

Abstract
The 10th type III module of fibronectin possesses a β-sandwich structure consisting of seven β-strands (A–G) that are arranged in two antiparallel sheets. It mediates cell adhesion to surfaces via its integrin binding motif, Arg78, Gly79, and Asp80 (RGD), which is placed at the apex of the loop connecting β-strands F and G. Steered molecular dynamics simulations in which tension is applied to the protein’s terminal ends reveal that the β-strand G is the first to break away from the module on forced unfolding whereas the remaining fold maintains its structural integrity. The separation of strand G from the remaining fold results in a gradual shortening of the distance between the apex of the RGD-containing loop and the module surface, which potentially reduces the loop’s accessibility to surface-bound integrins. The shortening is followed by a straightening of the RGD-loop from a tight β-turn into a linear conformation, which suggests a further decrease of affinity and selectivity to integrins. The RGD-loop therefore is located strategically to undergo strong conformational changes in the early stretching stages of the module and thus constitutes a mechanosensitive control of ligand recognition.
Cellular response to mechanical force has emerged to be a critical regulator for sensory functions as well as for cell signal transduction, proliferation, and gene expression (ref. 1 and references therein). The mechanical tension generated within the contractile microfilaments of the cytoskeleton pull inward on the cell membrane. These inward-directed forces are counterbalanced by cell adhesion to anchoring sites within the extracellular environment. Transmembrane integrins thereby act as structural links to couple the intracellular microfilaments to extracellular ligands—for example, to RGD-containing proteins of the extracellular matrix (ECM). Current research efforts seek to characterize the “inside-out” signaling pathway by which the adhesiveness of integrins to external ligands is regulated by intracellular events affecting the cytoplasmic part of integrins. Simultaneously, “outside-in” signaling pathways exist that are triggered by binding of external ligands to membrane-bound integrins, leading to the assembly of integrins and cytoplasmic proteins into large focal adhesion complexes. Models describing both pathways implicitly assume that the RGD-containing proteins, either surface-bound or integrated into the ECM, serve as static anchoring sites against which cells can build up tensile strength. As a result, the dynamic regulation of cell adhesiveness is attributed solely to activation processes involving integrins. Simulations of forced unfolding of the 10th type III module of fibronectin (FnIII10) that are reported below now suggest that FnIII10 itself acts as a mechanosensitive switch. Accessibility of its RGD-loop to integrins is reduced when the FnIII10 module is stretched beyond an extension threshold value. This may explain how cells retract from RGD-containing matrices, a process that is key to understanding cell motility. The speed of cell migration, for example, is limited by the retraction rate of the rear of the cell from adhesive substrates, which requires disruption of all bonds between the cell membrane and the ECM (2).
The functional role of the RGD sequence to bind to integrins was first identified for fibronectin (3). Fibronectin is composed of three repeating structural motifs, of which one is the FnIII module. The three modules form a linear sequence of multiple tandem copies connected by short linker peptides as shown in Fig. 1a. The secondary structure of the FnIII10 module, which is the only fibronectin module to possess the RGD motif, consists of two β-sheets containing the antiparallel β-strands ABE and DCFG, respectively, which fold up to form a β-sandwich (Fig. 1b). The RGD sequence is located in the loop connecting the β-strands F and G. Fibronectin is a glycoprotein of 450–500 kDa with multiple recognition sites, thereby constituting a major building block of the extracellular matrix. In addition to cell recognition, essential for fibronectin’s function as an ECM protein is its ability to self-assemble into fibrils and to exhibit binding sites to other ECM proteins, including collagen, fibrin, heparin, and fibrinogen (4). The fibronectin type III module also is encountered in many other proteins, including tenascin, as well as the neural cell adhesion protein neuroglian, the muscle protein titin, and in a variety of different cytokine receptors (5, 6).
Figure 1.

(a) Modular structure of the fibronectin family. Fibronectin consists of two monomeric strands linked by two disulfide bridges (S—S). Each monomer contains three types of modules, types I, II, and III, and/or variable modules V. Modules that are either inserted or missing in various spliced forms of fibronectin are shown as filled circles. The cell adhesion site and other recognition sites are indicated. (b–e) Forced unfolding sequence of FnIII10 for various extensions of the module. A different gray scale is used to highlight the RGD motif and strand G. Hydrogen bond pairs that are broken in clusters during the unfolding burst of the module are displayed by dotted (AB bond pairs) and dashed (FG bond pairs) lines. The ball-and-stick representation indicates the position of the RGD cell binding region along the FG loop of FnIII10. Water molecules that were included in the SMD simulation are omitted for clarity. The equilibrated structure of FnIII10 is shown in b with its fixed terminal end (×) and the applied external pulling force (gray arrow). Partial distortion of the module is evident in c with the hydrogen bonds between strand F and G being stretched. Finally, in d, strand G clearly is separated from β-sheet FCD with the hydrogen bonds between F and G broken apart. Only strands C, D, and E remain in e to form a structural unit whereas the other strands form a line along the pulling direction. Further pulling on the module leads to total disintegration of the module with all strands aligned. This figure was created with molscript (37).
Forced unfolding of single proteins containing modular repeats recently has been studied experimentally for titin (7–10) and tenascin (11) by using atomic force microscopy (AFM) and optical tweezers. In all cases, pulling on the entire protein resulted in the consecutive rupture of the tertiary structure of individual modules. Each peak in AFM force-extension curves could be correlated to the rupture of a single protein module. Our previous studies of the forced unfolding of the 27th Ig module belonging to the I-band of titin (I27) by steered molecular dynamics (SMD) have reproduced the occurrence of a single peak in a computed force–extension graph (12) in agreement with experimental results. SMD simulations furthermore have been applied successfully to analyze ligand-receptor unbinding reactions (13–17). SMD simulations possess the advantage of providing an atomic scale picture of the unraveling process and can reveal how the folding scaffold of a protein responds to external forces. The simulations of the stretching and unfolding of the FnIII10 module illustrate how the accessibility of a recognition site can be modulated by external forces, opening the door to future studies of the role of tensile forces in regulating biological activity.
METHODS
Molecular dynamics simulations were carried out by using the programs xplor (18) and namd (19) with the charmm (20) force field. The x-ray crystallographic structure of FnIII10 was obtained from the tetramer FnIII7–10 (Protein Data Bank code 1fnf) (21). All atoms, including hydrogens, are described explicitly. The simulations were performed with a time step of 1 fs, TIP3P water parameters (22), a uniform dielectric constant of 1, and a cut-off of Coulomb forces with a switching function starting at a distance of 10 Å and reaching zero at 13 Å. FnIII10 was solvated by a sphere of water covering the module by at least five layers of water molecules before the entire system gradually was heated up to 300 K and equilibrated at a temperature of 300 K. A detailed description of the equilibration procedure is given in ref. 12. Equilibrating for 50 ps before SMD simulations yielded a stable solvated structure of FnIII10 with temperature fluctuations of <5 K and an averaged rms coordinate deviation of 1.5 Å in reference to the crystal structure. In SMD simulations, a time-dependent harmonic restraint directed along a specified coordinate then was imposed on the simulated molecular system, resulting in a nonequilibrium simulation. In the simulations, the N-terminal Cα atom (Leu1) of FnIII10 was constrained to a fixed point whereas the C-terminal Cα atom (Thr94) was attached to one end of a harmonic spring with a force constant k of 10 kBT/Å2, limiting the spatial fluctuation of its displacement to δz = √(kBT/k) = 0.32 Å. The other end of the spring was moved at a nearly constant pulling speed v of 0.5 Å/ps, which was achieved by changing its position every 100 fs by a distance of 0.05 Å. The direction of the velocity vector v was chosen to point from the constrained Cα atom to the moving Cα atom. Thus, the two termini of the protein module were subjected to an external force F of the form:
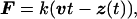 |
where z(t) is the displacement vector at time t of the C-terminal Cα atom. The unfolding events are independent of the pulling direction. SMD simulations on I27 (12) yielded similar results either when fixing one of the terminal Cα atoms and pulling on the other one or when pulling on both termini with half of the pulling speed. The extension of the module at any time t during the simulation is defined by the distance z(t). The simulation mimics an AFM experiment in which the cantilever represented by the harmonic spring is moving at constant speed with its tip attached to one end of the protein module and the other bound to a substrate.
RESULTS
Forced Unfolding of the FnIII10 Module.
FnIII10 was stretched from its initially compact and folded structure to its fully elongated configuration. A sequence of forced unfolding events is shown in Fig. 1 b–e. Details of the forced unfolding pathway can be correlated to characteristics of the force–extension curve. As seen in Fig. 2a, a force threshold of 1,808 pN must be overcome to rupture the tertiary structure of FnIII10, occurring at an extension of 24.6 Å. Two minor features were observed in close vicinity at an extension of 17.3 and 36.2 Å, with force peaks of 1,556 and 1,572 pN, respectively. The pulling force then steadily decreased until it leveled out to an average of ≈500 pN while the ruptured module was unraveling further. The module was fully elongated at an extension of 315 Å, at which time the force again rapidly increased and the simulation was halted. Simulations also were carried out for FnIII9 as well as at a pulling speed of 1.0 Å/ps, providing similar results to the ones discussed below.
Figure 2.

(a) Force–extension curve and (b–e) hydrogen bond data for FnIII10 at a pulling speed of 0.5 Å/ps. Hydrogen bond distances and energies are shown between β-strands FG (b and c) as well as AB (d and e), respectively. The region from 17 to 25 Å defines the bond breakage between strands F and G (dashed lines). For strands A and B, the bond breakage begins at 36 Å (dashed line). Energies were calculated from hydrogen oxygen interaction terms of the corresponding pairs; distances relate to the position of the hydrogen and oxygen atoms. Hydrogen bond pairs with residue and participant backbone atoms between β-strand F and G are (line pattern and color are solid and black unless otherwise noted in parenthesis) TyrO68—TyrH92, IleO70 —IleH90 (dashed), ValO72 —IleH88 (dot-dashed), IleO88 —ValH72 (long dashed), IleO90 —IleH70 (dashed, gray), and TyrO92 —TyrH68 (gray). Hydrogen bond pairs between β-strand A and B are SerO21 —GluH9 (gray), GluO9 —SerH21, LeuO19 —ValH11 (dashed), AlaO12 —LeuH9 (dot-dashed), and SerO17 —ThrH14 (dashed, gray).
The observation in SMD simulations that a force threshold must be overcome to unravel the module agrees well with experimental force–extension curves from the FnIII modules of tenascin (11). The computed force peak of 1,800 pN, however, is nearly an order of magnitude higher than the 200 pN force measured by AFM. This discrepancy results from differences in pulling speeds and spring constants: a pulling speed of 0.5 Å/ps had to be chosen for the SMD simulation because of the limitation in simulation time whereas the pulling speed of typical AFM experiments is several orders of magnitude smaller (23). However, reconstructing the potential of mean force along the SMD pulling coordinate allows one to estimate the force needed in AFM experiments. This method has been used successfully in the case of a one-dimensional potential, relating forces at different pulling speeds (24, 25).
A detailed analysis of the forced unfolding pathway revealed that the terminal ends of FnIII10, which possess no secondary structure, were straightened out first, followed by a slight rotation of the β-sheets ABE and DCFG such that their β-strands lined up in the direction of the applied force. The entire module was thus moving along the pulling direction (Fig. 1b). Strand G started moving past strand F in the force direction at an extension >17 Å (Fig. 1c), leading to a distortion of the six backbone hydrogen bonds connecting strand F and G, as seen in Fig. 2 b and c. These six hydrogen bonds collectively ruptured at ≈25 Å. The rupture of this set of backbone hydrogen bonds was correlated with the force peak and the first minor feature seen in the force–extension diagram (Fig. 2a). Coinciding with strand G sliding in the direction of the applied force, the 12-aa loop FG that contains the RGD sequence was dragged along (Fig. 1d) whereas strand F stayed closely bound to strand C. Distinct from this first rupture event that broke the backbone hydrogen bonds between strands G and F collectively, a second set of five backbone hydrogen bonds connecting strands A and B was distorted starting at an extension of 36 Å, as seen in Fig. 2 d and e. These hydrogen bonds did not, however, rupture collectively. Three broke apart at 40 Å followed by the rupture of the other two at ≈55 Å. At this point, strand A separated from strand B. After this last rupture, the module gradually unfolded, and the remaining strands, B through F, unraveled one by one. The hydrogen bond pairs between strands A and B, as well as F and G, were the only ones observed to break apart in clusters whereas all other inter-β-sheet backbone hydrogen bonds later broke individually during the extension of FnIII10. This behavior is expected to be independent of the amino acid sequence of those strands as long as mutations do not perturb the module’s tertiary structure.
The force required for further extension of the module was now mainly attributable to friction with the surrounding water molecules and surface tension of the water droplet used in the simulation. Finally, at full extension, all of the β-strands were aligned linearly (Fig. 1e). Further stretching beyond this point required changing the length and angle of covalent bonds along the module backbone, which resulted in the drastic increase in pulling force (Fig. 2a).
RGD-Loop Conformations Along the Unfolding Pathway.
Dramatic conformational changes of the RGD-loop were observed in the initial stages of the forced unfolding pathway of the module (Fig. 1 b–d). The loop initially was located at the apex of a hairpin β-turn connecting strands G and F. As illustrated in Fig. 1b as well as schematically in Fig. 3, this hairpin was bent at the Cα atoms of residues Thr76 and Ala83. On extension of the module past the rupture between strands G and F, this bend was straightened, as can be seen in the steep increase in the angle defined by the Cα atoms of residues Asp80, Ala83, and Ser84 at 20 Å (Fig. 3a). Subsequently, the apex of the loop was pulled closer to the surface of the module, i.e., to the plane defined by β-strands C and F. It was shortened from 12 to 8 Å whereas the module was extended from 25 to 35 Å (Fig. 3b). After this transformation, the RGD-loop opened up, which is indicated by a gradual change in the distance between the Cα atoms of residues Arg78 and Pro82 starting at 30 Å (Fig. 3c). Residue Asp80 of the RGD sequence initially defined the apex of the RGD-loop at extensions <31.4 Å. As the loop was dragged behind the G-strand, the apex of the loop moved closer to the F-strand. This resulted in a transition of the (φ, ψ) values of Asp80 from (−16°, −67°) to (−113°, −5°) at an extension of 31.4 Å, as illustrated in the Ramachandran plot (Fig. 4).
Figure 3.

Conformational changes of the RGD-containing FG loop during forced unfolding of FnIII10. (a) The angle formed by the Cα atoms of residues Asp80, Ala83, and Ser84 displays the change from a bent (≈113°) to a more planar (≈165°) conformation. A sharp increase is observed at an extension of 20 Å (dashed line). (b) The distance of the RGD segment to the FnIII10 core is measured from the Cα atom of Asp80 to the projected point on the plane defined by β-strand C and F. At an extension of 35 Å, the value drops to ≈8 Å, reducing the accessibility of RGD to membrane-bound integrins. Beyond an extension of 140 Å, the reference plane is distorted. (c) The width of the FG loop is measured by the distance between the Cα atoms of Arg78 and Pro82, which displays a drastic increase at an extension of 40 Å (dashed line).
Figure 4.

Transition states of forced unfolding of the RGD-loop apex. Three distinct regions of φ–ψ combinations of Asp80 are observed during the extension of FnIII10, leading from β-turn II′ (38) to a more linear conformation. The extension at the two transition points also is shown. The two-dimensional plot is continuous at the edges because −180° and +180° rotations are equivalent.
DISCUSSION
The observation that the β-strand G was pulled out of the FnIII10 module in an early phase of the forced unfolding pathway has intriguing consequences because the RGD sequence is located between strands G and F. In the unperturbed native state, the RGD-containing loop is located ≈12 Å away from the outer surface of the FnIII10 module and is thus accessible to cell surface integrins. Two events occurred in the SMD simulations immediately after the rupture of the hydrogen bonds stabilizing strands F and G: the RGD-containing loop was both shortened and straightened out. These events have been shown experimentally to decrease RGD accessibility to membrane-bound integrins and its selectivity to different members of the integrin family, respectively. For example, engineered antibodies obtained by inserting three repeats of RGD, namely (RGD)3, into the third complementary-determining loop (FG-loop) (26) of the heavy chain variable Ig module effectively bind to cell surface integrins (27). However, substitution of (RGD)3 with a single RGD shortened this loop, preventing integrin binding. Furthermore, experiments using RGD-peptides immobilized on a synthetic substrate have shown that binding to cell surface integrins depends on the distance of the RGD sequence from the underlying substrate surface. Optimal binding is obtained for distances ranging between 11 and 30 Å (28). This supports the notion derived from our SMD simulations of a force-regulated change in accessibility of the RGD segment of FnIII10 because a shortening of the loop from 12 to 8 Å was observed during the first 35 Å of the forced unfolding pathway (Fig. 3b).
The conformational geometry of the RGD-loop also has been shown to play an important role in defining the binding strength and selectivity with respect to different members of the integrin family (29). Synthetic RGD-peptides display a higher integrin selectivity and affinity if they are presented in a cyclic rather than a linear conformation (30–32). Furthermore, human lysozyme mutants that contain RGD sequences bind to integrins with an increased affinity if the RGD-containing loop is constrained by a disulfide bridge between two cysteines surrounding the RGD motif (33). Our SMD simulations illustrate how the RGD-loop looses its tight hairpin-like configuration if FnIII10 is stretched beyond the rupture of strands G and F. The SMD simulations, combined with experimental evidence, suggest that the accessibility and affinity of the RGD sequence for membrane-bound integrins can be altered if tension is applied to the terminal ends of FnIII10. The FnIII10 module thereby acts as a tensile molecular recognition switch that is activated under applied external force. On mechanical activation, the module’s affinity to membrane-bound integrins is reduced.
Reversible refolding after activation, however, is crucial to a tensile molecular recognition switch that has undergone a function-driven evolutionary process. The FnIII10 module is known to refold on a time scale of milliseconds after complete denaturization, which is considerably faster than the refolding of other FnIII modules (34, 35). Rapid refolding to the initial conformation is facilitated if only one β-strand needs to be pulled out to activate the switch while the remaining module experiences only minor structural perturbations. This brings up the question regarding how the β-sandwich motif of FnIII10 functionally differs from other β-sandwich motifs and why the RGD sequence is spliced predominately into FnIII10 rather than into other β-sandwich modules. SMD simulations forcing the unfolding of the C2 module of synaptotagmin I, a β-sandwich motif with the N- and C-termini in close proximity, and of cytochrome C6, an all α-helical protein, displayed no force peak during unfolding (36). This indicates that no structural element of these modules is protected from being readily unfolded by external force. These types of modules would not be suitable to include the RGD motif as a tensile molecular recognition switch because the loop would be disturbed too easily by applied forces.
A closer comparison of the two unrelated yet similar β-sandwich motifs, titin I27 and fibronectin FnIII10, which both show a single force peak early in the unfolding pathway, experimentally as well as in SMD simulations, provides even further insight. The seven β-strands of I27 also folds into a β-sandwich in which the upper and lower β-sheets are formed by β-strands ABED and A′GFC, respectively. Nevertheless, the backbone topology of I27 is slightly different from the one of FnIII10 (compare Fig. 1c and Fig. 5). For instance, β-strands A and A′ of I27, which are the equivalent to β-strand A of FnIII10, belong to different β-sheets. Backbone hydrogen bonds link β-strands A and B, as well as A′ and G in I27, whereas FnIII10 lacks hydrogen bonds between β-strands A and G. Previous SMD simulations of the forced unfolding of I27 under the same conditions as the ones described here have demonstrated that the I27 structure unraveled by sequentially breaking the hydrogen bonds between β-strands AB, A′G, and GF (12). Although, for FnIII10, the C-terminal β-strand G was pulled out first, in the case of I27, the N-terminal β-strands A and A′ were released first on applying tension to the module’s termini. The major advantage of functionalizing the GF-loop of FnIII10 thus could be that the G-strand forms backbone hydrogen bonds to only one neighboring strand (F) whereas the G-strand of I27 forms backbone hydrogen bonds to two neighboring strands (A′ and F). On activation of the recognition switch, the structural perturbation of the FnIII10 module therefore was minimized. This illustrates how the study of forced unfolding pathways may provide important clues for an understanding of the structure/function relations of this module.
Figure 5.

I27 module distortion at an extension of 17 Å. Hydrogen bond pairs between β-strand A and B as well as A′ and G (dashed lines) clearly are stretched beyond their breakage. In comparison, hydrogen bonds between strand F and G (dotted lines) remain close to their equilibrium value. Strands A and A′ are displayed in a different gray scale because both are the first to be separated from the module on forced unfolding. The single force peak for I27 occurs at 14.2 Å (12).
In summary, SMD simulations suggest that FnIII10 operates as a tensile molecular recognition switch if the module is extended beyond a threshold value to rupture the hydrogen bonds between strands F and G. The existence of a force threshold ensures the structural stability for the RGD motif to function properly unless the tensile force exceeds this threshold. Access to the RGD sequence is reduced after these hydrogen bonds are broken. The RGD sequence is, furthermore, located in a strategic position, namely, in the loop that is straightened out early in the forced unfolding process. The conformation of the RGD-loop thus is altered while introducing only minor structural disturbances to the remaining module. Identification of the FnIII10 module as a tensile molecular recognition switch by SMD simulations has important implications in cell biology and biotechnology. Experiments now can be designed to test whether cell binding to FnIII10 can be regulated if a tensile force is applied that initiates partial unfolding of FnIII10. The FnIII10 module within fibronectin, for example, is tightly linked to other repeats that recognize various extracellular matrix proteins. Tensile stress from the extracellular matrix potentially can be transmitted to the FnIII10 module, thereby modulating cell binding. FnIII10 thus constitutes a critical link in transmitting mechanical stress from the extracellular matrix to the cytoskeleton and vice versa by functioning not only as an anchor but also as a mechanosensitive regulator.
Acknowledgments
We thank G. Baneyx and Dr. T. P. Lybrand for fruitful discussions. This work was supported by grants from the National Institute of Health (Grants GM 49063 and PHS 5 P41 RR05969), by the National Science Foundation (Grants BIR 94-23827 EQ, NSF/GCAG BIR 93-18159, and MCA93S028), by the Roy J. Carver Charitable Trust, and the Whitaker Foundation.
ABBREVIATIONS
- FnIII
fibronectin type III
- I27
27th Ig module of titin’s I-band
- SMD
steered molecular dynamics
- ECM
extracellular matrix
- AFM
atomic force microscopy
References
- 1.Chicurel M E, Chen C S, Ingber D E. Curr Opin Cell Biol. 1998;10:232–239. doi: 10.1016/s0955-0674(98)80145-2. [DOI] [PubMed] [Google Scholar]
- 2.Palecek S P, Huttenlocher A, Horwitz A F, Lauffenburger D A. J Cell Sci. 1998;111:929–940. doi: 10.1242/jcs.111.7.929. [DOI] [PubMed] [Google Scholar]
- 3.Pierschbacher M D, Ruoslahti E. Nature (London) 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 4.Hynes R O. In: Fibronectins. Rich A, editor. New York: Springer; 1990. pp. 84–112. [Google Scholar]
- 5.Potts J R, Campbell I D. Matrix Biol. 1996;15:313–320. doi: 10.1016/s0945-053x(96)90133-x. [DOI] [PubMed] [Google Scholar]
- 6.Campbell I D, Spitzfaden C. Structure (London) 1994;2:333–337. doi: 10.1016/s0969-2126(00)00034-4. [DOI] [PubMed] [Google Scholar]
- 7.Rief M, Gautel M, Schemmel A, Gaub H E. Biophys J. 1998;75:3008–3014. doi: 10.1016/S0006-3495(98)77741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rief M, Gautel M, Oesterhelt F, Fernandez J M, Gaub H E. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 9.Kellermayer M S Z, Smith S B, Granzier H L, Bustamante C. Science. 1997;276:1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 10.Tskhovrebova L, Trinick J, Sleep J A, Simmons R M. Nature (London) 1997;387:308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- 11.Oberhauser A F, Marszalek P E, Erickson H P, Fernandez J M. Nature (London) 1998;393:181–185. doi: 10.1038/30270. [DOI] [PubMed] [Google Scholar]
- 12.Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Biophys J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kosztin D, Izrailev S, Schulten K. Biophys J. 1999;76:188–197. doi: 10.1016/S0006-3495(99)77188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isralewitz B, Izrailev S, Schulten K. Biophys J. 1997;73:2972–2979. doi: 10.1016/S0006-3495(97)78326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Biophys J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stepaniants S, Izrailev S, Schulten K. J Mol Model. 1997;3:473–475. [Google Scholar]
- 17.Grubmüller H, Heymann B, Tavan P. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 18.Brünger A T. x-plor (Version 3.1): A System for X-Ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 19.Nelson M T, Humphrey W, Gursoy A, Dalke A, Kale L V, Skeel R D, Schulten K. J Supercomputing Appl. 1996;10:251–268. [Google Scholar]
- 20.MacKerell A D, Jr, Bashford D, Bellot M, Dunbrack R L, Jr, Evanseck J D, Field M J, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 21.Leahy D J, Aukhil I, Erickson H P. Cell. 1996;84:155–164. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 22.Jorgensen W L, Chandrasekhar J, Madura J D, Impey R W, Klein M L. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 23.Evans E, Ritchie K. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gullingsrud, J. R., Braun, R. & Schulten, K. (1999) J. Comp. Phys., in press.
- 25.Balsera M, Stepaniants S, Izrailev S, Oono Y, Schulten K. Biophys J. 1997;73:1281–1287. doi: 10.1016/S0006-3495(97)78161-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandon C, Tooze F. Introduction to Protein Structure. New York: Garland; 1998. pp. 304–312. [Google Scholar]
- 27.Zanetti M, Filaci G, Lee R H, del Guercio P, Rossi F, Bacchetta R, Stevenson F, Barnaba V, Billetta R. EMBO J. 1993;12:4375–4384. doi: 10.1002/j.1460-2075.1993.tb06122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beer J H, Springer K T, Coller B S. Blood. 1992;79:117–128. [PubMed] [Google Scholar]
- 29.Carr P A, Erickson H P, Palmer A G., III Structure (London) 1997;5:949–959. doi: 10.1016/S0969-2126(97)00248-7. [DOI] [PubMed] [Google Scholar]
- 30.Pierschbacher M D, Ruoslahti E. J Biol Chem. 1987;262:17294–17298. [PubMed] [Google Scholar]
- 31.Scarborough R M, Naughton M A, Teng W, Rose J W, Phillips D R, Nannizzi L, Arfsten A, Campbell A M, Charo I F. J Biol Chem. 1993;268:1066–1073. [PubMed] [Google Scholar]
- 32.Nowlin D M, Gorcsan F, Moscinski M, Chiang S-L, Lobl T J, Cardarelli P M. J Biol Chem. 1993;268:20352–20359. [PubMed] [Google Scholar]
- 33.Yamada T, Song H, Inaka K, Shimada Y, Kikuchi M, Matsushima M. J Biol Chem. 1995;270:5687–5690. doi: 10.1074/jbc.270.11.5687. [DOI] [PubMed] [Google Scholar]
- 34.Plaxco K W, Spitzfaden C, Campbell I D, Dobson C M. J Mol Biol. 1997;270:763–770. doi: 10.1006/jmbi.1997.1148. [DOI] [PubMed] [Google Scholar]
- 35.Plaxco K W, Spitzfaden C, Campbell I D, Dobson C M. Proc Natl Acad Sci USA. 1996;93:10703–10706. doi: 10.1073/pnas.93.20.10703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu, H. & Schulten, K. (1999) Proteins Struct. Funct. Genet., in press.
- 37.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 38.Richardson J S. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]