

Short abstract
GoMiner, a program package that organizes lists of 'interesting' genes for biological interpretation in the context of the Gene Ontology, has been developed.
Abstract
We have developed GoMiner, a program package that organizes lists of 'interesting' genes (for example, under- and overexpressed genes from a microarray experiment) for biological interpretation in the context of the Gene Ontology. GoMiner provides quantitative and statistical output files and two useful visualizations. The first is a tree-like structure analogous to that in the AmiGO browser and the second is a compact, dynamically interactive 'directed acyclic graph'. Genes displayed in GoMiner are linked to major public bioinformatics resources.
Rationale
Gene-expression profiling and other forms of high-throughput genomic and proteomic studies are revolutionizing biology. That much is universally agreed. But the new technologies pose new challenges. The first is the experiment itself, the second is statistical analysis of results, the third is biological interpretation. That third challenge is often the most vexing and time-consuming. In gene-expression microarray studies, for example, one generally obtains a list of dozens or hundreds of genes that differ in expression between samples and then asks: 'What does all of this mean biologically?' The work of the Gene Ontology (GO) Consortium [1] provides a way to address that question. GO organizes genes into hierarchical categories based on biological process, molecular function and subcellular localization. In the past, this GO information was queried one gene at a time. Recently, batch processing has been introduced [2], but with a flat-format output that does not communicate the richness of GO's hierarchical structure.
We have developed, and present here, the program package GoMiner as a freely available computer resource that fully incorporates the hierarchical structure of the Gene Ontology to automate the functional categorization of gene lists of any length. GoMiner is downloadable free of charge from [3] or [4]. GoMiner was developed particularly for biological interpretation of microarray data; one can input a list of under- and overexpressed genes and a list of all genes on the array, and then calculate enrichment or depletion of categories with genes that have changed expression. GoMiner thus facilitates analysis and organization of the results for rapid interpretation of 'omic' [5,6] data. For concreteness, the descriptions in this article will focus on applications to microarray data, but the range of uses is obviously much broader.
Overview of GoMiner
GoMiner takes as input two lists of genes: the total set on the array and the subset that the user flags as interesting (for example, altered in expression level). GoMiner displays the genes within the framework of the Gene Ontology hierarchy, both as a directed acyclic graph (DAG) and as the equivalent tree structure. The latter is similar in format to the visualization in the AmiGO browser display [1]. However, each category is annotated to reflect the number of genes from the user's experiment assigned to that category plus the number assigned to its progeny categories (Figure 1a). This computation does not double-count genes that appear more than once along the traversal. The user has the option of designating each gene within the 'interesting gene' list as exhibiting under- or overexpression. If that is done, genes displayed in the tree-like view are tagged with green down-arrows or red up-arrows, respectively.
Figure 1.
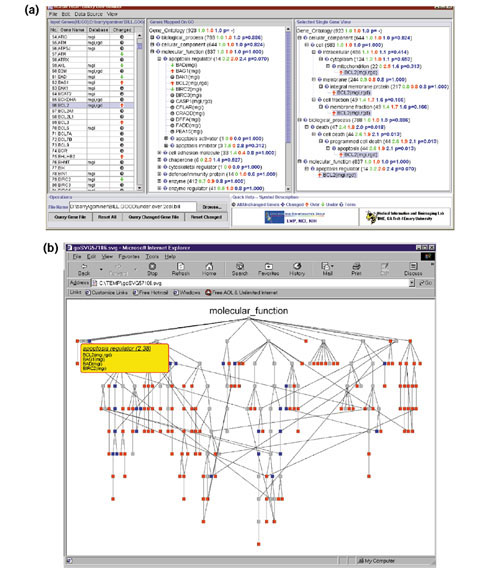
GoMiner displays for microarray gene-expression data on prostate cancer cell line DU145 and a subline (RC0.1) selected for resistance to a topoisomerase 1 inhibitor. (a) Tree-like display showing underexpressed genes (green down-arrows), overexpressed genes (red up-arrows), and unchanged genes (gray circles) in the GO 'Apoptosis Regulator' category and its subcategories. The blue number indicates a 2.4-fold enrichment of changed genes in this category. The p-value (Fisher's exact) indicates that, despite this degree of enrichment, the small total number of genes (14) in this category prevents statistical significance. (b) Dynamically generated SVG graphic of the 'Biological Process' DAG with genes in the GO 'Apoptosis Regulator' category opened in a pull-down list by mousing-over. Categories enriched more than 1.5-fold with flagged genes are color-coded red; those depleted more than 1.5-fold are blue. The rest of the categories are gray.
The most important parameter for purposes of interpretation is the enrichment (or depletion) of a category with respect to flagged genes (relative to what would have been expected by chance alone). This parameter will be discussed more extensively and more mathematically in the section on 'Statistical considerations'. In Figure 1a, the relative enrichment is indicated by blue numbers for total flagged genes and by red and green numbers for over- and underexpressed genes, respectively. The last number (blue) for each category is a two-sided p-value from Fisher's exact test.
In GoMiner, clicking on a gene of interest in the tree-structure opens a menu that can be used to submit that gene as a query to an external data resource. The number of such links is being expanded rapidly, but currently included are LocusLink [7], PubMed [8], MedMiner [9,10], GeneCards [11], the NCBI's Structure Database [12], and BioCarta and KEGG pathway maps as implemented by the NCI Cancer Genome Anatomy Project (CGAP) [13]. These external databases provide GoMiner with a rich set of resources for bioinformatic integration. For example, the links with CGAP and LocusLink provide interaction with pathway maps, chromosome visualizations, a database of single nucleotide polymorphism (SNP), and the Mammalian Gene Collection (MGC).
In GoMiner, clicking on a category instead of a gene brings up a second visualization (Figure 1b), a DAG programmed as a scalable vector graphic (SVG) that can be navigated fluently. Any of its nodes can be moused-over to list the flagged genes or clicked to highlight multiple pathways connecting it to the root. Detailed quantitative and statistical results are downloadable in several tab-delimited formats that can be read directly into a text file or a spreadsheet program for further analysis. For example, the spreadsheet data can be sorted by enrichment factor or p-value to focus attention on potentially interesting categories.
Development of GoMiner
GoMiner is based on a variety of open-source Java classes and developer tools, plus substantial in-house custom software engineering (Figure 2). We chose Java to achieve independence of operating system so that more researchers could use the tool. A custom graphical user interface (GUI) provides the user with flexibility and an intuitive view of biological relationships (Figure 1a). A complementary command-line version of GoMiner allows high-throughput applications and fluent integration with other programs.
Figure 2.
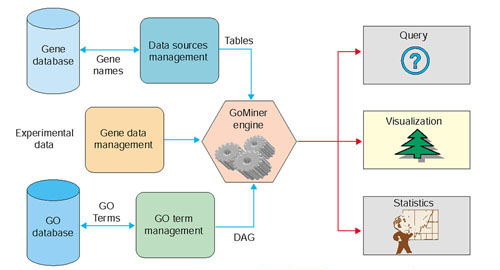
Schematic of GoMiner architecture and data flow.
The heart of GoMiner is its processing engine (Figure 2), which parses input gene lists and retrieves database entries for association with GO categories (also called 'terms'). The GO categories and gene associations are stored in a relational database. To enhance the speed of data manipulation, we model the information in memory using a DAG data structure. The root is the topmost node: 'Gene Ontology'. The other nodes represent gene categories, and the connections represent relationships between categories. Each category-node object contains its associated genes, functionality for counting genes, a flag for dereplication during counting, and results of statistical analyses. The gene-category associations are displayed in the form of a tree (Figure 1a) or, alternatively, in the form of a DAG (Figure 1b).
We have developed GoMiner as a client-server application. The client, a Java application, communicates with a server-side database through JDBC. The client can run on platforms with Java run-time environment version 1.3 or higher. The primary client-user GUI, written using the Java Swing API, takes the form of a three-panel window in which the user can inspect GO categories and genes. The left-hand panel lists the genes, the databases from which their identities were derived, and optional up- and down-arrows to indicate under- or over-expression; the middle panel shows a tree visualization of categories in the style of the AmiGO browser [1] and, in addition, provides a visualization of the flagged genes in the particular microarray experiment. The right-hand panel shows all appearances within the GO hierarchy of any gene selected from the left or middle panel. The gene and category names are implemented as links to facilitate navigation of the data structures and access to public resources.
A second type of visualization, the DAG (programmed as an SVG) shows in compact form the spanning hierarchy for all flagged genes. Optionally, it can include only nodes below a specified level if the entire DAG would be too large for easy visualization. The client application uses several open source components: the Berkeley Drosophila Genome Project (BDGP) Java Toolkit [14] for utility classes; Browser Launcher [15] for cross-platform web browser integration; Jakarta-ORO [16] for text processing; the Jena Semantic Web Toolkit [17] for manipulating RDF models; MySQL Connector/J [18] for database connectivity; and Xerces [19] for parsing XML. The back-end is a relational database server, which stores all gene ontology data. It includes an implementation in MySQL [20] of the GO Consortium database.
In addition to the deployed components, we have introduced a number of open-source tools to enhance the development environment. In particular, the Concurrent Versions System (CVS) tool [21] coordinates program development at the Georgia Institute of Technology with that at the NCI, and also coordinates development within each of the groups. jUnit [22] automates unit- and system-level testing of the application.
Statistical considerations
The two-sided Fisher's exact test p-value for a category reflects a test of the null hypothesis that the category is neither enriched in, nor depleted of, flagged genes with respect to what would have been expected by chance alone. That is, it reflects the null hypothesis that, for each category, there is no difference between the proportion of flagged genes that fall into the category and the proportion of flagged genes that do not fall into the category. The two groups of genes are mutually exclusive, as required for Fisher's exact test. Note that the predicate of the null hypothesis does not include 'the flagged genes that fall into the union of the rest of the categories'. That predicate would not ensure mutual exclusivity. The statistical question can be framed in terms of a classical 2 × 2 contingency table (Table 1).
Table 1.
Two-by-two contingency table for flagged and unflagged genes in a GO category
| Flagged genes | Non-flagged genes | Total | |
| In category | nf | n - nf | n |
| Not in category | Nf - nf | (N - n) - (Nf - nf) | N - n |
| Total | Nf | N - Nf | N |
nf is the number of flagged genes in the category, n is the total number of genes in the category, Nf is the number of flagged genes on the microarray, and N is the total number of genes on the microarray. All numbers are those obtained after dereplicating multiple instances of the same gene.
The null hypothesis can be formulated as:
H0:p1 - p2 = 0,
where p1 = nf/n and p2 = (Nf - nf)/(N - n). The two-sided p-value for Fisher's exact test is the sum of probabilities of observing tables that give at least as many extreme values as the one actually observed, given that the null hypothesis is true [23-25]. The use of Fisher's exact test implies that we are conditioning on fixed marginal totals (n, N - n, Nf, N - Nf) under the null hypothesis. For a discussion of the implications of fixed marginal values, see for example [23-25].
Note that the 2 × 2 table does not require any information about the topology of the hierarchy or about how many genes are included in any category other than the one to which the test is being applied. We used the two-sided version of the test, which detects a significant difference in the proportions in either direction (that is, when the proportion of flagged genes in the category is either higher or lower than would be expected by random chance). Clearly, calculations analogous to the ones used here for all flagged genes can also be applied to test separately the equivalent null hypotheses for under- and overexpressed genes. Unlike the Z-statistic with the hypergeometric distribution, and tests based on it, Fisher's exact test is appropriate even for categories containing a small number of genes. Our Java implementation of the Fisher's exact test is based on Javascript by Øyvind Langsrud [26].
The following limitations of this statistical formulation should be borne in mind, and the p-values should be interpreted judiciously.
Random experimental and categorization error
Experimental error and any uncertainties in the classification of genes in GO are not included in the statistical model. Perhaps, given enough information (which we essentially never have) about those sources of error, they could be included in the statistical model, for example through a resampling technique.
Gene representation bias
The microarray gene set (or set from some other type of genomic or proteomic experiment) will generally be a biased representation of all genes. Therefore, enrichments and depletions, of necessity defined in terms of the genes studied, may be biased with respect to biological significance as well. An alternative is to replace the list of the total set of genes on the microarray with a list of the total set of genes in the genome (or a representative sample), but that approach introduces another source of bias: genes not on the microarray are counted in determining N and n but have no chance to be flagged.
GO consortium database bias for human gene associations
The GO Consortium [1] provides a set of flat files that indicate the association between gene names and GO categories for several species [27]. Although the flat files for human are quite comprehensive, we found a low hit rate for GO annotation of human genes using the database created by the GO Consortium's downloaded MySQL script files [28]. The hit rates were low both when the gene names were used in the format of HUGO names and when the gene names were used in the format of 'HUGO_HUMAN.' We tried the latter format because the flat files often contained '_HUMAN' appended to the human gene names. In contrast, when we used a combination of mouse (MGI) and rat (RGD) association files, there were reasonable numbers of hits. Therefore, we now routinely use mouse and rat annotations for human data. We are currently augmenting the human associations in the GO Consortium database to provide a richer annotation of human gene names. This goal will be achieved by using the MatchMiner database to integrate the information in the GO Consortium database [27] and the Swiss-Prot, TrEMBL and TrEMBLnew databases [29], and GoMiner will implement this database for human data in the near term. The MySQL script files will be freely available and should represent an improvement over what is currently available to program developers and end-users.
Non-independence of gene data
Gene-expression values within a category may be correlated for any of several reasons. They may represent the same gene, close family members with similar functions, genes in the same pathway or genes in alternative pathways for performing a biological function. Gene classifications in GO may be correlated for analogous reasons. How do such relationships affect the statistics? The answer is most easily seen by imagining a category containing nothing but five instances of the same gene (perhaps because five different identifiers were used and not recognized as representing the same gene). That category might appear either to be strikingly enriched (with five out of five genes flagged) or strikingly depleted (with none out of five genes flagged). But the appropriate value of n for determining statistical significance in those cases would be 1, not 5. GoMiner's companion program MatchMiner [30,31] handles this problem by identifying replicates of the same gene, even if they are represented by different identifiers.
What about possible sources of correlation other than 'same-gene'? Do we want to dereplicate them as well? Generally, the answer is 'no'. Correlation of genes in the same pathway is precisely the phenomenon we are often trying to identify. We would not want a statistical test to adjust for (and, in effect, null out) the effect of such relationships. Close family members might be considered an intermediate case. The statistical model implemented in GoMiner assumes, as our state of prior knowledge, that we know when two 'genes' are identical but nothing about their relationship if they are not identical. That seems the only available course. However, for each category, GoMiner provides the gene identities and the numbers given in Table 1 – sufficient information for the knowledgeable user to decide to eliminate close family members or pathway partners if desired.
The multiple comparisons problem
If one has not decided before analysis which particular gene category is to be examined, a correction should be made for the multiple opportunities to obtain a p-value indicating statistically significant enrichment or depletion. For example, with 1,000 categories, we would expect approximately 1,000 × 0.05 = 50 false positives simply by chance if we set the critical value at p = 0.05. The most common way to correct for this problem is that of Bonferroni (see, for example [32]), in which the critical value is divided by the number of trials (in this case, 1,000). However, that approach assumes independence of categories and is so conservative that it becomes extremely hard to detect true positives. A number of less conservative statistical methods have also been developed, but it is beyond the scope of this paper to review them here. An approach based on resampling will be incorporated into GoMiner in the coming months.
Overall, the p-values quoted should be considered as heuristic measures, useful as indicators of possible statistical significance, rather than as the results of formal inference. The p-values can be used, for example, to sort categories to identify those of the most potential interest.
As another useful measure, we have calculated the relative enrichment factor, Re, defined as
Re = (nf/n)/(Nf/N)
and shown as blue numbers in Figure 1a. The analogous quantities for overexpressed (red numbers) and underexpressed (green numbers) are also shown. Depletion is, of course, represented by an enrichment factor less than unity.
Benchmarking GoMiner on a biological problem
As a test, GoMiner was applied to the results of our cDNA microarray study of the molecular mechanisms by which drug resistance develops [33]. The DAG shown in Figure 1a was generated from that study, which used quadruplicate 'Oncochip' microarrays (Microarray Facility, Advanced Technology Center, NCI [34]) to compare gene expression profiles in a prostate cancer cell line (DU145) and a subline (RC0.1) selected from it for resistance to the topoisomerase 1-inhibitor 9-nitro-camptothecin. The microarray included 1,399 cancer-interesting genes. 181 of those genes differed in expression according to a threshold criterion (>1.5-fold difference). MatchMiner was used to translate IMAGE clone Ids for the 1,399 genes into HUGO names for input to GoMiner. Figure 1a shows that the category 'apoptosis regulator' was enriched 2.4-fold in genes with altered expression levels. More specifically, it was enriched 3.2-fold with underexpressed genes and 2.0-fold with overexpressed genes. Flow cytometric annexin V and TUNEL assays verified important differences in apoptotic potential between the cell lines, and analysis generated a novel hypothesis (the 'permissive apoptosis-resistance' hypothesis) for the relationship between apoptotic and cell-proliferation pathways in the development of drug resistance. Figure 1a provides more detailed information, indicating that these differences were focused in particular subcategories of apoptosis. Thus, GoMiner can help the user in at least two ways: it identifies categories enriched in, or depleted of, genes of interest; and it generates hypotheses to guide further research.
Unfortunately for us, interpretive analysis of the DU145/RC0.1 study was initially done one gene at a time before development of GoMiner (and, in fact, motivated that development). Performing the GO analysis one gene at a time would have taken more than two solid hours at the computer for the 181 genes before getting to the much harder parts of the task: doing the same for the entire array (nominally > 15 hours), then collating and organizing the information for each GO category. In contrast, operating on a 266 MHz PC with 250 MB RAM, it took 90 seconds to browse for and load the files, then 30 seconds for GoMiner to process the entire array of 1,399 genes and display the flagged and unflagged genes in their hierarchical context. In another test, running 900 flagged genes and all of HUGO (15,000 genes) took 4 minutes and 40 seconds on the same computer. Overall, the processing time was essentially linear with respect to the total number of genes (time in minutes = 0.0003 × genes + 0.0656; R2 = 0.998).
Comparison of GoMiner with related programs
Several other programs related to GoMiner have recently appeared. These include MAPPFinder [35,36], FatiGO [37], Onto-Express [2,38], and GoSurfer [39]. The following represents our best attempt at comparison, based on review of the available implementations and associated documentation as of January 2003.
FatiGO is a web application. The current implementation is very restrictive in that the user must specify ahead of time one particular level of the GO hierarchy that is to be used for analysis of the data. The other available applications, including GoMiner, process data for the entire GO hierarchy and allow the user to select views of the results dynamically. In a trial using FatiGO's recommended search criteria with our standard test gene files, FatiGO did not find any GO categories with clusters of differentially expressed genes.
Onto-Express is also implemented as a web application. Although more flexible than FatiGO, it is largely limited to a flat view of the biological world. Whereas GoMiner provides both tree and DAG views of the genes embedded within the GO hierarchy, Onto-Express does not provide any hierarchical structure (the fundamental defining feature of GO). Onto-Express lists enriched and depleted categories, but it does not provide a statistical analysis of the results to aid understanding. 'Version 2,' recently announced (at a price of $1,500 – $5,000), provides a p-value (computed by a method not specified in the announcement).
GoSurfer is implemented as a Windows application. As such, it lacks the flexibility of platform-independence that Java confers upon GoMiner. GoSurfer is also rather inflexible in that the input identifiers are required to be specific Affymetrix probe sets. It is not clear whether other identifier types suggested in a figure on the web site have been implemented. In contrast, GoMiner uses HUGO gene names as input. These gene names are more convenient for human interpretation, and GoMiner's companion program MatchMiner [30,31] allows many other types of identifiers (listed at the end of this section) to be converted easily into HUGO gene names. The visual output of GoSurfer is in the form of a DAG. GoMiner uses a text-based tree as its primary visual output because the nodes of the DAG are inherently more difficult to label without creating unacceptable screen clutter. The DAG gives an intuitive feel for the overall complexity of the categorizations, but it is not particularly useful for detailed dynamic navigation or for examination of categorized genes. The tabular output of GoSurfer does not include the HUGO names, which we consider to be the most useful key to gene identity. In contrast to GoMiner, it appears that GoSurfer does not provide complete quantitative and statistical summary data.
MAPPFinder is a pioneering project that integrates GO analysis and biological pathway maps. GoMiner also provides the potential for this type of integration, since each gene in the GoMiner tree classification is dynamically linked to the corresponding set of BioCarta and KEGG biological pathway maps. In addition to providing integration with biological pathway maps, GoMiner provides integration with chromosomal information via dynamic linking to LocusLink's chromosome viewer. GoMiner also provides dynamic linking to SNPs and MGC databases via LocusLink. MAPPFinder provides the fundamental tree representation of the GO hierarchy, with summary and statistical data in line with each category. However, unlike the tree implementation in GoMiner, it shows only the categories; the genes themselves are shown in an auxiliary table. In GoMiner, both the categories and the genes are seamlessly shown as integral components of the tree.
MAPPFinder does not appear to include a DAG representation. In GoMiner, the DAG view provides a qualitative and quantitative picture of the often-complex, multiple parenthood of some categories. In our opinion, this type of visualization is complementary to the tree form and important to an appreciation of the complex, highly nonlinear relationships within biological systems and gene networks. This complexity is not easy for a human to infer from the tree representation. The GO consortium selected the DAG as its fundamental data structure (though not its visualization), in part because it includes the characteristics of a network that are not included in a tree.
MAPPFinder is written in Microsoft's Visual Basic and is therefore restricted to running on PCs under Windows. In contrast, GoMiner is written in Java and runs on multiple operating systems. We have tested it on Windows XP, 2000, NT, and 98, as well as on Mac OS X, Solaris, Linux (Red Hat distribution), IRIX (SGI), and FreeBSD. See the GoMiner website for specific operating-system issues.
We recently implemented an alternative command-line interface for GoMiner (S.N., M.S., D.W.K. and B.R.Z., unpublished work) to complement the GUI version. The command-line interface allows GoMiner to be integrated with other tools via scripts or pipes. Our website will post updated versions of the documentation and program as soon as comprehensive testing of this interface has been completed. In preliminary trials with the new interface we have routinely processed more than 2,000 datasets at a time through GoMiner. This high-throughput capability has made two further developments possible: first, randomization studies are being done to address the multiple-comparisons problem (that is, to estimate the fraction of false positives among the selected categories); second, the output data stream is being coupled with integrated downstream analysis for automated recognition of interesting results buried within a large number of exploratory experiments. The user can explore and visualize these interesting results with GoMiner's graphical user interface.
The command-line interface also allows GoMiner to interact flexibly with its companion program MatchMiner. With MatchMiner as a 'preprocessor', GoMiner can take input data organized on the basis of 'omic' identifiers other than the HUGO names central to GO. MatchMiner currently resolves IMAGE clone ids, UniGene clusters, GenBank accession numbers, Affymetrix ids, chromosome locations, gene common names, and FISH clone ids, and greatly facilitates the preparation of microarray data for analysis in GoMiner.
In conclusion, GoMiner will continue in development with a view to integration with other bioinformatic resources being generated by the NCI and NIH for use by the biomedical research community. GoMiner is flexible both because it is coded in Java to be platform-independent and because it can accommodate either the default GO hierarchy and gene associations or customized versions. The default is the GO Consortium's database of categories and gene associations as implemented on our server. However, the user can, if desired, edit categories and gene memberships using DAG-Edit, the BDGP Gene Ontology Editor Tool [40]. The edited database can then be accessed by GoMiner from a local server to accommodate domain- and expertise-specific applications. Another important type of flexibility is the wide range of uses. In this report, we have presented GoMiner in the context of microarray data, but the variety of applications is clearly much broader; it embraces the full range of genomic and proteomic studies.
Acknowledgments
Acknowledgements
GoMiner is being developed jointly by groups from the National Cancer Institute (NCI), the Georgia Institute of Technology, and Emory University. This project has been supported by a contract funded by the NCI's Center for Cancer Research and by The Wallace H. Coulter Biomedical Engineering Department of Georgia Tech and Emory University academic funds for Professor May D. Wang. Its user features, statistical repertoire, and links to external resources will continue to be expanded through the contract funded by the NCI's Center for Cancer Research and through Professor Wang's academic funds.
References
- Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Draghici S, Ostermeier G, Krawetz S. Profiling gene expression using Onto-Express. Genomics. 2002;79:266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- GoMiner http://discover.nci.nih.gov/gominer
- GoMiner http://www.miblab.gatech.edu/gominer
- Weinstein JN. Fishing expeditions. Science. 1998;282:628–629. doi: 10.1126/science.282.5389.627g. [DOI] [PubMed] [Google Scholar]
- Weinstein JN. 'Omic' and hypothesis-driven research in the molecular pharmacology of cancer. Curr Opin Pharmacol. 2002;2:361–365. doi: 10.1016/s1471-4892(02)00185-6. [DOI] [PubMed] [Google Scholar]
- LocusLink http://www.ncbi.nlm.nih.gov/LocusLink
- PubMed http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=PubMed
- MedMiner http://discover.nci.nih.gov
- Tanabe L, Scherf U, Smith LH, Lee JK, Hunter L, Weinstein JN. MedMiner: an internet text-mining tool for biomedical information, with application to gene expression profiling. BioTechniques. 1999;27:1210–1217. doi: 10.2144/99276bc03. [DOI] [PubMed] [Google Scholar]
- GeneCards http://thr.cit.nih.gov:8081/cards/index.html
- NCBI Entrez Structure http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Structure
- The Cancer Genome Anatomy Project http://cgap.nci.nih.gov/Pathways
- Berkeley Drosophila Genome Project: developers' resources http://www.fruitfly.org/developers
- BrowserLauncher http://browserlauncher.sourceforge.net
- The Apache Jakarta Project http://jakarta.apache.org/oro/index.html
- HP Labs Semantic Web Research http://www.hpl.hp.com/semweb/index.html
- MySQL Connector/J downloads http://www.MySQL.com/downloads/api-jdbc.html
- Xerces2 Java Parser Readme http://xml.apache.org/xerces2-j/index.html
- MySQL http://www.MySQL.com
- Concurrent Versions System http://www.cvshome.org
- jUnit http://www.junit.org
- Agresti A. Categorical Data Analysis. New York: John Wiley; 1990. [Google Scholar]
- Agresti A. A survey of Exact inference for contingency tables. Stat Sci. 1992;7:131–177. [Google Scholar]
- StatXact 5 for Windows User Manual. Cambridge, MA: Cytel Software Corporation; 2002. [Google Scholar]
- Fisher's Exact Test http://www.matforsk.no/ola/fisher.htm
- GO Database http://www.geneontology.org/#godatabase
- GO downloads http://www.godatabase.org/dev/database/archive/
- Swiss-Prot, TrEMBL and TrEMBLnew database ftp://ftp.ebi.ac.uk/pub/databases/sp_tr_nrdb/
- Bussey JK, Kane D, Sunshine M, Narasimhan S, Nishizuka S, Reinhold WC, Zeeberg B, Ajay , Weinstein JN. MatchMiner: a tool for batch navigation among gene and gene product identifiers. Genome Biol. 2003;4:R27. doi: 10.1186/gb-2003-4-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MatchMiner http://discover.nci.nih.gov/matchminer/
- Bonferroni http://home.clara.net/sisa/bonhlp.htm
- Reinhold WC, Kouros-Mehr H, Kohn KW, Maunakea AK, Lababidi S, Roschke A, Stover K, Alexander J, Pantazis P, Miller L, et al. Apoptotic susceptibility of cancer cells selected for camptothecin resistance: gene expression profiling, functional analysis, and molecular interaction mapping. Cancer Res. 2003;63:1000–1011. [PubMed] [Google Scholar]
- NCI human Oncochip genes http://nciarray.nci.nih.gov/gi_acc_ug_title.shtml
- Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2002;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAPPFinder http://www.genmapp.org/MAPPFinder.html
- FatiGO http://fatigo.bioinfo.cnio.es/
- Onto-Express http://vortex.cs.wayne.edu/Projects.html
- GoSurfer http://biosun1.harvard.edu/complab/gosurfer/
- BDGP Gene Ontology Editor Tool http://www.godatabase.org/dev/editor.html