

Short abstract
MatchMiner is a freely available program package for batch navigation among gene and gene product identifier types commonly encountered in microarray studies and other forms of 'omic' research.
Abstract
MatchMiner is a freely available program package for batch navigation among gene and gene product identifier types commonly encountered in microarray studies and other forms of 'omic' research. The user inputs a list of gene identifiers and then uses the Merge function to find the overlap with a second list of identifiers of either the same or a different type or uses the LookUp function to find corresponding identifiers.
Rationale
One of the more painful tasks in 'omic' research [1,2] is navigating among different gene or gene product identifiers. After a cDNA microarray experiment, for example, one usually must translate from IMAGE clone ids to GenBank accession numbers, HUGO names, common names, or chromosome locations for a list of genes. As we generate more and more data from diverse platforms and species, such translations will become increasingly complex but also more important to the synthesis of a coherent biological picture. Beyond simply looking up additional information about a list of genes, such synthesis will require the ability to find the intersection between two lists of genes that are designated by the same or a different identifier type.
Currently, the basic translations can be done on a gene-by-gene basis using public databases such as UniGene, LocusLink, OMIM (Online Mendelian Inheritance in Man), and the working draft of the human genome (from the University of California Santa Cruz (UCSC) or the National Center for Biotechnology Information) [3-5] or else in batch through Source [6] or GeneLynx [7]. However, no single data source contains all the necessary information about every gene and, to complicate matters further, the relationships among identifiers are often not one-to-one. For example, there may be several GenBank accession numbers and multiple IMAGE clone ids for the same gene, and a single gene symbol may be an alias for multiple different genes. Therefore, any high-throughput solution to the problem must take these challenges into account and respond with an approach that minimizes the need for human intervention. At the same time, those instances when human intervention is necessary must be flagged and enough metadata must be provided for accurate decision-making without extensive further research.
Motivated by many days spent at the computer doing these tedious, time-consuming translations for our own experimental data, we developed MatchMiner [8] as a freely available public resource that automates the process for collections of genes. MatchMiner provides two primary functions. The first, LookUp, translates an input list of gene identifiers into a matching output list of identifiers of a different type; the second, Merge, combines two separate lists of either the same or different types of identifiers into one list that details all one-to-one, one-to-many, and many-to-many relationships between corresponding gene identifiers in the two lists.
Identifier navigation with MatchMiner
As shown schematically in Figures 1 and 2, MatchMiner leverages information from the four public databases listed above, and from Affymetrix, by parsing them into relational tables for use in doing translations. The LookUp function can operate interactively on single identifiers or in batch mode on a list of identifiers in a file. When used interactively for one or a few genes, it saves the user the trouble of querying five different databases and collating the data. More important, however, is batch querying of a list file, for instance a list of the dozens or hundreds or thousands of genes that show interesting differences between samples in a microarray experiment. In this mode, the user specifies the input and output identifier types, as well as the search algorithms to be used in traversing the various data sources (Table 1). The program is context-sensitive in that it will search only the pertinent data sources (for example, only UniGene to identify IMAGE clone ids, which are not found in the other sources). An important feature is the optional output of diagnostic metadata that tell the user in which source (s) the identifier was found and whether an input identifier corresponds to more than one gene. This feature enables the user to judge the reliability of matches. The results can be displayed in HTML format or downloaded as tab-delimited text suitable for direct entry into a spreadsheet program. A summary indicates the number of successful and unsuccessful translations.
Figure 1.

Information Flow in MatchMiner. Input identifier lists are first translated into unique internal gene indices to form a translation table. The translation table is then either converted into another set of identifiers using the LookUp function or compared with another such table using the Merge function to generate a report showing the intersection of two separate identifier lists. The resulting output can be displayed as HTML or else saved as text for import into other programs.
Figure 2.
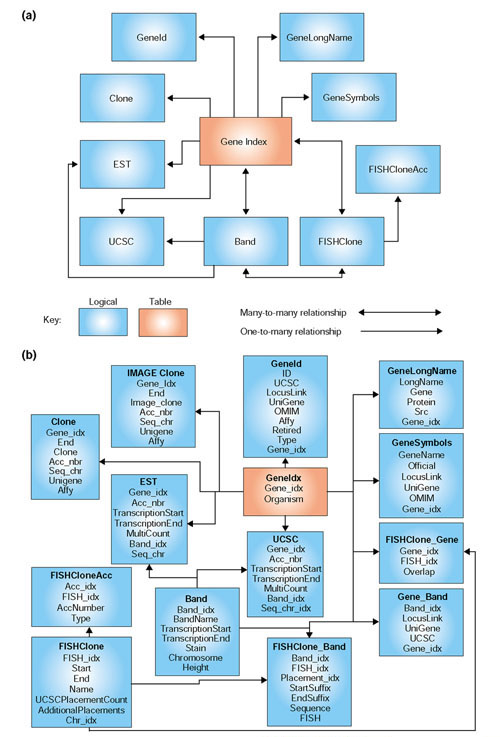
Database relational table schema for MatchMiner. (a) Logical database representation. Data are incorporated from the UCSC Human Genome Build, LocusLink, UniGene, OMIM, and the Affymetrix annotation sets for HU95 and HU133 chips. Each candidate gene is assigned a gene index in the GeneIdx table. These gene indexes are used as keys for all of the MatchMiner operations. The number of many-to-many relationships in the model illustrates the complexity of the data. (b) Physical representation of the database. The implementation currently includes 14 tables with about 12 million rows.
Table 1.
MatchMiner LookUp search options
| Identifier type | Input algorithm | Output algorithm |
| Name (Gene symbol, alias or descriptive name) | HUGO then Alias | HUGO then Alias |
| Starts with Official. If not found, proceeds through all other sources. | Returns the HUGO name. If no name is flagged as HUGO, returns all aliases. | |
| ALL (HUGO and Alias) | ALL (HUGO and Alias) | |
| Searches all data sources for all matches to the symbol and flags those that are HUGO. | Returns all gene symbols and flags the HUGO symbol. | |
| Official | Official | |
| Searches all data sources for match as the HUGO name. | Returns the HUGO name. If not found, nothing is returned. | |
| Long | Long | |
| Searches all data sources for descriptive name. | Returns all descriptive names. | |
| GenBank accession number | ALL | ALL |
| Searches all data sources starting with UCSC known genes, then LocusLink, UniGene and UCSC ESTs until match found. | Returns accession number from UCSC known genes. If not found, proceeds through UniGene then UCSC EST. | |
| Data-source specific | Data-source specific | |
| Look up input in a specific data source. | Returns accession numbers found in a particular data source. | |
| IMAGE clone | UniGene | UniGene |
| Only data source with IMAGE clone ids. | Returns all IMAGE clone IDs associated with the UniGene | |
| Cytogenetic location | ALL | ALL |
| Searches all gene indexes for matching chromosome band. | Returns chromosome band from UCSC sequence to band translation. If not found, proceeds through all other sources with multiple bands listed separately. | |
| UCSC | ||
| Returns chromosome band from UCSC sequence to band translation. | ||
| Database id | UniGene | UniGene |
| Searches gene index for matching UniGene id. | Returns UniGene id. | |
| Affymetrix | Affymetrix | |
| Searches gene index for matching Affymetrix probe set identifier. | Returns Affymetrix probe set identifier. | |
| Sequence location number (bp) | Not implemented | Transcription Start |
| Returns transcription start from UCSC Known Genes. If not found, proceeds to UCSC EST. | ||
| FISH clone | UCSC | UCSC |
| Searches UCSC FISH clones for match to gene index based on sequence position overlap with UCSC known genes. | Returns FISH clone id. |
The Merge function, the most powerful function of MatchMiner, identifies which genes are common to two input lists of identifiers and gives detailed output of the one-to-one, one-to-many, or many-to-many relationships between corresponding identifiers in the two lists. This function is used, for example, to compare datasets of different experiment types (for example, transcript expression, protein expression, array-based comparative genomic hybridization (CGH)) by identifying the genes in common between them. The output includes summary tallies as well as a gene-by-gene listing of items matched, unmatched and not found. As with the LookUp function, diagnostic resource information is provided. Any identifier with an ambiguous gene assignment (for example, an IMAGE clone id that belongs to two different UniGene clusters) is flagged for user intervention, with all possible assignments returned.
Performance
In one illustrative case that motivated development of MatchMiner, we (X. Lee, K.J.B., F.G. Gwadry, W.C.R., G. Riddick, S. L. Pelletier, S.N., and J. N.W., unpublished data) had to match up as many as possible of 9,706 cDNA microarray clones [9,10] with HU6,800 Affymetrix chip oligonucleotide sets [11], having run both platforms on the same 60 human cancer cell samples (the NCI-60). To do so, we developed an early form of MatchMiner. The particular task was to identify all relationships between the 9,706 IMAGE clone ids and 7,129 GenBank accession numbers based on UniGene cluster membership. To complete the task manually, one gene at a time at maximum speed (about 30 seconds per gene) would take over 140 hours - even if one could keep accurate track of the results. In contrast, the current version of MatchMiner took 10 minutes on a 750 MHz Pentium III PC with 320 MB RAM to generate the merged list, specifying all possible matches between IMAGE clone ids and GenBank accession numbers. When we compared MatchMiner Merge results with those obtained using the LookUp function for a random sample of the genes, there were no discrepancies. The same task with Source required translating both lists into UniGene clusters and then further processing the data. After identification and reformatting of entries with multiple UniGene cluster associations, the resulting lists were imported into Microsoft Access and queried to create the appropriate matches. The entire procedure gave results similar to those of MatchMiner but took approximately one hour, most of that user time.
With the exception of MatchMiner, tools that can do some kind of translation are geared toward research dealing with expressed sequence, either at the RNA or protein level. However, many interesting questions can be asked from the perspective of genomic sequence. One example relates to the identification of genes represented in an array CGH experiment in which the targets on the chip are fluorescent in situ hybridization (FISH)- and site-tagged sequence (STS)-mapped bacterial artificial chromosome (BAC) clones. The challenge is to begin to interpret array CGH results in the context of the biological literature and of other classes of data. BAC clones are not generally annotated by the genes they span, but rather by their position in the cytogenetic and sequence-based maps. Therefore, an association between the BAC clones and genes must be made. MatchMiner provides this function with the ability to search on the FISH clone ids. Mapping of the FISH clones to genes is done by sequence alignment of the BAC ends during off-line construction of the overall MatchMiner database (Figure 3). MatchMiner takes 5 minutes to return the gene symbol for a list of 100 FISH-mapped BACs [12]. Such a search is not possible using other tools. A summary of commonly used analogous tools and their capabilities can be found in Table 2.
Figure 3.

Associating FISH-mapped BACs with genes. Schematic view of FISH-mapped BACs from 1p36.33 near the PITSLRE kinase genes (UCSC Genome Browser, June 2002 freeze). Note that a single BAC can encompass one or more genes. In MatchMiner, the FISH-mapped BAC table from UCSC is imported, and chromosomal positions are read from the table for comparison with the transcriptional start positions of UCSC 'Known Genes'. If a transcriptional start is contained within the bounds of a BAC, that BAC is associated with the corresponding gene index. Thus, a BAC containing several genes will be associated with each of those genes.
Table 2.
Comparison of the capabilities of gene identifier translation tools
| Program | Implementation | Search Types | Batch | Translation path traceable in interactive (single-gene) mode? | Translation path traceable in batch(gene-list) mode? | Multiple input associations flagged? | Output in form suitable for automated processing? |
| MatchMiner | Command line, Web application | LookUp, Merge | Yes | Yes | Yes | Yes | Yes |
| Source | Web application | LookUp | Yes | Yes | No | Yes, if "Show all Cluster Ids if Multiple Clusters" option selected | Yes |
| Genelynx | Web application | LookUp | Yes | Yes | No | Yes | No |
As noted previously, identifiers are not always unique or uniquely assigned. For example, GenBank accession numbers are specific to a sequence, but the assignment of that sequence to a gene may change over time. Even more disconcerting, common gene names or aliases are often used by different investigators for different genes. Therefore, it is important to look in detail at the results of searches to check for correspondences other than one-to-one and to examine the data source tags to get a sense of the strength of the association between identifiers.
One non-obvious advantage of MatchMiner is that it can combine information from more than one of the data sources to show matches that could not be made on the basis of any single source. The gene ACVR2B, which has aliases ACTR-IIB and ACTRIIB, provides an example. LocusLink and OMIM both reference the HUGO symbol ACVR2B, but LocusLink does not reference ACTRIIB, and OMIM does not reference ACTR-IIB. Therefore, if one of the aliases were used as input, the success of any search outside of MatchMiner would be data-source dependent.
Algorithm and software development
MatchMiner was written in Java and can be deployed as either a web or command-line application, the latter suitable for high-throughput pipeline purposes. In its design and implementation, we leveraged a variety of open-source tools and libraries, including jUnit (unit testing framework), CVS (configuration management), Xerces (XML parser) and Ant (build tool). Before run-time, data from UniGene, LocusLink, OMIM, UCSC and Affymetrix are downloaded and parsed to generate an integrated database implemented under MySQL. If an entry from the imported data matches a candidate gene that was already identified, it is assigned the same gene index. If an entry does not match any of the candidate genes, then a new gene index is generated. Import begins with data from the UCSC's 'Known Gene' table, followed by UCSC's EST (expressed sequence tag) table, LocusLink, UniGene, OMIM and Affymetrix. Different identifiers are stored in different tables and several tables are required to resolve the many-to-many relationships between identifiers (Figure 2a,b). The central algorithm for resolving identifiers uses an instantiation of the ChainOfResponsibility pattern [13], which combines different searches sequentially in a logical manner. In MatchMiner, it maximizes the likelihood of correctly translating back and forth from identifiers to gene indices using the different databases. The algorithm is non-trivial. For each identifier type, we establish a ChainOfResponsibility hierarchy of the data sources based on their respective abilities to match the user input (Table 3). The search algorithms then use this ranking. For example, when an input list of gene names is processed using the 'ALL (HUGO, Alias)' search algorithm, the list is scanned for HUGO names, and each one found is associated with the corresponding unique internal gene index. The remaining unmatched gene names are then scanned again, this time matching aliases (Table 1). The rationale is that an official HUGO name is more likely to be the desired match, but any match is better than none. A similar approach is used when going from the unique index to an output list. For instance, if the desired output is cytogenetic location, MatchMiner first scans the UCSC build of the human genome. If the location is not found there, LocusLink and Unigene are searched (Table 1). The ChainOfResponsibility approach enables us to combine the precision of highly focused algorithms with the greater coverage of more broadly based ones.
Table 3.
ChainOfResponsibility hierarchies for data sources in MatchMiner
| Identifier type | Hierarchy of source reliability |
| Cytogenetic location | UCSC Known Genes, LocusLink, UniGene, UCSC EST, OMIM |
| GenBank accession number | UCSC Known Genes, LocusLink, UniGene, UCSC EST, OMIM |
| HUGO gene symbol | LocusLink, OMIM |
| IMAGE clone id | UniGene |
| Long gene name | UCSC Known Genes, LocusLink, UniGene, UCSC EST, OMIM |
| Affymetrix probe id | Affymetrix |
| UniGene cluster id | UniGene |
Although currently human-specific, MatchMiner will be expanded in the near future to incorporate data from other species, with emphasis on mouse. Additional features to be implemented include the ability to handle lists of mixed types of identifiers, the ability to request multiple types of identifiers within a single search, and the incorporation of additional public sources for use in making translations. We will continue to enhance and develop MatchMiner under a contract funded by the Center for Cancer Research of the US National Cancer Institute.
Download
MatchMiner is available as a web-application or as a command line jar file at [8]. The MatchMiner database is maintained on our server and updated at approximately 6-month intervals. Detailed documentation for both implementations is available at the site.
In summary, MatchMiner is an efficient application for navigating the complex world of gene and gene product identifiers. It can batch search publicly available databases to convert between identifier types and can determine the intersection of two gene lists with different identifiers. MatchMiner will greatly enhance the ability of the research community to annotate and compare 'omic' datasets.
References
- Weinstein JN. Fishing expeditions. Science. 1998;282:628–629. doi: 10.1126/science.282.5389.627g. [DOI] [PubMed] [Google Scholar]
- Weinstein JN. 'Omic' and hypothesis-driven research in the molecular pharmacology of cancer. Curr Opin Pharmacol. 2002;2:361–365. doi: 10.1016/s1471-4892(02)00185-6. [DOI] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2002;30:17–20. doi: 10.1093/nar/30.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DL, Church DM, Lash AE, Leipe DD, Madden TL, Pontius JU, Schuler GD, Schriml LM, Tatusova TA, Wagner L, et al. Database resources of the National Center for Biotechnology Information: 2002 update. Nucleic Acids Res. 2002;30:13–16. doi: 10.1093/nar/30.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Source http://source.stanford.edu
- GeneLynx: a portal to the human genome http://www.genelynx.org
- MatchMiner http://discover.nci.nih.gov/matchminer
- Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, Kohn KW, Reinhold WC, Myers TG, Andrews DT, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de RM, Waltham M, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- Staunton JE, Slonim DK, Coller HA, Tamayo P, Angelo MJ, Park J, Scherf U, Lee JK, Reinhold WO, Weinstein JN, et al. Chemosensitivity prediction by transcriptional profiling. Proc Natl Acad Sci USA. 2001;98:10787–10792. doi: 10.1073/pnas.191368598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nowak N, Jang W, Kirsch IR, Zhao S, Chen XN, Furey TS, Kim UJ, Kuo WL, Olivier M, et al. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409:953–958. doi: 10.1038/35057192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamma E, Helm R, Johnson R, Vlissides J. Design Patterns. Boston: Addison-Wesley; 1995. [Google Scholar]