

Abstract
Cloning whole animals with somatic cells as parents offers the possibility of targeted genetic manipulations in vitro such as “gene knock-out” by homologous recombination. However, such manipulation requires prolonged culture of nuclear donor cells. Previous successes in cloning have been limited to the use of cells collected either fresh or after short-term culture. Therefore, demonstration of genetic totipotency of cells after prolonged culture is pivotal to combining site-specific genetic manipulations and cloning. Here we report birth of six clones of an aged (17-year-old) Japanese Black Beef bull using ear skin fibroblast cells as nuclear donor cells after up to 3 months of in vitro culture (10–15 passages). We observed higher developmental rates for embryos derived from later passages (10 and 15) as compared with those embryos from an early passage (passage 5). The four surviving clones are now 10–12 months of age and appear normal, similar to their naturally reproduced peers. These data show that fibroblasts of aged animals remain competent for cloning, and prolonged culture does not affect the cloning competence of adult somatic donor cells.
Genetic manipulation of mouse embryonic stem cells has revolutionized mouse genetic research. However, embryonic stem cells are not available in other species. Fortunately, animal cloning using cultured somatic cells offers the possibility of targeted genetic manipulations like those performed in the mouse, should those somatic cells remain competent for cloning after prolonged culture. Live clones have been obtained from adult somatic cells in sheep (1), mice (2), and cows (3, 4). Furthermore, transgenic animals have been produced by cloning gene-transfected fetal somatic donor cells (5, 6). However, to date, successful somatic cell cloning has been largely limited to the use of the donor cells either fresh (2) or after short-term (under 10 passages) in vitro culture (1, 3–6), which would not allow targeted gene manipulations.
A recent report (7) indicates that Dolly, the cloned sheep, inherited the shortened telomeres of the adult nuclear donor animal. Moreover, the telomeres of Dolly were further shortened during the brief in vitro culture of the donor cells. These observations raise the questions of whether healthy clones may be obtained from aged donor animals, particularly after long-term cultures of the “aged” donor cells. This study was conducted to test the cloning competence of skin fibroblast cells after prolonged in vitro culture, using an aged (17-year-old) elite bull. In this paper, we report that normal live clones were produced from cultured adult somatic cells in a cattle model after up to 3 months of culture (passage 15). Our finding offers promise for producing site-specific genetically modified animals such as “gene knockout” animals by somatic cell cloning. Additionally, success in cloning live, aged animals opens the possibility to compare the telomere lengths, aging, and the “biological age” of the cloned animals.
Materials and Methods
Adult Somatic Cell Collections and Culture.
A skin biopsy was obtained from the ear of a high genetic merit 17-year-old Japanese Black Beef bull. The tissue biopsy was cut into small pieces (3 mm2), and the pieces as tissue explants were cultured in DMEM (GIBCO, catalog no. 12100-061) plus 10% FBS and antibiotics (GIBCO, catalog no. 15240-013) at 37.5°C in a humidified atmosphere of 5% CO2 and 95% air. After a week in culture, fibroblast cell monolayers had formed around the tissue explants. The explants were then removed, and the fibroblast cells were cultured to confluency. The cell strain was routinely maintained on dishes until passage 17 and then were stored frozen as described below. For each passage (estimated two cell doublings per passage), cells were cultured until confluent, were disaggregated by incubation in a 0.1% (wt/vol) trypsin (Difco) and EDTA (Nacalai Tesque, Kyoto) solution for 1 min at 37°C, and were allocated to three new dishes for further passaging. Normally, each passage lasted about 6 days. For long-term storage, the cells at different passages were collected after trypsin treatment, frozen in 10% dimethyl sulfoxide (Sigma), and stored in liquid nitrogen.
Cell-Specific Markers.
Conformation of fibroblast phenotype of donor cells was conducted by immunocytochemical staining with monoclonal antibodies directed against the cytoskeletal filaments vimentin (for fibroblasts) or cytokeratin (for epithelial cells). In brief, cells were grown to confluency in Lab-Tek chamber slides (Nalge Nunc). Cells were washed with PBS and were fixed in methanol at 4°C for 20 min. After fixation, the cells were washed in PBS (139 mM NaCl/2.7 mM KCl/4.3 mM Na2HPO4·7H2O/1.48 mM KH2PO4)and were blocked with 3% BSA in PBS for 15 min at 37°C. Block was removed, and 100 μl of either a 1:40 dilution antivimentin clone V9 (Sigma, catalog no. 6630) or a 1:400 dilution of antipan cytokeratin clone-11 (Sigma, catalog no. 2931) was added. Slides were incubated for 1 h at 37°C. Cells were washed with PBS and were incubated for 1 h with 100 μl of a 1:300 dilution of FITC-labeled anti-mouse IgG. Cells were washed in PBS, were covered with 50% glycerol in PBS under a coverslip, and were observed by fluorescence microscopy. Appropriate controls for autofluorescence and secondary antibodies were included.
Cell Cycle Analyses.
Analysis of cell cycle stage was performed as described (8). In brief, cell cultures at different passages were either grown to confluency, were serum starved or nonstarved, and were trypsinized. After trypsinization, cells were washed with DMEM plus 10% FBS and were resuspended to a concentration of 5 × 105 cells/ml in 1 ml of PBS with glucose (6.1 mM) and 0.5 mM EDTA at 4°C. Cells were fixed overnight by adding 3 ml of ice-cold ethanol. For nuclear staining, cells were then pelleted, were washed with PBS, and were resuspended in PBS containing 30 μg/ml propidium iodide (Sigma) and 0.3 mg/ml RNase A (Sigma). Cells were allowed to incubate for 1 h at room temperature in the dark before being filtered through a 30-μm mesh (Spectrum Laboratories). Ten-thousand cells were collected on a Becton Dickinson FACs Caliber and were analyzed by using cell quest 3.1 software (Becton Dickinson).
Cell Proliferation Assay.
To examine whether the serum-starved donor cells were at quiescent stage, the ability of cells to proliferate was measured by immunofluorescence assay to detect 5-bromo-2′-deoxy-uridine (BrdUrd) incorporation into cellular DNA (9, 10). Confluent cells at passages 5, 10, and 15 were serum starved (0.5% FBS), and cell proliferation was measured on days 1, 2, 3, 4, 5, and 6 by BrdUrd incorporation. Nonconfluent cells were also included as controls (data not shown). In brief, BrdUrd labeling medium (Boehringer Mannheim, catalog no. 1296-736) was added to cell culture for 24 h at 37°C. Cells were harvested by trypsinization and were fixed by Carnoy's fixative. Fixed cell suspension was placed on clean microscopic slides overlaid with anti-BrdUrd solution and was incubated for 30 min at 37°C. After washing three times with anti-mouse Ig-fluorescein solution and an additional incubation at 37°C, the slides were mounted and examined by using a fluorescence microscope. Confluent cells at passage 5 without serum starvation were similarly measured as controls. Additionally, nonconfluent cells at passages 5, 10, and 15 at day 1 of culture were also treated, and cells that had incorporated BrdUrd were counted.
Chromosome Analysis.
To examine the ploidy of the cultured somatic donor cells at various passages, chromosome counts were determined at passages 5, 10, and 15 of culture by using standard preparation of metaphase spreads (11). In brief, 24 h after plating, cells were treated with hypotonic KCl (0.075M) for 15 min at 37°C. The cells were then fixed in acetic methanol (vol/vol = 1:3), and drops of cell suspension were spread on clean microscopic slides. The chromosomes were stained with 5% Giemsa for 10 min. The numbers of well spread chromosomes within a clear cell boundary were counted under a light microscope at 1,000× magnification under oil. At least 100 metaphase spreads/group were counted.
Donor Cells and Recipient Oocyte Preparation and Nuclear Transfer.
Donor cells either were subjected to serum starvation (0.5% FBS) for 5 days after reaching confluency (passages 5, 10, and 15) or were allowed to grow for an additional 5 days in 10% serum upon confluency (cells at passage 5 only) (1, 12). Immediately before nuclear transfer, donor cells were trypsinized, washed by centrifugation, and resuspended in PBS supplemented with 0.5% FBS. Recipient oocyte collection, maturation, and enucleation were as described (13) at ≈24 h after maturation culture. Successful enucleation was confirmed by Hoechst 33342 staining. Cells with an approximate diameter of 10–15 μm (14) were transferred to the perivitelline space of the recipient cytoplast using our standard procedure (13). After transfer, the cell-cytoplast complexes were induced to fuse with two pulses of direct current of 2.5 kV/cm for 10 μsec each by an Electrocell Manipulator 200 (BTX, San Diego). These electrical pulses also simultaneously induced initial oocyte activation. Fusion was then confirmed by microscopic examination. All fused embryos were further activated by culturing with cycloheximide (10 μg/ml; Sigma) in CR1aa medium (15) for 5 additional h.
In Vitro Culture of Cloned Embryos and Embryo Transfer.
The nuclear transferred embryos were cultured in CR1aa medium for 48 h at 38.5°C in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2. Cleavage rates were recorded, and cleaved embryos were cultured further in CR1aa medium supplemented with 5% FBS with cumulus-cell coculture for 5 days. On day 7, blastocyst development was recorded, and one or two good quality blastocysts were transferred nonsurgically into the uterus of each synchronized recipient. Recipients were examined for pregnancy by rectal palpation or ultrasonography on days 40, 60, 90, and 120 of gestation.
Genotyping of Microsatellite Markers.
To confirm the clonal status of the newborns, individual identification and parentage diagnosis were performed with 23-microsatellite markers (16). Total genomic DNA from the donor bull, cells of passages 5, 10, and 15, the six cloned animals, and the six foster mothers were prepared from peripheral blood leukocytes using a QIA-amp blood kit (Qiagen, Chatsworth, CA). PCR primers for microsatellite markers were labeled with fluorescent dyes [6-FAM, HEX, and TET (Applied Biosystems/Perkin–Elmer)], and the DNA typing was conducted as described (16). Genotypes were determined by polyacrylamide gel electrophoresis using the ABI373A DNA sequencer (Applied Biosystems/Perkin–Elmer) and were analyzed by genscan 672 and genotyper software (Applied Biosystems/Perkin–Elmer).
Statistical Analysis.
Embryo development experiments were repeated at least three times. Differences among treatment groups were analyzed by χ2 test.
Results
Effect of Donor Cell Passage Number on Cloning Competence.
To test the cloning competence of adult somatic cells after prolonged culture, skin fibroblast cells from a 17-year-old bull were cultured for 5, 10, and 15 passages followed by nuclear transfer assays. The fusion rates were low (36–43%, P > 0.05), regardless of the donor cell treatment or passage number. At passage 5, the effect of serum starvation on nuclear transfer was tested. The rates of cleavage (66 vs. 78%) and blastocyst development (21 vs. 28%) were not different between embryos derived from serum-starved and nonstarved donor cells (Table 1; P > 0.05). No pregnancy, however, was established from the nonserum-starved cells (n = 10 recipients) whereas a 30% pregnancy rate (n = 10 recipients; Table 2) was established from the serum-starved cells. Therefore, for donor cells of passages 10 and 15, serum starvation treatment was applied to all donor cells. When compared retrospectively, significantly higher rates of blastocyst development were obtained from donor cells of passages 10 and 15 than those of passage 5 (37 and 33% vs. 21%, P < 0.05). After embryo transfer, nine pregnancies (n = 14 recipients) from cells at passage 10 and three pregnancies (n = 12 recipients) from cells at passage 15 were obtained, resulting in term development of four and two normal clone calves, respectively. The bull calves were born on December 21, 23, 24, and 30, 1998 and February 7 and 8, 1999, respectively (Table 3; Fig. 1). Overall, we obtained a higher pregnancy and calving rate from embryos derived from cells at passage 10 (64 and 29%) than from those at passage 15 (25 and 17%), but these differences were not statistically significant.
Table 1.
In vitro developmental rates of nuclear transfer embryos derived from adult fibroblast cells at different passages
| Passage no. | Serum-starved | No. of oocytes injected | No. (%) fused | No. (%) cleaved | No. (%) of blastocysts |
|---|---|---|---|---|---|
| 5 | No | 282 | 102 (36) | 79 (78) | 28 (28)a |
| 5 | Yes | 288 | 114 (40) | 75 (66) | 24 (21)a |
| 10 | Yes | 269 | 115 (43) | 72 (63) | 43 (37)b |
| 15 | Yes | 264 | 109 (41) | 81 (74) | 36 (33)b |
Summaries of 4–6 replicates. Groups with different superscripts differ (P < 0.05).
Table 2.
Embryo transfer and pregnancy rates of cloned embryos from adult fibroblast cells at different passages after serum starvation
| Cell passage | No. of embryos | No. of recipients | No. (%) pregnant | No. (%) aborted | Days abortions observed |
|---|---|---|---|---|---|
| 5 | 15 | 10 | 3 (30) | 3 (100) | 61, 88, 123 |
| 10 | 22 | 14 | 9 (64) | 5 (56) | 39, 67, 69, 76, 119 |
| 15 | 17 | 12 | 3 (25) | 1 (33) | 113 |
| Total | 54 | 36 | 15 (42) | 9 (60) |
Table 3.
Birth data of full-term cloned calves
| Calf ID | Gestation length, days | Birth weight, kg | Current status |
|---|---|---|---|
| Pass 10-A | 291 | 32.5 | Died |
| Pass 10-B | 293 | 35.5 | Live |
| Pass 10-C | 295 | 42.5 | Live |
| Pass 10-D | 299 | 30.7 | Died |
| Pass 15-A | 292 | 42.5 | Live |
| Pass 15-B | 293 | 32.5 | Live |
| Average | 294 | 36.0 | – |
Figure 1.

The cloned calves and the 17-year-old bull used for cloning. Cloned calves from left to right: Pass 15-A, Pass 15-B, Pass 10-B, and Pass 10-C.
Analysis of the Clones.
Among the six clones born, two from passage 10 died shortly after birth. One of the deaths was caused by an infection of Akabane Virus, and the other was caused by dystocia at parturition. Postmortem autopsy revealed no gross or histopathological abnormalities in these two clones. The gestation periods for the cloned pregnancies (average, 294 days; range, 291–299 days) were 9 days longer than the average gestation period for the breed (285 days). The birth weights of the clones (average, 36 kg; range, 30.7–42.5 kg) were 20% heavier than the average birth weight for male calves of this breed (30 kg). The four surviving clones and the donor bull are shown in Fig. 1. These cloned calves are now 10–12 months of age. Veterinary examination of them indicates that they are healthy and normal compared with their age-matched peers derived from conventional reproduction. We also checked their growth curves plus ≈30 blood parameters, indicative of the health status of the calves, and found no difference between the clones and their age-matched peers.
To confirm the clone status of these calves, we conducted DNA typing on the six clones, the donor bull, and donor cells at passages 5, 10, and 15 by using 23 microsatellite markers (16). The respective foster mothers of the clones were also included in the assays. We found that all six clones were identical to the donor bull, to the nuclear donor cells, and to each other with respect to all 23 DNA markers analyzed (Fig. 2). Previously, it has been shown that these 23 microsatellite markers could distinguish between 31 trillion individuals and exclude 37 million sires (16).
Figure 2.

A representative DNA microsatellite assay. Lanes: 1, donor bull; 2–4, donor cells at passages 5, 10, and 15; 5–10, the six clones; 11–16, the six foster mothers. Eleven sets of identical microsatellite markers were observed in lanes 1–10. Red bands, DNA molecular weight markers (in base pairs). PCR primers for microsatellite markers were labeled with fluorescent dyes: 6-FAM (blue), HEX (yellow), and TET (green).
Characterization of Donor Cells.
The cells used for cloning were systematically characterized by (i) cell type-specific marker staining, (ii) cell cycle analysis, (iii) cell proliferation assays, and (iv) chromosomal analysis. To examine the specific cell type of the somatic donor cells, we stained the cultured cells by cell-specific markers (cytokeratin 18 and vimentin) at passages 2, 5, 10, and 15 (Fig. 3). All skin cells at passages 10 and 15 were vimentin-positive, but cytokeratin-negative, demonstrating that they were fibroblast cells (Fig. 3). A majority of cells at passages 2 and 5 were vimentin-positive and cytokeratin-negative, but a small portion of cells exhibited positive staining for cytokeratin, suggesting contamination of skin epithelial cells in the early passages. This may be partly responsible for the relatively poor development of the cloned embryos derived from cells at passage 5.
Figure 3.
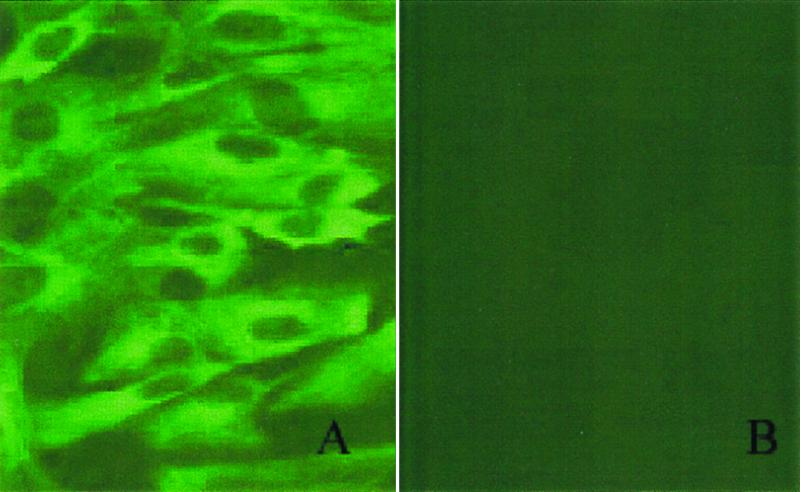
Characterization of bovine fibroblasts by immunocytochemistry. (a) Vimentin-FITC. (b) Cytokeratin-FITC-labeled bovine fibroblast cells.
Fig. 4 shows representative cell-cycle FACs histograms depicting passage 5 cells from either serum-starved vs. nonserum-starved cultures. In the nonstarved culture, 64–66% of the cells was at G0+G1 stage; upon serum starvation, the percentage of cells in G0+G1 was significantly increased to 83–85%. At passages 10 and 15, between 82% and 90% of cultured confluent somatic donor cells were in the G0+G1 phase of the cell cycle, regardless of cell passage number or serum starvation treatment.
Figure 4.

FACs histograms of passage 5 serum-starved (a) or non-serum-starved (b) bovine fibroblast cultures. Percentages of cells at each cell cycle stage are as follows: (a) G0+G1 = 84.5 ± 8.1%; S = 1.1 ± 0.8% and G2+M = 7.6 ± 2.0%. (b) G0+G1 = 64.9 ± 1.0%; S = 7.3 ± 0.6% and G2+M = 19.7 ± 0.3%.
To determine the response of the cultured cells to serum starvation, we examined cell proliferation rate by BrdUrd incorporation. Confluent cells at passages 5, 10, and 15 were subjected to serum starvation and were examined for BrdUrd incorporation daily until day 6 of starvation. Serum starvation to prevent BrdUrd incorporation was more effective for cells at passages 10 and 15 than those at passage 5 for both confluent and nonconfluent cultures (Table 4). Although a complete inhibition of BrdUrd incorporation was observed between 3 and 6 days of starvation in cells at passages 10 and 15, a portion (≈4%) of cells at passage 5 showed no response to serum starvation for at least 6 days, which again suggests possible contamination of other cell types in the early passages.
Table 4.
Cell proliferation rate (BrdUrd incorporation) of confluent cells at different passages and with or without serum starvation
| Cell passage | Serum starvation | Total cells
counted (percent of cells with BrdUrd incorporation)
|
|||||
|---|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | ||
| 5 | No | 656 (17) | 673 (11) | 453 (4) | 457 (8) | 124 (7) | 437 (5) |
| 5 | Yes | 455 (3) | 271 (4) | 369 (4) | 389 (4) | 200 (2) | 321 (4) |
| 10 | Yes | 436 (2) | 298 (3) | 379 (0) | 262 (0) | 278 (0) | 333 (0) |
| 15 | Yes | 301 (2) | 278 (1) | 269 (0) | 161 (0) | 143 (0) | 263 (0) |
We also conducted chromosomal analyses of the cultured skin fibroblast cells at various passages. A majority of the cells (70–80%) showed a normal chromosomal complement (60 chromosomes including X and Y chromosomes) regardless of passage number examined (Table 5).
Table 5.
Chromosomal analysis of donor cells at different passages
| No. of cell passage | No. (%) cells with
chromosomes of
|
No. of spreads counted | ||
|---|---|---|---|---|
| <60 | 60 | >60 | ||
| 5 | 8 (8) | 77 (75)* | 17 (17) | 102 |
| 10 | 7 (6) | 93 (82)* | 14 (12) | 114 |
| 15 | 19 (16) | 84 (72)* | 13 (11) | 116 |
*No significant differences (P > 0.05).
Discussion
In the present study, we investigated the possibility of producing normal, cloned cattle by using adult somatic donor cells after long-term culture. Our results demonstrated that long-term culture (up to 15 passages or estimated over 45 cell doublings) of skin fibroblast cells derived from an aged bull did not seem to compromise their cloning competence. This finding is significant because it offers the possibility of using gene-targeted somatic donor cells for cloning. Currently, we have cultured adult cattle skin fibroblast cells for >30 passages, and the cloning competence of these cells is being evaluated. To our knowledge, these were the first normal clones born by using adult skin fibroblast cells after long-term culture. Somatic donor cells after short-term culture (under 10 passages) have been used previously for nuclear transfer (1, 3, 4, 17, 18). However, the cell passage effect on the outcome of cloning cannot be concluded from those studies because they were conducted by different groups using different cell types and passage numbers, different protocols of cell preparation, nuclear transfer, and activation, and different culture systems. In the present study, we used the same standardized procedures in our nuclear transfer experiments, and all micromanipulations were performed by the same skilled microinjectionist. This particular setup makes direct comparison of cell passage effects more meaningful.
We observed that the developmental competence of reconstructed embryos derived from cells after long-term culture (10 and 15 passages) was actually better than those from cells of short-term culture (five passages) by two criteria used in this study: embryo development to blastocyst stage and pregnancy results. The exact explanation to this observation is unclear at this moment, but it may be partially explained by the possibility that cells at passage 5 were mixed populations of fibroblasts and other cell types, such as epithelial cells. Our ongoing research showed that mammary epithelial cells were inferior to skin fibroblast cells for cloning (C.K. and X.Y., unpublished observation). While in the cell populations of later passages, the other cell types are most likely outgrown by fibroblast cells. This explanation is supported by our cell-specific marker staining and BrdUrd examination of donor cells of different passages. Another possible explanation might be that the person manipulating cells was on a learning curve and had acquired more experience at later passages. This is unlikely because this person had extensive experience with nuclear transfer, including using adult somatic cells as nuclear donors. Further, we have shown that fresh somatic cells were not as good as those after culture for cloning (C.K. and X.Y., unpublished observation). However, it is not clear how culture improves the nuclear totipotency of donor cells. Seasonal factors are an unlikely explanation because all nuclear transfer manipulations reported in the present study were performed in the spring of 1998.
This study is also significant in the use of an aged (17-year-old) bull for cloning. Shiels et al. (7) reported that Dolly not only inherited her mother's short telomeres, but her telomere lengths were further shortened during the brief in vitro culture. This indicated that the reduced lengths of telomeres in the adult somatic donor cells may not be restored by the nuclear transfer procedure and thus raises the question of whether normal clones can be obtained from an aged animal. Although the telomere lengths of our clones remain to be determined, our data showed that the genome of skin fibroblast cells from aged animals may be completely reactivated and reprogrammed to support normal development to term. The capacity of cells from an aged adult to continue cell doubling in vitro and their effectiveness for cloning compared favorably to those reported for fetal cells (5, 6) or embryo-derived cells (12, 14). The overall full-term development of the cloned embryos from this aged bull is 15% (n = 39), which is comparable to the 14% survival reported for fetal cell clones (6) and the 10% survival from follicular cell clones of a young heifer (4). The four surviving clones from this study are approaching puberty at the preparation of this manuscript and appeared normal compared with their peers reproduced by conventional breeding. Our findings suggest that the life span of cells may be altered by reprogramming and that age of the donor animal may not correlate with the cell's doubling capacity in vitro. The fact that the donor bull is still alive merits a direct comparison of the aged donor and his clones on the possible “premature” aging of the clones. The telomeres from the clones, the donor cells, and the donor bull are being analyzed, and various physiological parameters indicative of aging are also being monitored to determine the biological ages of these clones.
Cloning by using skin cells offers the advantage of easy accessibility and noninvasiveness without animal sex or age limitations. Previously, successful cloning of adult animals has largely been limited to the use of female reproductive system cells: e.g., mammary epithelial cells (1), cumulus cells (3, 4), or oviductal epithelial cells (3). Although skin cells from a 2-week-old calf were successfully cloned, the single calf produced from that study survived only 7 weeks and died of lymphoid hypoplasia (19). In mice, tail-tip cells from a 10- to 12-week-old mouse have been used for cloning, and only one viable clone was produced from 274 embryos (20). The findings of the current study, therefore, have important implications for tissue banking and preservation of endangered species.
In the present study, we directly compared the effect of serum starvation treatment of donor cells on nuclear transfer. As shown in Table 4, serum starvation of cells at passage 5 did not improve in vitro developmental competence of the reconstructed embryos. This phenomenon has also been observed by Shiga et al. (18) using cultured adult muscle cells. The importance of serum-starvation of the donor cells to induce quiescence was emphasized in the generation of Dolly (1). However, pregnancies and live calves have been reported from fetal fibroblasts (6, 21) without serum starvation, indicating that serum starvation is not essential for the success of nuclear transfer. This is very likely attributable to the fact that more than half of an actively dividing cell population in culture is in the G0+G1 stage (ref. 6 and this study). As shown by Boquest et al. (8), an even larger portion of fetal fibroblast cells are in the G0+G1 stage in the small-cell populations. Therefore, if small cells are selected as nuclear donors during micromanipulation, virtually all cells will be at G0+G1 stage, which is in essence equivalent to using a synchronized G1 cell population. The fact that the donor cells were subjected to serum starvation or not after they reached confluency in the present study pointed to the possibility that the two populations of cells were not very different after all. This is because they both were induced to G0+G1 stage by independent mechanisms. In the present study, however, no pregnancy was established from embryos derived from cells without serum starvation. This could be attributable to the relatively small number of transfers performed in this treatment group because, in a recent separate study, we have produced several clones from nonconfluent cycling adult somatic cells (C.K. and X.Y., unpublished data).
To date, the overall cloning efficiency using somatic cells has been low, with the reported efficiency ranging from 0 to nearly 10%. This could be partially explained by the high embryonic loss during the first half of gestation. Our observation of a high rate of embryonic loss between days 60 and 120 of gestation is consistent with previous reports for fetal fibroblast clones (5, 6, 17, 22) and adult cumulus cell-derived clones (4). Although low embryonic loss and high calving rates were reported in a previous study (3) using oviductal or cumulus cells for cloning, a high neonatal mortality (50%) was noted. The exact mechanisms of early or late embryonic loss and neonatal death of clones are still not clear; however, incomplete reprogramming of the donor cell genome in the current cloning scheme may be partially responsible. Abnormal placenta development for the cloned fetus has recently been reported (23). To improve the cloning efficiency, the exact mechanism for embryonic loss and high neonatal mortality needs to be investigated systematically.
In this study, we demonstrated that adult somatic cells remained totipotent for cloning after long-term culture. This suggests the feasibility of targeted genetic manipulations such as gene knockout using cultured somatic cells before cloning to produce knockouts or other types of genetically engineered cloned animals. Cloning using site-specific genetically manipulated cells would be a valuable tool with applications in agriculture, medicine, and basic biological research.
Acknowledgments
We are grateful to Sueyoshi Meat Inspection Center and Minami Kyusyu Chikusan Kogyo Co., Ltd. for the donation of ovaries, Ohsumi Rakunougyou Kyoudou Kumiai for embryo transfer, and Kagoshima Central Animal Health Hygiene Center and Dr. Masumi Sato for pathological inspection. We also thank Drs. Hiroshi Imai, Yoshiaki Izaike, Seiya Takahashi, and Michio Harada for technical assistance and Drs. X. Cindy Tian, R. H. Foote, and T. Wagner for critical editing of this manuscript. This manuscript is a scientific contribution (no. 1919) of the Storrs Agricultural Experiment Station at the University of Connecticut.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 956.
References
- 1.Wilmut I, Schnieke A E, McWhir J, Kind A J, Campbell K H S. Nature (London) 1997;385:810–813. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 2.Wakayama T, Perry A C F, Zuccotti M, Johnson K R, Yanagimachi R. Nature (London) 1998;394:369–374. doi: 10.1038/28615. [DOI] [PubMed] [Google Scholar]
- 3.Kato Y, Tani T, Sotomaru Y, Kurokawa K, Kato J, Doguchi H, Yasue H, Tsunoda Y. Science. 1998;282:2095–2098. doi: 10.1126/science.282.5396.2095. [DOI] [PubMed] [Google Scholar]
- 4.Wells D N, Misica P M, Tervit H R. Biol Reprod. 1999;60:996–1005. doi: 10.1095/biolreprod60.4.996. [DOI] [PubMed] [Google Scholar]
- 5.Schnieke A E, Kind A J, Ritchie W A, Mycock K, Scott A R, Ritchie M, Wilmut I, Colman A, Campbell K H. Science. 1997;278:2130–2133. doi: 10.1126/science.278.5346.2130. [DOI] [PubMed] [Google Scholar]
- 6.Cibelli J B, Stice S L, Golueke P J, Kane J J, Jerry J, Backwell C, de Leon F A P. Science. 1998;280:1256–1258. doi: 10.1126/science.280.5367.1256. [DOI] [PubMed] [Google Scholar]
- 7.Shiels P, Kind A J, Campbell K H S, Waddington D, Wilmut I, Colman A, Schnieke A E. Nature (London) 1999;399:316–317. doi: 10.1038/20580. [DOI] [PubMed] [Google Scholar]
- 8.Boquest A C, Day B, Prather R S. Biol Reprod. 1999;60:1013–1019. doi: 10.1095/biolreprod60.4.1013. [DOI] [PubMed] [Google Scholar]
- 9.Gratzner H G. Science. 1982;218:474–475. doi: 10.1126/science.7123245. [DOI] [PubMed] [Google Scholar]
- 10.Ellwart J, Dormer P. Cytometry. 1985;6:513–520. doi: 10.1002/cyto.990060605. [DOI] [PubMed] [Google Scholar]
- 11.Verma R S, Babu A. Human Chromosomes. New York: Pergamon; 1989. pp. 26–27. [Google Scholar]
- 12.Campbell K H S, McWhir J, Ritchie W A, Wilmut I. Nature (London) 1996;380:64–66. doi: 10.1038/380064a0. [DOI] [PubMed] [Google Scholar]
- 13.Kubota C, Yang X, Dinnyes A, Todoroki J, Yamakuchi H, Mizoshita K, Inohae S, Tabara N. Mol Reprod Dev. 1998;51:281–286. doi: 10.1002/(SICI)1098-2795(199811)51:3<281::AID-MRD7>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 14.Wells D N, Misica P M, Day A M, Tervit H R. Biol Reprod. 1997;57:385–393. doi: 10.1095/biolreprod57.2.385. [DOI] [PubMed] [Google Scholar]
- 15.Rosenkrans C R, First N L. Theriogenology. 1991;35:266. [Google Scholar]
- 16.Inoue M, Hirano T, Watanabe T, Mizoshita K, Yamakuchi H, Nakane S, Sugimoto Y. Animal Sci Technol. 1997;68:443–449. [Google Scholar]
- 17.Baguisi A, Behboodi E, Melican D T, Pollock J S, Destrempes M M, Cammuso C, Williams J L, Nims S D, Porter C A, Midura P, et al. Nat Biotechnol. 1999;17:456–461. doi: 10.1038/8632. [DOI] [PubMed] [Google Scholar]
- 18.Shiga K, Fujita T, Hirose K, Sasae Y, Nagai T. Theriogenology. 1999;52:527–535. doi: 10.1016/S0093-691X(99)00149-1. [DOI] [PubMed] [Google Scholar]
- 19.Renard J P, Chastnat S, Chesne P, Richard C, Marchal J, Cordonnier N, Chavette P, Vingnon X. Lancet. 1999;353:1489–1491. doi: 10.1016/S0140-6736(98)12173-6. [DOI] [PubMed] [Google Scholar]
- 20.Wakayama T, Yanagimachi Y. Nat Genet. 1999;22:127–128. doi: 10.1038/9632. [DOI] [PubMed] [Google Scholar]
- 21.Vingnon X, Chesne P, LeBourhis D, Flechon J E, Heyman Y, Renard J P. C R Acad Sci. 1998;321:735–745. doi: 10.1016/s0764-4469(98)80014-0. [DOI] [PubMed] [Google Scholar]
- 22.Zakhartchenko V, Gurcova-Hills G, Stojkovic M, Schernthaner W, Prele K, Steinborn R, Muller M, Brem G, Wolf E. J Reprod Fertil. 1999;115:325–331. doi: 10.1530/jrf.0.1150325. [DOI] [PubMed] [Google Scholar]
- 23.Hill J R, Roussel A J, Cibelli J B, Edwards J F, Hooper N L, Miller M W, Thompson J A, Looney C R, Westhusin M E, Robl J M, Stice S L. Theriogenology. 1999;51:1451–1465. doi: 10.1016/s0093-691x(99)00089-8. [DOI] [PubMed] [Google Scholar]