, 11, 12), and the switch between both forms, which is controlled by the glycolytic phosphometabolite fructose 1,6-bisphosphate (FBP) (8), regulates the glycolytic flux in tumor cells.
In humans, cancer of the cervix is linked to infection by human papillomaviruses (HPV) of the high-risk group, whereas viruses of the low-risk group are not associated with cancers in vivo and fail to transform human cells in vitro (13). The E7 oncogene of HPV-16, a high-risk HPV type, cooperates with the HPV-16 E6 gene to immortalize human keratinocytes (14) and with an activated ras gene to transform rodent fibroblasts (15). The transforming activity of E7 is sensitive to mutations in both the N-terminal (15) and C-terminal (16) domains. Although the E7 N terminus mediates binding to proteins of the retinoblastoma gene family and thereby contributes to deregulation of the cell cycle (reviewed in ref. 17), the function of the C-terminal domain remains to be disclosed.
MATERIALS AND METHODS
Two-Hybrid Screen.
Yeast strain EGY48/pSH1834/pLexA-16 E7(39–98)∷HIS3 (18) was transformed with a human cDNA library derived from human serum-starved WI-38 fibroblasts (19), according to Gietz et al. (20). Primary transformants (3.9 × 106) were plated on 2% galactose/1% raffinose Ura−, His−, Trp− Leu− plates. Leu+ colonies were analyzed on selection media as described (19), to identify clones in which both the LEU2 and LacZ reporter genes display galactose-dependent transcription. Helper plasmids pSH1834 and pLexA-16 E7(39–98) were eliminated from positive clones by genetic selection (21); subsequently, plasmids encoding the selected B42 fusion proteins were rescued from the respective yeast strains as described (21) and sequenced. For quantitative interaction analysis, pLexA-E7 and B42-M2-PK fusion proteins were coexpressed in yeast strain EGY48/pSH1834, containing a LexA operator-lacZ gene (lexAo8-Gal1-lacZ∷URA3), and β-galactosidase activity was measured as described (18). The values were corrected by subtracting the intrinsic transactivation activity of each individual LexA fusion protein.
In Vitro Interaction Analysis.
Purified glutathione S-transferase (GST) or GST fusion proteins (15 ng/μl each) immobilized on glutathione Sepharose 4B beads were incubated with 1.0 mg of whole-cell extract from yeast cells, and the amount of B42-HA1-M2PK protein that was retained on the beads was determined by direct immunoblotting as described (22), by using a mAb to the HA1 epitope (YPYDVPDYA) (kindly provided by T. Nilson, European Laboratory for Molecular Biology, Heidelberg, Germany).
E7-Expressing Cell Lines.
Early passage NIH 3T3 cells, which have a high aerobic glycolytic rate, a low glutaminolytic flux rate, and a high amount of the tetrameric form of M2-PK (S.M., unpublished data), were used after stable transfection with the pMo expression vector (M/1 cells; ref. 23) or expression vectors encoding wild-type HPV-16 E7 (E7/2 cells; ref. 23) or a C-terminal deletion mutant (Δ79–83; ref. 24). The 14/2 cell line was derived from baby rat kidney cells by stable expression of an activated ras oncogene, combined with glucocorticoid-inducible expression of the HPV-16 E7 gene (25).
Immunoprecipitations.
Cells were extracted in hypotonic lysis buffer as described (22); precleared extracts (1 mg each) were either adjusted to 125 mM NaCl and 0.1% Nonidet P-40 and subjected to immunoprecipitation with mAbs to HPV-16 E7 (D6, kindly provided by R. Tindle, Queensland University, Brisbane, Australia), and GST (GST-2; Sigma) or adjusted to 50 mM NaCl and 0.1% Nonidet P-40 and immunoprecipated with a mAb to human M2-PK (DF4, ScheBo Tech, Wettenberg, Germany). Immunoprecipitates then were analyzed by Western blotting, using antibodies to HPV-16 E7 (Ciba Corning, Alameda, CA) or M2-PK (DF4, ScheBo Tech).
Gel Filtration Analysis of M2-PK Activity.
Cells (14/2) were synchronized by removal and readdition of dexamethasone as described (26). Whole-cell extracts were prepared from mock-treated 14/2 cells or cells treated with dexamethasone for 4 hr. Extracts from 2.4 × 107 cells were extracted in 3 ml of lysis buffer (100 mM NaPi, pH 7.4/1 mM DTT/1 mM NaF/1 mM β-mercaptoethanol/1 mM ɛ-aminocaprone acid/0.2 mM phenylmethylsulfonyl fluoride/10% glycerol), lysed in a dounce tissue grinder, and centrifuged (20 min at 20,000 g) to remove cell debris. Extracts were passed over a gel filtration column, and the activities of various glycolytic enzymes were analyzed as described (12).
Isoelectric Focusing Analysis of M2-PK Complexes.
Extracts were prepared from E7-expressing or control 14/2 cells, as described above, by using a lysis buffer containing 10 mM Tris (pH 7.4), 1 mM NaF, 1 mM EDTA-Na2, and 1 mM mercaptoethanol. Homogenates were subjected to isoelectric focusing, and enzyme activities were determined in the individual fractions as described (27).
Modulation of M2-PK Activity in Vitro.
Fractions derived from uninduced 14/2 cell extracts were incubated for 1 hr at 37°C with 90 ng/μl of GST or 90 ng/μl of GST-HPV-16E7 protein, followed by a determination of catalytic activity. Analysis of the data in a Lineweaver–Burk plot was used to determine the Km values. To determine the influence of recombinant E7 on M2-PK quarternary structure in vitro, extracts of M/1 cells were incubated with recombinant GST or GST-16E7 (90 ng/μl) protein, followed by gel filtration experiments similar to those described above.
Determination of Metabolite Concentrations.
The concentrations of carbohydrates in cellular supernatants (8, 12), intracellular concentrations of metabolites (8, 12), and nucleotides (28) were determined as described and used for the calculation of the relative conversion rates.
RESULTS
Identification of M2-PK as Cellular Target for HPV-16 E7.
To identify cellular targets for the carboxyl-terminal domain of the HPV-16 E7 oncoprotein, we used the two-hybrid system in yeast. The C-terminal part of HPV-16 E7 was fused in-frame to the DNA-binding domain of the bacterial LexA-repressor, to yield LexA-16E7(39–98) (18), which can bind to the LexA-binding sites of a synthetic LEU 2 reporter gene and thereby allows us to monitor the interaction of E7 with a second hybrid protein that contains the B42 transactivation domain fused in-frame to a heterologous cDNA (Fig. 1A). When a galactose-inducible human cDNA library (Fig. 1A) (19) was coexpressed with LexA-16E7(39–98) in yeast strain EGY48/pSH1834 and screened for transformants that grow on leucine-deficient media, the cDNA encoding the glycolytic enzyme M2-PK (Fig. 1B) was repeatedly isolated. Coexpression of the M2-PK-B42 fusion protein with pLexA-16 E7(39–98) rendered yeast strain EGY48/pSH1834 able to grow on galactose but not glucose minimal plates, indicating that leucine prototrophy depends on expression of the M2-PK-B42 fusion protein. The inability of two unrelated fusion proteins, LexA-Myc(C-term) and LexA-Bicoid to bind to B42-M2-PK in this assay (Fig. 1C) indicates that the E7 sequences are essential for binding to M2-PK. When lacZ was used as a reporter gene (Fig. 2A), high β-galactosidase activity was observed when B42-M2-PK was coexpressed with LexA-16 E7(1–98) but not with the unrelated fusion proteins LexA-Bicoid and LexA-Myc(C-term) (Fig. 2B), confirming the specific in vivo interaction between E7 and M2-PK. When yeast extracts were incubated with GST-HPV-16 E7 in vitro, the isolated B42 domain was unable to bind to GST-HPV-16 E7 (Fig. 2C); in contrast, the B42-M2-PK fusion protein was efficiently bound by GST-HPV-16 E7, whereas no binding was observed for GST alone (Fig. 2D). Deletion of amino acids 79–83 within the C terminus of HPV-16 E7, as in the mutant Δ79–83, considerably reduces the transforming potential of E7 (24). We found that this mutant displays reduced affinity for M2-PK (Fig. 2D), suggesting that the ability of E7 to bind M2-PK may play a role in cell transformation. This conclusion is further strengthened by our finding that the E7 protein of HPV-11, a nononcogenic HPV subtype (13), binds only weakly to M2-PK, in both the two-hybrid assay (Fig. 2B) and the GST pull-down experiment (Fig. 2D).
Figure 1.

Identification of M2-PK as E7-binding protein. (A) Plasmid pLexA-16 E7(39–98)∷HIS3 (18) encodes the DNA binding domain of LexA fused in-frame to the C-terminal part of HPV-16 E7 and the HIS3 selectable marker. The structure of the expression library, derived from vector pJG4–5 (19) by insertion of cDNA fragments from a WI-38 cDNA library, is indicated. B42-TAD refers to the B42 transactivation domain. B42 fusion proteins are expressed from the inducible GAL1 promotor, and these plasmids contain the TRP1 gene as selectable marker. (B) The plasmid designated pB42-M2-PK∷TRP1 was isolated during the interaction screen; it contains the full-length human cDNA for M2-PK, as described by Tani et al. (43). (C) Derivatives of yeast strain EGY48/pSH1834, expressing various LexA fusion proteins as indicated, were transformed with the plasmid pB42-M2PK∷TRP1. pJG4–5, expressing the unfused B42 trans-activation domain, was used as negative control. Transformants were selected for uracil, histidine, and tryptophane prototrophy and grown in glucose minimal medium (Ura−, His−, Trp−). Yeast cells then were streaked out onto each of three plates and incubated for 4 days at 30°C under the following nutrient conditions (19). Control: glucose minimal medium with leucine; all strains grow. Glucose: glucose minimal medium without leucine; selection for B42 fusion protein independent activation of the LexAo6-LEU2 reporter. Galactose: galactose minimal medium without leucine, selecting for B42 fusion protein dependent activation of the LexAo6-LEU2 gene.
Figure 2.

E7 domains required for M2-PK binding. (A) Structure of the E7 proteins. Conserved domains 1 and 2 (cd1 and cd2) and the putative zinc finger motifs (CXXC) of the E7 proteins of HPV-11 and HPV-16 (44) are indicated. (B) Saccharomyces cerevisiae strain EGY48/pSH1834, containing a LexA operator-lacZ gene (LexAo8-Gal1-LacZ∷URA3), was transformed with the plasmid pB42-M2PK∷TRP1. Plasmids encoding LexA fusion proteins were coexpressed, as indicated. β-galactosidase activity was measured in cellular extracts. LexA-Bicoid and LexA-C-Myc(C-term) were used as negative controls. Extracts from yeast strains expressing the B42-HA1 (C) or B42-HA1-M2PK (D) fusion proteins were incubated with purified GST or GST-E7 fusion proteins and immobilized on glutathione Sepharose 4B beads. Bound proteins were separated on a 12.5% SDS-polyacrylamide gel and analyzed by Western blotting using anti-HA1 antibodies. Lysate refers to 5 μg of yeast whole-cell extract that was loaded as input control.
Physical and Functional Interaction of E7 with M2-PK in E7-Expressing Cells.
To address a potential interaction between E7 and M2-PK in E7-expressing mammalian cells, immunoprecipitation experiments were performed. As shown in Fig. 3, M2-PK was specifically coprecipitated with E7 in extracts from E7-expressing NIH 3T3 cells (subclone E7/2; ref. 23) but not control NIH 3T3 (M/1) cells (Fig. 3 A-C). Antibodies to M2-PK precipitated similar amounts of M2-PK from extracts of both E7/2 and M/1 cells (Fig. 3D), whereas coprecipitation of E7 was observed only in E7/2 extracts (Fig. 3E). These data strongly suggest that M2-PK can form a complex with the E7 oncoprotein in mammalian cells. To determine effects of E7 on the quarternary structure of M2-PK, extracts from E7/2 and M/1 cells were analyzed by gel filtration, followed by a determination of M2-PK activity in the individual fractions. We found that a substantial part of tetrameric M2-PK is shifted to the dimeric form in E7-expressing cells (Table 1), suggesting that E7 alters the equilibrium between tetrameric M2-PK and the dimeric form in favor of the dimer. In keeping with the current model (9, 29), the shift of M2-PK to the low-affinity form was accompanied by a substantial increase of the intracellular FBP levels (Table 1). We also analyzed the status of M2-PK in a cell line expressing the Δ79–83 mutant of E7, which is deficient for M2-PK binding (see above, Fig. 2). The protein encoded by this E7 mutant is stable in mammalian cells (24) and well expressed in the NIH 3T3 cell line derived by us (Fig. 3F). Expression of Δ79–83 did not significantly affect the activity of M2-PK in cell extracts or the intracellular levels of FBP (Table 1), indicating that the ability of E7 to bind M2-PK is required for modulation of M2-PK activity. The relatively large variation of FBP concentrations observed in this experiment is probably the result of subtle oscillations of the FBP levels, which appear to be an intrinsic property of all cells (9).
Figure 3.

Interaction of M2-PK and HPV-16 E7 in mammalian cells. (A-E) Extracts were prepared from E7/2 and M/1 cells and subjected to immunoprecipitation by mAbs to HPV-16 E7, human M2-PK, and GST (control). Total cell lysate (60 μg) was loaded as input control. (A and B) Immunoprecipitation with monoclonal anti-HPV-16 E7 antibodies; Western blot was performed with anti-M2-PK antibodies (A) and antibodies to HPV-16 E7 (B). (C) Immunoprecipitation with monoclonal control antibodies; Western blot was performed with monoclonal anti-M2-PK antibodies. (D and E) Immunoprecipitation with monoclonal anti-M2-PK antibodies; Western blot was performed with anti-M2-PK (D) and anti-HPV-16 E7 (E) antibodies. (F) Western blot analysis of the expression of the Δ79–83 mutant of HPV-16 E7 in NIH 3T3 subclone Δ79–83. IgGh, immunoglobulin G heavy chain; IgGl, immunoglobulin G light chain.
Table 1.
FBP levels and the quarternary structure of M2-PK in E7-expressing NIH3T3 cells
| Cell type | FBP, nmole/ mg protein | Tetramer, % | Dimer, % |
|---|---|---|---|
| M/1 | 0.22 ± 0.04 | 63.5 ± 0.3 | 36.5 ± 0.6 |
| Δ79-83 | 0.40 ± 0.10n.s. | 59.9 ± 1.6n.s. | 40.1 ± 1.6n.s. |
| E7/2 | 0.40 ± 0.04 | 47.2 ± 0.9 | 52.8 ± 0.8 |
| (P < 0.05) | (P < 0.001) | (P < 0.001) |
Control and E7-expressing NIH3T3 subclones were used for gel
filtration analysis of M2-PK as indicated; the results are given as the
relative amounts of the tetrameric and dimeric forms of M2-PK
(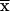 ± SEM, n = 4). FBP levels were determined
in perchloric acid extracts.
± SEM, n = 4). FBP levels were determined
in perchloric acid extracts. 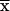 ± SEM, n = 5.
n.s., not significant.
± SEM, n = 5.
n.s., not significant.
Kinetic Studies on E7-Induced Alterations of M2-PK Quarternary Structure.
To address the mechanism by which E7 expression can
modulate the function of M2-PK, we made use of 14/2 cells, a cell
line derived from normal rat kidney cells by constitutive expression of
an activated ras gene together with glucocorticoid-inducible
expression of HPV-16 E7 (25). We found that expression of M2-PK is
up-regulated in 14/2 cells as compared with normal rat kidney cells,
whereas expression of E7, induced by dexamethasone treatment for 4 hr,
did not affect the expression level of M2-PK (Fig.
4A); apparently, the
observed up-regulation of M2-PK depends on the ras oncogene,
as was suggested by others (30). As in the case of E7/2 cells, M2-PK
could be coprecipitated from 14/2 cells with anti-E7 antibodies,
indicating that both proteins also interact in this cell line (data not
shown). To determine the influence of E7 on the quarternary structure
of the enzyme, extracts were subjected to gel filtration, followed by a
determination of M2-PK activity in the individual fractions. Whereas in
mock-treated 14/2 cells the bulk of M2-PK activity was found in
fractions corresponding to the tetrameric form of the enzyme, the peak
of M2-PK activity was shifted to the dimeric form on short-term
expression of E7 (Fig. 4B). Accordingly, a significant
amount of the M2-PK protein was relocated from fractions 19–23 to
fractions 31–34 in E7-expressing cells, as determined by direct
immunoblotting (Fig. 4B). The relative amount of the
tetramer was 51% ± 9% in control 14/2 cells and 29% ± 1% in
E7-expressing 14/2 cells (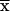 ± SEM,
n = 5, P < 0.05). In control
experiments, we found that dexamethasone had no effect on the
equilibrium between the tetrameric and dimeric forms of M2PK in normal
rat kidney cells (data not shown). Similarly, activation of E7 gene
expression in 14/2 cells did not affect the quarternary structure or
catalytic activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH),
enolase, or lactate dehydrogenase, assayed as controls (Fig.
4B and data not shown). These results suggest that
expression of HPV-16 E7 triggers a specific change in the quarternary
structure of M2-PK.
± SEM,
n = 5, P < 0.05). In control
experiments, we found that dexamethasone had no effect on the
equilibrium between the tetrameric and dimeric forms of M2PK in normal
rat kidney cells (data not shown). Similarly, activation of E7 gene
expression in 14/2 cells did not affect the quarternary structure or
catalytic activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH),
enolase, or lactate dehydrogenase, assayed as controls (Fig.
4B and data not shown). These results suggest that
expression of HPV-16 E7 triggers a specific change in the quarternary
structure of M2-PK.
Figure 4.

Modulation of M2-PK activity by HPV-16 E7. (A) Overexpression of M2-PK in 14/2 cells. Expression of E7 was induced in 14/2 cells by addition of dexamethasone for 4 hr. In a control experiment, asynchronously growing NRK cells were either mock-treated or treated with dexamethasone for 4 hr. Extracts were prepared from these cells as indicated, and the abundance of M2-PK in the cellular extracts was analyzed by direct immunoblotting. (B Upper) Gel filtration analysis. Extracts from E7-expressing or control 14/2 cells were subjected to gel filtration, followed by determination of M2-PK activity in individual fractions, by using a PEP concentration of 2 mM. The peaks of main activities also are indicated for GAPDH, enolase (EN), and lactate dehydrogenase (LDH), assayed in parallel. (Lower) The abundance of M2-PK in each fraction was determined by direct immunoblotting. (C and D) Isoelectric focusing. Extracts from E7-expressing or control 14/2 cells were subjected to isoelectric focusing, and M2-PK activity was determined in the presence of 2 mM PEP (C) or 0.2 mM PEP (D) as indicated. The peak of main activity also is indicated for GAPDH, assayed in parallel. pI values of selected fractions are given for reference.
To further characterize E7-induced changes of M2-PK complexes, we used another method, isoelectric focusing, which allows us to detect the association of M2-PK with other glycolytic enzymes (27). In this experiment, three peaks of M2-PK activity could be detected in E7-nonexpressing 14/2 cells, when measured at saturating substrate concentration, i.e., 2 mM PEP (Fig. 4C). When measured at a substrate concentration of 0.2 mM PEP (Fig. 4D), only the fractions corresponding to the middle peak (pI = 6.0) showed significant M2-PK activity, indicating that these fractions contain the tetrameric (high affinity) form of M2-PK (Fig. 4D). These findings confirm the conclusion that on expression of E7 the equilibrium of M2-PK is shifted to the dimeric state. This experiment also revealed that expression of E7 considerably reduces the M2-PK activity at low PEP concentrations (0.2 mM; Fig. 4D). The M2-PK present in the smallest peak (leftmost peak in Fig. 4B; pI = 4.5) cofocuses with GAPDH, enolase, and phosphoglycero mutase (Fig. 4C) and therefore represents the multienzyme complex as described (27). Apparently, the major fraction of M2-PK in 14/2 cells is not associated with this complex, and the composition of this complex is not altered on E7 expression.
Modulation of M2-PK Catalytic Activity by E7.
To analyze
whether the activity of M2-PK is altered by E7 in living 14/2 cells,
we determined the intracellular concentrations of the reactants and
products of PK. Expression of E7 resulted in a decrease of the
ATP/ADP ratio and an increase in FBP levels (Table
2), and the ratio of products to reactant
Γ = ([pyruvate]⋅[ATP]):([PEP]⋅[ADP]) decreased from
19.3 to 6.8 on expression of E7, indicating that M2-PK activity is
modulated by E7 in vivo. To investigate whether E7 may
directly influence the activity of M2-PK in vitro, protein
extracts derived from E7 nonexpressing 14/2 cells were incubated with
recombinant HPV-16 E7 protein. We found that addition of GST-16 E7 to
tetrameric M2-PK led to an increase of
Km from 0.25 ± 0.04 mM to
0.46 ± 0.04 mM (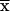 ± SEM; P
< 0.05; n = 3), whereas addition of GST did not induce
significant changes of the Km value.
The data indicate that E7 reduces the PEP affinity of M2-PK in
vitro, probably by changing the equilibrium between the tetrameric
and the dimeric forms of M2-PK. In support of this conclusion, we found
that addition of recombinant E7 protein to extracts from M/1 cells
induced a reproducible shift from the tetrameric to the dimeric forms
of M2PK; in this experiment, the bulk of the recombinant E7
protein was associated with the dimeric form of M2-PK (Fig.
5).
± SEM; P
< 0.05; n = 3), whereas addition of GST did not induce
significant changes of the Km value.
The data indicate that E7 reduces the PEP affinity of M2-PK in
vitro, probably by changing the equilibrium between the tetrameric
and the dimeric forms of M2-PK. In support of this conclusion, we found
that addition of recombinant E7 protein to extracts from M/1 cells
induced a reproducible shift from the tetrameric to the dimeric forms
of M2PK; in this experiment, the bulk of the recombinant E7
protein was associated with the dimeric form of M2-PK (Fig.
5).
Table 2.
E7-dependent alterations of intracellular concentrations of glycolytic metabolites in 14/2 cells
| Metabolites |
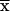 ± SEM
± SEM
|
||
|---|---|---|---|
| E7 off | E7 on | Significance | |
| FBP, nmole/mg protein | 13.4 ± 0.6 | 28.3 ± 5.9 | P < 0.5 |
| PEP, nmole/mg protein | 0.9 ± 0.1 | 1.7 ± 0.2 | P < 0.05 |
| IMP, nmole/mg protein | 0.2 ± 0.4 | 2.0 ± 1.7 | P < 0.05 |
| [ATP] : [ADP] | 5.2 ± 1.4 | 3.5 ± 0.6 | P < 0.05 |
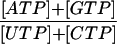 |
4.1 ± 0.9 | 1.9 ± 0.2 | P < 0.001 |
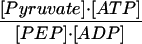 |
19.3 | 6.8 | |
The concentration of various metabolites is given for mock-treated and dexamethasone-treated 14/2 cells. In each case, the significance of dependence on E7 expression was calculated as described (8, 12). n = 5. The term (pyruvate⋅ATP):(PEP⋅ADP) was calculated from the concentrations of the individual metabolites.
Figure 5.

Modulation of M2-PK by E7 in vitro. (Upper) Extracts from M/1 cells were incubated with GST (90 ng/μl) or GST-HPV16E7 (90 ng/μl) for 1 hr at 4°C and subjected to gel filtration. Activity of M2-PK was measured in all fractions in the presence of 2 mM PEP. On addition of GST-HPV16E7, the proportion of tetrameric M2PK was reduced from 63% to 54%, and the proportion of dimeric enzyme increased from 37% to 46%. For calibration of the column, the maximal activities of GAPDH, lactate dehydrogenase (LDH), and enolase (EN) were determined in the individual fractions. (Lower) The distribution of M2-PK and GST-16E7 in the fractions was monitored by direct immunoblotting using antibodies against M2-PK and GST, respectively.
DISCUSSION
The loss of the tissue-specific isoenzyme of PK, i.e., PK type L in liver or type M1 in brain, and the subsequent expression of the M2 isoenzyme is one of the first steps in multistep carcinogenesis (31). At later stages, expression of M2-PK is up-regulated, and subsequently the enzyme is shifted to the dimeric state. In all tumors so far investigated the low-affinity dimeric form of the enzyme, termed tumor M2-PK (Tu M2-PK) (32), is accumulated. Although the reasons for the prevalence of the dimeric form in tumors remain obscure, it has been hypothesized (9) that the expansion of phosphometabolite pools resulting from the down-regulation of M2-PK substrate affinity is required for the increased biosynthetic activity of rapidly proliferating tumor cells. How are changes of M2-PK activity brought about in E7-transformed cells? The results reported here suggest that the E7 protein physically interacts with and stabilizes the dimeric form of M2-PK. This observation implies that part of the E7 protein is localized in the cytoplasmic compartment, consistent with previous studies applying biochemical fractionation and indirect immunofluorescence analysis (33–35).
The results shown in this report establish the enzymatic activity of M2-PK as in vivo target for E7. Because the binding of E7 seems to favor the accumulation of the dimeric enzyme, E7 may mimic the effect of inhibitory amino acids, e.g., alanine or leucine, on M2-PK activity (29, 36, 37). We found that expression of E7 increases the intracellular concentrations of PEP and FBP, two glycolytic metabolites upstream of M2-PK. Increased levels of FBP are known to favor the reassociation of the dimeric to the tetrameric form. However, in E7-expressing cells a significant part of M2-PK remains in the dimeric form although FBP levels increase more than 2-fold (Table 2), in agreement with the conclusion that E7 acts to stabilize the dimeric form of M2-PK. The glycolytic metabolites between PEP and FBP are essential for biosynthetic processes. Concomitant with the E7-driven increase of these precursor metabolites in 14/2 cells, we also found an E7-dependent decrease in the ratio of ([ATP]+[GTP]):([UTP] + [CTP]) (calculated in Table 2), which is an indicator of altered nucleotide biosynthesis (38).
What might be the significance of the interaction between E7 and M2-PK? Although it was convincingly demonstrated that the interaction of E7 with nuclear proteins, e.g., the members of the retinoblastoma protein family, is essential for its transforming potential (reviewed in ref. 39), our current observation that both HPV-11 E7 and the transformation-deficient mutant Δ79–83 (24) of HPV-16 E7 fail to bind M2-PK with high affinity (Fig. 2) raises the possibility that the interaction of E7 with M2-PK, a cytoplasmic enzyme, also may contribute to cell transformation. This hypothesis is in agreement with our finding that expression of E7 in 14/2 cells leads to a significant increase of the total glycolytic rate and an increased conversion of glucose into lactate (Table 3), which implies that expression of E7 insures the channeling of glucose carbons to synthetic processes and at the same time reduces the cell’s requirement for oxygen, two important properties of tumor cells (3, 40, 41). Because of the complex regulation of glycolysis it is difficult to prove that all of the alterations found in our model system are directly correlated with the alterations in M2-PK. Although the analysis of all glycolytic enzymes, including their metabolite levels and equilibrium constants (42), would be required to finally prove this cause-effect relation, the present results suggest that the E7-expressing 14/2 cells are an interesting model to further characterize the role of M2-PK and the glycolytic phosphometabolites in cellular growth control.
Table 3.
E7-dependent alterations of nutrient fluxes in 14/2 cells
| Metabolite conversions, nmole/h × 105 cells | E7 off | E7 on | Significance |
|---|---|---|---|
| Glucose consumption | 85 ± 3 | 121 ± 3 | P < 0.001 |
| Lactate production | 149 ± 3 | 219 ± 6 | P < 0.001 |
| Glutamine consumption | 9 ± 4 | 8 ± 4 | n.s. |
| Glutamate production | 6 ± 0.3 | 6 ± 0.5 | n.s. |
The consumption and production of various key nutrients and
metabolites is given for mock-treated and dexamethasone-treated 14/2
cells. In each case, the significance of dependence on E7 expression is
indicated by the P value.  ± SEM,
n = 5. n.s., not significant.
± SEM,
n = 5. n.s., not significant.
Acknowledgments
We thank R. Brent for the WI-38 cDNA library, R. Tindle and J. Braspenning for antibodies to HPV-16 E7, and M. Tommasino for critical reading of the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (S.M.: Ma 1760/1–2; P.J-D.: Ja 427/5–2), the European Union (Biomed 2), and the Land Hessen (S.M.: Hessisches Ministerium für Wissenschaft und Kunst, HSP III).
ABBREVIATIONS
- PK
pyruvate kinase
- M2-PK
type M2 PK
- HPV
human papillomavirus
- FBP
fructose 1,6-bisphosphate
- GST
glutathione S-transferase
- PEP
phosphoenolpyruvate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
References
- 1.Tokiwa G, Tyers M, Volpe T, Futcher B. Nature (London) 1994;371:342–345. doi: 10.1038/371342a0. [DOI] [PubMed] [Google Scholar]
- 2.Belenguer P, Pelloquin L, Oustrin M L, Ducommun B. Biochem Biophys Res Commun. 1997;232:204–208. doi: 10.1006/bbrc.1997.6253. [DOI] [PubMed] [Google Scholar]
- 3.Mazurek S, Boschek B, Eigenbrodt E. J Bioenerg Biomembr. 1997;29:315–330. doi: 10.1023/a:1022490512705. [DOI] [PubMed] [Google Scholar]
- 4.Tian W N, Braunstein L D, Pang J, Stuhlmeier K M, Xi Q C, Tian X, Stanton R C. J Biol Chem. 1998;273:10609–10617. doi: 10.1074/jbc.273.17.10609. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan O, Navon G, Lyon R C, Faustino P J, Straka E J, Cohen J S. Cancer Res. 1990;50:544–551. [PubMed] [Google Scholar]
- 6.Mathupala S, Rempel A, Pedersen P L. J Bioenerg Biomembr. 1997;29:339–343. doi: 10.1023/a:1022494613613. [DOI] [PubMed] [Google Scholar]
- 7.Board M, Humm S, Newsholme E A. Biochem J. 1990;265:503–509. doi: 10.1042/bj2650503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazurek S, Michel A, Eigenbrodt E. J Biol Chem. 1997;272:4941–4952. doi: 10.1074/jbc.272.8.4941. [DOI] [PubMed] [Google Scholar]
- 9.Eigenbrodt E, Reinacher M, Scheefers-Borchel U, Scheefers H, Friis R. Crit Rev Oncog. 1992;3:91–115. [PubMed] [Google Scholar]
- 10.Netzker R, Hermfisse U, Wein K H, Brand K. Biochim Biophys Acta. 1994;1224:371–376. doi: 10.1016/0167-4889(94)90270-4. [DOI] [PubMed] [Google Scholar]
- 11.Ashizawa K, Willingham M C, Liang C M, Cheng S Y. J Biol Chem. 1991;266:16842–16846. [PubMed] [Google Scholar]
- 12.Hugo F, Mazurek S, Zander U, Eigenbrodt E. J Cell Physiol. 1992;153:539–549. doi: 10.1002/jcp.1041530315. [DOI] [PubMed] [Google Scholar]
- 13.zur Hausen H. Virology. 1991;184:9–13. doi: 10.1016/0042-6822(91)90816-t. [DOI] [PubMed] [Google Scholar]
- 14.Münger K, Phelps W C, Bubb V, Howley P M, Schlegel R. J Virol. 1989;63:4417–4421. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phelps W C, Yee C L, Münger K, Howley P M. Cell. 1988;53:539–547. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- 16.Jewers R, Hildebrandt P, Ludlow J, Kell B, McCance D. J Virol. 1992;66:1329–1335. doi: 10.1128/jvi.66.3.1329-1335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jansen-Dürr P. Trends Genet. 1996;12:270–275. doi: 10.1016/0168-9525(96)81455-7. [DOI] [PubMed] [Google Scholar]
- 18.Zwerschke W, Joswig S, Jansen-Dürr P. Oncogene. 1996;12:213–220. [PubMed] [Google Scholar]
- 19.Gyuris J, Golemis E, Chertkov H, Brent R. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- 20.Gietz D, St. John A, Woods R A, Schiestl R H. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ausubel F M, Kingston R, Moore D, Seidman J, Smith J, Struhl K, editors. Current Protocols in Molecular Biology. New York: Wiley; 1988. [Google Scholar]
- 22.Zerfass-Thome K, Zwerschke W, Mannhardt B, Tindle R, Botz J, Jansen-Dürr P. Oncogene. 1996;13:2323–2330. [PubMed] [Google Scholar]
- 23.Davies R, Hicks R, Crook T, Morris J, Vousden K. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massimi P, Pim D, Banks L. J Gen Virol. 1997;78:2607–2613. doi: 10.1099/0022-1317-78-10-2607. [DOI] [PubMed] [Google Scholar]
- 25.Crook T, Morgenstern J P, Crawford L, Banks L. EMBO J. 1989;8:513–519. doi: 10.1002/j.1460-2075.1989.tb03405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zerfass K, Schulze A, Spitkovsky D, Friedman V, Henglein B, Jansen-Dürr P. J Virol. 1995;69:6389–6399. doi: 10.1128/jvi.69.10.6389-6399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazurek S, Hugo F, Failing K, Eigenbrodt E. J Cell Physiol. 1996;167:238–250. doi: 10.1002/(SICI)1097-4652(199605)167:2<238::AID-JCP7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 28.Wice B M, Trugnan G, Pinto M, Rousset M, Chevalier G, Dussaulx E, Lacroix B, Zweibaum A. J Biol Chem. 1985;260:139–146. [PubMed] [Google Scholar]
- 29.Oude Weernink P A, Rijksen G, Mascini E M, Staal G E. Biochim Biophys Acta. 1992;1121:61–68. doi: 10.1016/0167-4838(92)90337-d. [DOI] [PubMed] [Google Scholar]
- 30.Gregory S H, Kumari H L, Lakshmi M V, Bose S K. Arch Biochem Biophys. 1976;175:644–653. doi: 10.1016/0003-9861(76)90555-5. [DOI] [PubMed] [Google Scholar]
- 31.Hacker H J, Steinberg P, Bannasch P. Carcinogenesis. 1998;19:99–107. doi: 10.1093/carcin/19.1.99. [DOI] [PubMed] [Google Scholar]
- 32.Eigenbrodt E, Basenau D, Holthusen S, Mazurek S, Fischer G. Anticancer Res. 1997;17:3153–3156. [PubMed] [Google Scholar]
- 33.Smotkin D, Wettstein F O. J Virol. 1987;61:1686–1689. doi: 10.1128/jvi.61.5.1686-1689.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato H, Watanabe S, Furuno A, Yoshiike K. Virology. 1989;170:311–315. doi: 10.1016/0042-6822(89)90386-3. [DOI] [PubMed] [Google Scholar]
- 35.Kanda T, Zanma S, Watanabe S, Furuno A, Yoshiike K. Virology. 1991;182:723–731. doi: 10.1016/0042-6822(91)90613-g. [DOI] [PubMed] [Google Scholar]
- 36.Oude Weernink P, Rijksen G, Staal G E. Tumor Biol. 1991;12:339–352. doi: 10.1159/000217735. [DOI] [PubMed] [Google Scholar]
- 37.Eigenbrodt E, Gerbracht U, Mazurek S, Presek P, Friis R. Biochem Mol Aspects Selected Cancers. 1994;2:311–385. [Google Scholar]
- 38.Ryll T, Wagner R. Biotechnol Bioeng. 1992;40:934–946. doi: 10.1002/bit.260400810. [DOI] [PubMed] [Google Scholar]
- 39.Alani R M, Munger K. J Clin Oncol. 1998;16:330–337. doi: 10.1200/JCO.1998.16.1.330. [DOI] [PubMed] [Google Scholar]
- 40.Kim C Y, Tsai M H, Osmanian C, Graeber T G, Lee J E, Giffard R G, DiPaolo J A, Peehl D M, Giaccia A J. Cancer Res. 1997;57:4200–4204. [PubMed] [Google Scholar]
- 41.Brand K A, Hermfisse U. FASEB J. 1997;11:388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- 42.Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell D A, Veech R L, Passonneau J V. J Biol Chem. 1994;269:25502–25514. [PubMed] [Google Scholar]
- 43.Tani K, Yoshida M C, Satoh H, Mitamura K, Noguchi T, Tanaka T, Fujii H, Miwa S. Gene. 1988;73:509–516. doi: 10.1016/0378-1119(88)90515-x. [DOI] [PubMed] [Google Scholar]
- 44.Ullman C G, Haris P I, Galloway D A, Emery V C, Perkins S J. Biochem J. 1996;319:229–239. doi: 10.1042/bj3190229. [DOI] [PMC free article] [PubMed] [Google Scholar]