

Abstract
Nuclear hormone receptors (NRs) are potential targets for therapeutic approaches to many clinical conditions, including cancer, diabetes, and neurological diseases. The crystal structure of the ligand binding domain of agonist-bound NRs enables the design of compounds with agonist activity. However, with the exception of the human estrogen receptor-α, the lack of antagonist-bound “inactive” receptor structures hinders the rational design of receptor antagonists. In this study, we present a strategy for designing such antagonists. We constructed a model of the inactive conformation of human retinoic acid receptor-α by using information derived from antagonist-bound estrogen receptor-α and applied a computer-based virtual screening algorithm to identify retinoic acid receptor antagonists. Thus, the currently available crystal structures of NRs may be used for the rational design of antagonists, which could lead to the development of novel drugs for a variety of diseases.
Members of the nuclear hormone receptor (NR) family are under the control of a wide variety of hormones and ligands, such as steroids, retinoids, thyroid hormone, 1,25-dihydroxy-vitamin D3, and prostanoids. Many of these NRs are potential targets for the therapy of a variety of diseases: antagonists of estrogen receptor-α (ERα) (e.g., tamoxifen) are clinically used for the treatment of breast cancer (1) whereas retinoic acid receptor (RAR) agonists and antagonists block the growth of a number of neoplastic cells including breast tumor cells (2, 3). Agonists for retinoid X receptors (RXRs) and peroxisome proliferator-activated receptor γ (PPARγ) are potential candidates for use in the treatment of cancer and diabetes (PPARγ is the receptor for the antidiabetic drug thiazolidinedione) (4–7), whereas Nurr1 ligands may be useful for treatment of Parkinson's disease (8). Thus, designing molecules that selectively activate or inhibit specific NRs is of considerable biological significance and will likely have the potential for use in important clinical applications.
The crystal structures of the ligand binding domain (LBD) of many members of the NR family recently have been solved, and the ligand-dependent structural changes involved in transcriptional activation have been clarified, enabling the structure-based design of specific agonists (9, 10). Recent studies on ERα also have shed light on the LBD structural changes mediated by NR antagonists (11, 12): ERα agonists and antagonists superimpose well and engage in a very similar network of hydrophobic and electrostatic contacts with the receptor. However, in the agonist-bound conformation, the C-terminal helix H12 sits like a lid on top of the ligand (11) (a similar observation was made for virtually all of the NR LBD structures solved so far; ref. 9). In contrast, the two ERα antagonists present a protruding arm that is not compatible with the “closed lid” conformation (11, 12) (Fig. 1A). As a result, helix H12 is pushed away from the ligand binding site and relocates in the coactivator-binding pocket of the receptor (Fig. 1B) (11). Moreover, the LxxML motif (where L is a leucine, M a methionine, and x any residue) of the ERα helix H12 mimics, and probably competes with, a LxxLL helical peptide found in a wide variety of coactivator proteins. The alignment of the LBD of various NRs (13) suggests that a common structural mechanism would be for the antagonists to induce the relocation of helix H12 into the hydrophobic coactivator-binding groove of the receptor. The observation that the progesterone receptor antagonist RU486 superimposes with the natural hormone progesterone, but presents a protruding arm similar to that of tamoxifen (14, 15) provides support for the universality of this mechanism of antagonistic activity.
Figure 1.

Modeling of the antagonist-bound structure of RAR. Agonist (white) and antagonist (cyan) superimpose in the binding pocket of ERα, but the antagonist presents an additional protruding arm that pushes helix 12 (H12, green) away (A). As a result, H12 relocates in the coactivator binding pocket of the receptor (H12, red) (B). Based on the ERα structure, helix H12 of RARγ (red) was docked to the coactivator binding pocket of the RARγ-LBD (critical hydrophobic residues are displayed in magenta) (C), and the C terminus of the protein was remodeled from its agonist-bound conformation (green) to its antagonist-bound conformation (red) (D).
Our goal in this study is to provide further evidence for this hypothesis by building a model of the antagonist-bound conformation of RARα, a NR that plays an important role in the differentiation and proliferation of a wide variety of cell types and for which only the agonist bound conformation is known (16–18), and to rationally and rapidly identify new antagonists for this receptor. We built a model of the antagonist-bound structure of RAR, based on the ERα/tamoxifen complex (12). The model was used for the virtual screening of a database of ≈150,000 available compounds, and antagonist candidates were tested in vitro. Two novel antagonists and a novel agonist were discovered. The ligands were specific for RAR, confirming the validity of our model and the potential therapeutic application of our strategy.
Materials and Methods
Building of the Model of Antagonist-Bound RAR.
A helical peptide PLIREMLENP corresponding to helix H12 of RARγ was docked into the putative coactivator binding pocket of another RARγ molecule. We hypothesized that the IxxML motif contacts the coactivator binding site of the receptor, and an automatic docking procedure was carried out toward this site, with flexible protein and peptide side chains, according to a biased probability Monte Carlo energy minimization procedure (19, 20). Two critical features of the interaction between the LBDs of NRs and their coactivators were used to carry out the docking: (i) The “charge clamp,” initially observed in the complex between SRC-1 and peroxisome proliferator-activated receptor γ (21), where a conserved glutamate (E414 in RARγ) and lysine (K246 in RARγ) at opposite ends of the hydrophobic cavity of the receptor contact the backbone of the coactivator's LxxLL box, enabled the orientation of the helical peptide. (ii) The finding that the leucines of the LxxLL motif of SRC-1 are buried in the hydrophobic cavity of the receptor determines which side of the helix faces the receptor. Here, the isoleucine, methionine, and leucine of the IxxML motif were buried in the binding site of RARγ. Loose distance restraints were set between the charge clamp of the receptor (21) (i.e., E414 and K246) and backbone nitrogen and oxygens of the peptide (nitrogen of the isoleucine on one end, and carbonyl of the methionine, leucine, and asparagine in the MLEN motifs, respectively). The energy of the complex was minimized in the internal coordinate space by using the modified ECEPP/3 potentials. The subset of the variables minimized with the icm method (19, 20, 22, 23) included the side chains of the receptor, six positional variables of the helix, and the side-chain torsion angles of the helix.
After the icm docking procedure, we built a model of antagonist-bound RARγ. The structure of the receptor was kept rigid but for the side chains and backbone of the 25 C-terminal residues (corresponding to the last 10 residues of helix H11, the loop from H11 to H12, and H12), and for the side chains of the putative coactivator binding site (within 6 Å of the previously docked helical peptide). Tethers then were set between the C terminus of the receptor and the corresponding residues of the docked helical peptide, and the energy of the receptor was minimized by a stochastic global energy optimization in the internal coordinate space (22, 23).
The last step was, from the resulting model of antagonist-bound RARγ, to derive the structure of the antagonist-binding pocket of RARα: the three nonidentical residues in the vicinity of the binding pocket (A234, M272, and A397) were changed to the RARα isoform (S234, I272, and V397, respectively) and energy-minimized. Another possibility would have been to introduce the mutations before remodeling the C terminus of the receptor. We preferred to proceed as described here to preserve the integrity of the receptor during the critical remodeling of the C-terminal end.
Receptor-Ligand Docking.
An initial docking was carried out with a grid potential representation of the receptor and flexible ligand (24). The resulting conformation then was optimized with a full atom representation of the receptor, flexible receptor side chains, and flexible ligand, by an icm stochastic global optimization algorithm as implemented in the MolSoft icm 2.7 program (23, 24).
Screening of a Virtual Library of Compounds.
The flexible-ligand/grid-potential-receptor docking algorithm (23, 24) was carried out automatically on the Available Chemicals Directory library of 153,000 available chemical compounds (MDL Information Systems, San Leandro, CA). The screening took less than a month on 10 194-MHz IP25 processors. Each compound was assigned a score, according to its fit with the receptor, which took into account continuum as well as discreet electrostatics, hydrophobicity, and entropy parameters (25). The distribution of the compounds according to their score is presented at http://abagyan.scripps.edu/PNAS/MS2000/. All compounds scoring better (i.e., lower) than −32 were screened further for the number of hydrogen bonds engaged with the receptor. The 134 compounds that made at least two hydrogen bonds with the receptor were preselected. The 609 compounds scoring better than −37 also were preselected, regardless of the hydrogen bonding network. This preselection pool then was further minimized with a full atom representation of the receptor, as described above. The quality of the fit of the 500 best-scoring compounds then was visually estimated, and 32 compounds were selected for biological testing. These compounds are not necessarily the ones with the best final scores, but the ones we thought, after careful visual inspection, presented the best characteristics, such as Van der Waals fit or hydrogen bonding (see http://abagyan.scripps.edu/PNAS/MS2000/).
It occurred to us that during the selection by the MolSoft virtual screening procedure, it was preferable to set up an initial cut-off value poorly selective (i.e., −32) to recover a large pool of preselected compounds and to apply to this pool subsequent screens specific for the system, such as number of hydrogen bonds (used here) or presence of a hydrogen bond acceptor (for example) at a specific point of space. As a result, we derived the value −32 as a good initial threshold (this value generates an initial pool of 3,000–4,000 compounds).
Biological Activity of the Antagonist and Agonist Candidates.
HeLa cells were transfected by calcium phosphate precipitation using 1 μg of the Gal4-responsive chloramphenicol acetyltransferase (CAT) reporter pMC110 and 1 μg of Gal4-hRARα-LBD or 1 μg of Gal4-hRXRβ-LBD. Studies also were performed with the three wild-type hRAR isoforms (hRARα, hRARβ, and hRARγ) by using a ΔMTV-IR-CAT reporter as described (26, 27). Cell cultures were supplemented with indicated ligands immediately after addition of the calcium phosphate/DNA precipitate. Media and ligands were replaced after 24 h, and cells were harvested and essayed for CAT activity 24 h later.
Results
Modeling of the RAR Antagonist Binding Pocket.
The x-ray structure of RARγ bound to the agonist all-trans RA is available (18); however, the conformation of the receptor bound to an antagonist is not known. We used the observations made from the structure of ERα bound to an agonist, 17β-estradiol (11), and two antagonists, tamoxifen and raloxifene (11, 12), to build a model of antagonist-bound RAR (Fig. 1 A and B). We docked helix H12 of RARγ into the putative coactivator binding pocket of the receptor as described (27) (see Materials and Methods for details) (Fig. 1C) and remodeled the 25 C-terminal residues, starting near the end of helix 11, through an extensive global energy minimization procedure (Fig. 1D).
Docking of Known RAR Antagonists into the Modeled Receptor.
A few RAR antagonists have been described in the literature; and several of them are serious candidates for cancer therapy (2, 28). A well-characterized ligand is AGN193109, which inhibits the three RAR isoforms at nanomolar concentrations (29). Another very potent antagonist is MX781, which is effective against ERα-positive and -negative breast cancer cells, with no apparent toxicity (2). The activity of these two ligands has been presented in detail, but no structural information has been reported on their mode of interaction with the receptor. We built a model of RARγ complexed either with AGN193109 or MX781, by using our flexible docking algorithm (24) (Fig. 2 A and B). In both cases, the antagonist superimposed with the agonist all-trans RA. As observed for ERα, the antagonists also presented a protruding arm, which was absent in RAR agonists. Very importantly, this protruding arm coincided exactly with the single opening in the ligand binding pocket of our modeled receptor, generated by the displacement of helix H12 (Fig. 2 A and B), and made stabilizing hydrophobic contacts with the protein. It is very unlikely that this perfect fit, observed for both antagonists, was fortuitous. On the contrary, this feature mimics the inactivation mechanism revealed by the crystal structure of ERα bound to tamoxifen and raloxifene. Therefore, our docking results of AGN193109 and MX781 very strongly suggest that: (i) the structural mechanisms of antagonist activity for ERα are shared by other NRs, and (ii) our model of the RAR antagonist binding pocket could be used to design novel antagonists.
Figure 2.
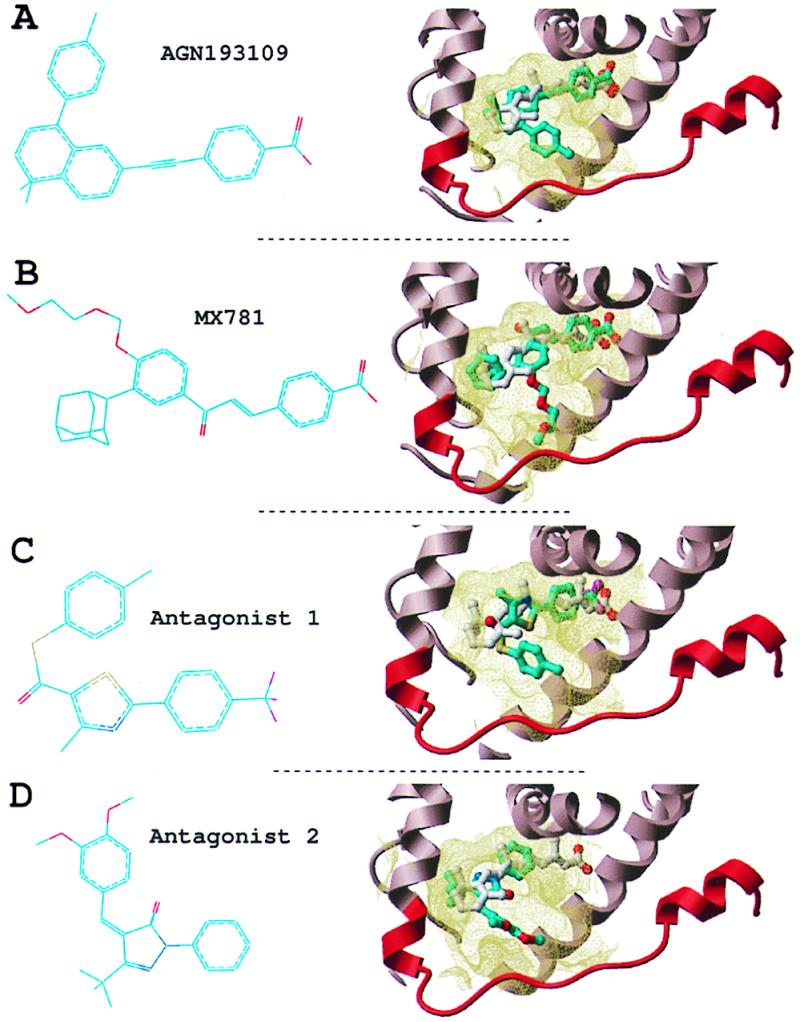
RAR antagonists. Two known antagonists (A and B) and two novel antagonists (C and D). (Left) Chemical structure. (Right) Conformation docked into the receptor (part of the receptor is displayed as a ribbon representation, and the binding pocket boundary is displayed in yellow). Cyan, carbons; red, oxygen; blue, nitrogen; magenta, fluorine; yellow, sulfur. Hydrogens are not represented for clarity.
Screening of a Virtual Library and Discovery of Novel RAR Antagonists.
High throughput functional screening currently is the most used method for the discovery of receptor-specific ligands. Although efficient, it requires the physical availability and management of hundreds of thousands of chemical compounds. In the present work, we used a virtual library composed of the predicted structure of more than 150,000 available compounds (see Materials and Methods). Each compound was automatically docked in a grid representation of the modeled RARα antagonist binding pocket. Five grid potentials carried information on the shape, hydrophobicity, electrostatics, and hydrogen-bonding availability of the receptor, and enabled a rapid docking simulation (24, 25). RARα was selected over the other two isoforms (RARβ and RARγ) because recent data suggests it could be a medically more relevant target (28). After an automatic selection procedure with flexible ligands, and optimization of the selected candidates with flexible protein side chains (see Materials and Methods for details), 32 compounds were considered as potential antagonists of RARα and ordered.
To test these compounds in vitro, HeLa cells were transfected with a Gal4-hRARα-LBD expression vector and a Gal4-CAT reporter gene (26). Studies also were performed with the three wild-type hRAR isoforms and a ΔMTV-IR-CAT reporter (26, 27). These gave similar results as those found with Gal4-hRARα-LBD (data not shown). The cells were incubated with all-trans RA to stimulate CAT activity, and the effect of each antagonist candidate on inhibiting CAT stimulation by all-trans RA was examined. Possible toxicity of the compounds was deduced from the amount of cellular protein extract after 2 days of incubation. Two antagonist candidates inhibited CAT activity by 55% and 33% at 20 μM with no apparent toxicity (Fig. 3). The Gal4-hRARα activity illustrated in Fig. 3 was equivalent for the other two RAR isoforms (data not shown). No inhibition was observed when CAT expression was under the control of a Gal4-mRXRβ-LBD fusion construct, indicating that: (i) the antagonists are specific for RAR, and (ii) the inhibition is caused by an interaction with the Gal4-RAR-LBD fusion protein and does not result from some nonspecific effect on CAT activity (data not shown).
Figure 3.

Functional assays of the novel antagonists. HeLa cells were transfected with a Gal4-hRARα-LBD expression vector and a Gal4-CAT reporter gene (results were similar in studies using the three hRAR isoforms). The cells were incubated with 5 nM all-trans RA to stimulate CAT activity, and the effect of each antagonist on inhibiting CAT was examined at 2 and 20 μM concentration (the known antagonist RO-41–5253 was used as a positive control).
The two RAR antagonists dock into the ligand binding pocket of the receptor (Figs. 2 C and D and 4). As observed for AGN193109 and MX781, they fit in the same binding pocket as the natural agonist all-trans RA, but present an additional arm, which protrudes out of the pocket. Antagonist 1 has a tri-fluoro group where the retinoid receptor ligands usually carry a carboxylate group (in antagonist 2, the corresponding domain is truncated). In our model, antagonist 2 engages in a hydrogen bond with Ser-234 of the hRARα (Fig. 4B). However, the S234A mutation in the other two isoforms does not alter the ligand antagonist activity, suggesting that this hydrogen bond is not essential for the interaction. An obvious way to increase the affinity of these antagonists would be to substitute the tri-fluoro group by a carboxylate in antagonist 1 or elongate and add a carboxylate to antagonist 2, which would result in more stabilizing interactions with two conserved arginines of the receptor. However, the purpose of this work is to provide evidence that the rational design of antagonists from the model of the inactive receptor is feasible and not to optimize the affinity of the compounds. The in vitro functional assays provide evidence that our modeling scheme is relevant and can be used to design novel antagonists of NRs.
Figure 4.

Novel RAR antagonists. (A and B) Stereo representation of antagonists 1 and 2 docked into the binding site of the receptor. The ligands make extensive hydrophobic interactions with residues from helix 3, helix 5, and helix 11. Antagonist 2 (B) is engaged in an additional hydrogen bond with Ser-234 of helix 3 and contacts the remodeled C terminus (red) at Pro-405. (C and D) The fit of antagonists 1 and 2 into the receptor binding pocket is shown.
We applied the same strategy to discover agonists, by using the crystal structure of the active conformation of RARγ (18), and could discover three novel agonists 10–25% active at 200 nM and fully active at 20 μM, of 30 compounds tested (data not shown).
Screening of a Database of Known Ligands.
To assess the quality of our setup of the icm screening algorithm (23), we built a small virtual database made up of antagonists and agonists for different members of the NR family (Table 1). We screened this database with our model of antagonist-bound RAR, as we did for the Available Chemicals Directory database. The screening was repeated four times, to test the reproducibility of our method. Table 1 shows that for each ligand the score varies a lot from one screening to the other. This finding reflects the generation of different ligand conformations from one docking simulation to another (data not shown) and represents the limitation of our method, as discussed below.
Table 1.
Control screening of known NR ligands
| Ligand | Activity | Score 1 | Score 2 | Score 3 | Score 4 | Selected | Binding | References |
|---|---|---|---|---|---|---|---|---|
| First series | ||||||||
| AGN193836 | RAR_agonist | −19.9 | −9.04 | −20.6 | −19.7 | − | + | (33) |
| ATRA | RAR pan-agonist | −46.4 | −41 | −41.7 | −41. | + | + | (34) |
| Ro415253 | RAR_antagonist | −25.5 | −22. | −28.3 | −28.6 | − | + | (28) |
| MX781 | RAR antagonist | −28. | −23.9 | −27.1 | −36.4 | + | + | (2) |
| CD2366 | RAR pan-antagonist | −28.5 | −23.3 | −30.9 | −32.3 | + | + | (34) |
| Targretin | RXR pan-agonist | −17.9 | −18.1 | −19.1 | −18.6 | − | − | (4) |
| SR11203 | RXR pan-agonist | −27.5 | −27. | −27. | −27.2 | − | − | (34) |
| Tamoxifen | ER modulator | −29.3 | −27.5 | −29.8 | −28.3 | − | − | (23) |
| Raloxifene | ER modulator | −23.4 | −20.8 | −26.7 | −34.6 | + | − | (22) |
| RU486 | Progest Rec antag. | −21.2 | −21.3 | −21.4 | −21.3 | − | − | (25) |
| 9cisRA | RAR/RXR agonist | −32.5 | −32.6 | −32.9 | −16.9 | + | + | (34) |
| AGN193109 | RAR pan-antagonist | −39.2 | −56. | −57.4 | −39.4 | + | + | (29) |
| AGNpartia | RAR partial agonist | −54.4 | −54.3 | −49.5 | −29.1 | + | + | (29) |
| Am580 | RAR_agonist | −34.2 | −34.4 | −34.8 | −34.5 | + | + | (34) |
| EM652 | ER antagonist | −27. | −27.4 | −21.7 | −28.8 | − | − | (35) |
| Antagonist 1 | Novel RAR antag. | −28.5 | −28.1 | −28.7 | −28.8 | − | + | (35) |
| Antagonist 2 | Novel RAR antag. | −27.6 | −38.9 | −40.2 | −26.3 | + | + | (35) |
| Second series | ||||||||
| AGN193836 | RAR_agonist | −37.2 | −36.5 | −36.7 | −35.3 | + | + | (33) |
| ATRA | RAR pan-agonist | −51.7 | −52.6 | −51.8 | −52.0 | + | + | (34) |
| Ro415253 | RAR_antagonist | −28.9 | −24.4 | −39.0 | −46.6 | + | + | (28) |
| MX781 | RAR antagonist | −45.3 | −48.0 | −40.2 | −45.6 | + | + | (2) |
| CD2366 | RAR pan-antagonist | −50.7 | −50.8 | −29.3 | −29.3 | + | + | (34) |
| Targretin | RXR pan-agonist | −25.4 | −23.0 | −22.2 | −31.0 | − | − | (4) |
| SR11203 | RXR pan-agonist | −28.2 | −22.7 | −22.1 | −27.5 | − | − | (34) |
| Tamoxifen | ER modulator | −26.4 | −24.6 | −30.3 | −23.4 | − | − | (23) |
| Raloxifene | ER modulator | −15.6 | −23.7 | −18.4 | −17.4 | − | − | (22) |
| RU486 | Progest Rec antag. | −21.4 | −20.6 | −20.3 | −20.1 | − | − | (25) |
| 9cisRA | RAR/RXR agonist | −38.8 | −39.5 | −33.5 | −38.7 | + | + | (34) |
| AGN193109 | RAR pan-antagonist | −55.1 | −55.5 | −41.2 | −54.8 | + | + | (29) |
| AGNpartia | RAR partial agonist | −61.4 | −61.3 | −61.4 | −61.0 | + | + | (29) |
| Am580 | RAR_agonist | −46.6 | −47.2 | −46.6 | −46.5 | + | + | (34) |
| EM652 | ER antagonist | −26.3 | −23.1 | −23.7 | −27.3 | − | − | (35) |
| Antagonist 1 | Novel RAR antag. | −32.1 | −32.1 | −31.7 | −31.6 | + | + | (35) |
| Antagonist 2 | Novel RAR antag. | −33.3 | −29.7 | −33.8 | −33.8 | + | + | (35) |
First series: A similar screening as the one performed on the ACD database was carried out four times on a small database made of known RAR antagonists, agonists, as well as ligands for other NRs and the two novel RAR antagonists. The ligands that met at least once with the criteria for selection used during the ACD screening are listed as Selected. The ligands that are experimentally binding to RAR are listed as Binding. Second series: Screening of known ligands after adjustment of the receptor's binding pocket conformation. The RAR antagonist Ro415253 was docked into our model of antagonist-bound RAR with flexible receptor side chains and ligand. The resulting receptor conformation was used for a novel screening.
Table 1 lists as “selected” the ligands that met with the criteria for preselection and final inspection during the Available Chemicals Directory screening (i.e., score better than −37 or score better than −32 and at least two hydrogen bonds with the receptor; see Materials and Methods for details). Seven of the nine known RAR ligands (i.e., ≈80%) and one of the six non-RAR ligands (i.e., ≈16%) were selected. The fact that RAR agonists, as well as antagonists, produced good scores was expected, because the binding pocket used for the screening is equivalent to the agonist binding pocket, with an additional opening generated by the remodeling of the C terminus of the receptor. The two false negatives, AGN193836 and Ro415253, were missed because of steric clashes, as discussed below. Antagonist 1 was not found either, reflecting its rather low affinity for the receptor. It is important to underline here that we do not expect to detect all of the true binders. The algorithm was rather designed to minimize the number of false positives, which correlates with the number of unnecessary in vitro experiments (25).
In that respect, the presence of one false positive of six nonbinders could be alarming, because such a ratio would represent about 25,000 false positives of a database of 150,000 compounds. However, the binding pockets of the NRs represented in this database are close in size and shape; as a result, the database used for this benchmark was composed of molecules presenting strong similarities with RAR ligands. Therefore, we believe this ratio is not representative. The fact that we needed to test only 32 molecules to discover three novel RAR ligands confirms this assumption.
Next, we tried to address why some ligands, such as Ro415253, were repeatedly missed by our screening algorithm (Ro415253 was still not selected after 10 docking simulations, data not shown). We hypothesized that the ligand could not fit into the potential maps generated from our model and carried out a docking simulation with a full atom representation of the receptor, according to a Monte Carlo energy minimization of the complex, with both flexible ligand and flexible receptor side chains (24). This docking simulation produced a solution were the ligand fits into the binding pocket; the core of the ligand (from the carboxylate to the internal sulfone) superimposes with agonists such as all-trans RA, whereas the alkyl arm sticks out of the pocket, as previously described for the other antagonists (data not shown). The conformation of several receptor side chains was modified during the docking simulation, to accommodate the size of the ligand, and this solution would not have been found with rigid side chains. This finding suggests that Ro415253 could not fit into the potential maps generated from the original receptor conformation, which we used for the screening. We generated a new series of potential maps from the optimized receptor structure and screened the small database of known ligands with these maps four times as above (Table 1). The score assigned to Ro415253 was twice lower (i.e. better) than the threshold. Surprisingly, this new series of potential maps totally eliminated the presence of both false positive and false negative (all RAR ligands and only RAR ligands were selected).
Discussion
In this study, we presented a strategy for the discovery of antagonists, as well as agonists, for NRs, which are very important targets for drug design. An important aspect of our approach was to exclude any preconceived pharmacophore bias from our database screening. Most drug design strategies impose chemical constraints on the selected molecule to conserve the functional groups believed to be most important in existing ligands, preventing the discovery of novel ligand types. In the present work, we avoided pharmacophore constraints thanks to a robust flexible docking program and scoring function: the only filters used for screening were a good fit with the receptor and reasonable bioavailability parameters (30). As a result, we discovered novel original ligands that could be further optimized into potent RAR-selective antagonists and agonists.
A limitation of our method, which leaves room for further improvement, is that a compromise must be made between the time allocated for each ligand (less than 2 min on one processor here) and the reliability of the sampling of the conformational space. Indeed, Table 1 shows that four runs for each ligand are necessary to minimize efficiently missed hits (the remaining missed positives were not selected because of inappropriate receptor side-chain conformations and not because of an insufficient sampling). Improvement of the computing power, the docking algorithm, and the scoring function all could result in a more robust virtual database screening.
Another drawback is that the conformation of the receptor is not necessarily unique, but can vary from one ligand to another. As a result, a ligand that fits in receptor conformation A will never be found if receptor conformation B is used for the screening. The case of Ro415253 illustrates this issue well: this known antagonist was never selected, even after 10 trials, because the binding pocket used for the screening was too narrow. The potential maps used for the screening have a smoother van der Waals profile than the atomic representation of the receptor; as a result, the maps are more tolerant regarding steric clashes with the ligand. However, the degree of tolerance is limited and cannot accommodate important conformational changes of the receptor side chains (or backbone, obviously). When new potential maps generated from a model of RAR bound to Ro415253 were used for screening, the three RAR ligands missing from the first screening were selected (Table 1). This finding confirms that the initial conformation of the receptor prevented the selection of, or reduced the chances of selecting, some known RAR ligands. The false positive raloxifene (Table 1) was making extensive van der Waals interactions with the narrow RAR binding pocket, which compensated for the lack of stabilizing electrostatic interactions. However, in the new conformation of the receptor (Table 1), the binding pocket is wider and the fit not as tight. As a result, raloxifene was not selected. This observation emphasizes, if necessary, that virtual screening is very sensitive to the conformation of the receptor.
In that respect, it is interesting to note that the topology of the remodeled C-terminal loop is probably not unique, and that the conformation used to generate the receptor potential maps was one among many others. It is therefore legitimate to wonder whether novel antagonists could not be discovered as efficiently from a structure of the receptor where the C terminus, instead of being remodeled, was truncated. This brings up a fundamental question: is the role of antagonists only to antagonize the “closed lid” conformation where helix H12 sits on top of the ligand binding pocket, or are they also stabilizing the inactive conformation of the receptor? It is important to keep it mind that the C-terminal tail of RAR (as well as for other NRs) is a very dynamic entity when no ligand is bound to the receptor and probably oscillates between active and inactive conformations. Once bound in the ligand binding pocket, agonists contact the H12 helix and lock the receptor in its coactivator-binding conformation. Likewise, it is reasonable to speculate that antagonists would contact the C-terminal tail of the receptor and stabilize the inactive state. However, it is probable that the conformation of the receptor varies from one ligand to another; indeed, recent results on ERα show that different ligands induce distinct conformational change of the receptor (31). We used the crystal structure of ERα bound to tamoxifen to build our model of inactive RAR and could find two specific antagonists, one of which contacts the remodeled tail of the receptor. Although the conformation we used for the C-terminal tail was probably not the only possible one, we believe that its presence was important to bias the screening toward compounds that actually do contact the flexible arm of RAR, as well as to impose a reasonable boundary on the antagonist binding pocket, and prevent the ligands from drifting out of the pocket during the docking simulations.
An important point was to demonstrate that we could discover novel antagonists for a NR other than ERα, provided that the structure of the agonist-bound active form of the protein was known. Rational design of ligands from a model of a receptor is thought by many to yield very low success rates. The present study demonstrates that this strategy can be successfully undertaken with appropriate biological systems and robust modeling tools. Moreover, targeting models of diverse members of the NR family could be further justified by the wealth of structural and sequence information (9, 13), as well as the finding that NR family members share similar mechanisms of transcriptional activation and inhibition (9).
The recent publication of the crystal structures of medically relevant receptor targets, such as peroxisome proliferator-activated receptor γ (21), RAR (18), RXR (32), ERα (11), or progesterone receptor (15), has created an exciting opportunity for the discovery of novel ligands. This study demonstrates that the rational design of both antagonists and agonists, by using computer-generated models based on these structures, is possible.
Acknowledgments
We thank M. Totrov for helpful discussion. We thank MolSoft LLC for making the latest version of the icm program available for this research project. This research was supported by Department of Defense Grant DAMD179818133, a Kaplan Comprehensive Cancer Center grant from New York University Medical Center, National Institutes of Health Grant GM5541801, and Department of Energy Grant DEFG0296ER62268 to M.S. and R.A., and National Institutes of Health Grant DK16636 and New York State Empire Award C015710 to H.H.S.
Abbreviations
- NR
nuclear hormone receptor
- RAR
retinoic acid receptor
- ER
estrogen receptor
- LBD
ligand binding domain
- RXR
retinoid X receptor
- CAT
chloramphenicol acetyltransferase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Dees E C, Kennedy M J. Curr Opin Oncol. 1998;10:517–522. doi: 10.1097/00001622-199811000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Fanjul A N, Piedrafita F J, Al-Shamma H, Pfahl M. Cancer Res. 1998;58:4607–4610. [PubMed] [Google Scholar]
- 3.Shiohara M, Dawson M I, Hobbs P D, Sawai N, Higuchi T, Koike K, Komiyama A, Koeffler H P. Blood. 1999;93:2057–2066. [PubMed] [Google Scholar]
- 4.Bischoff E D, Moon T E, Heyman R A, Lamph W W. Cancer Res. 1998;58:479–484. [PubMed] [Google Scholar]
- 5.Mukherjee R, Davies P J, Paterniti J R, Jr, Heyman R A. Nature (London) 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 6.Spiegelman B M. Diabetes. 1998;47:507–514. doi: 10.2337/diabetes.47.4.507. [DOI] [PubMed] [Google Scholar]
- 7.Elstner E, Muller C, Koshizuka K, Williamson E A, Park D, Asou H, Shintaku P, Said J W, Heber D, Koeffler H P. Proc Natl Acad Sci USA. 1998;95:8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zetterstrom R H, Solomin L, Jansson L, Hoffer B J, Olson L, Perlmann T. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- 9.Moras D, Gronemeyer H. Curr Opin Cell Biol. 1998;10:384–391. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 10.Klaholz B P, Renaud J P, Mitschler A, Zusi C, Chambon P, Gronemeyer H, Moras D. Nat Struct Biol. 1998;5:199–202. doi: 10.1038/nsb0398-199. [DOI] [PubMed] [Google Scholar]
- 11.Brzozowski A M, Pike A C, Dauter Z, Hubbard R E, Bonn T, Engstrom O, Ohman L, Greene G L, Gustafsson J A, Carlquist M. Nature (London) 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 12.Shiau A K, Barstad D, Loria P M, Cheng L, Kushner P J, Agard D A, Greene G L. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 13.Wurtz J M, Bourguet W, Renaud J P, Vivat V, Chambon P, Moras D, Gronemeyer H. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 14.Cadepond F, Ulmann A, Baulieu E E. Annu Rev Med. 1997;48:129–156. doi: 10.1146/annurev.med.48.1.129. [DOI] [PubMed] [Google Scholar]
- 15.Williams S P, Sigler P B. Nature (London) 1998;393:392–396. doi: 10.1038/30775. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald P, Teng M, Chandraratna R A, Heyman R A, Allegretto E A. Cancer Res. 1997;57:2642–2650. [PubMed] [Google Scholar]
- 17.Giannini G, Dawson M I, Zhang X, Thiele C J. J Biol Chem. 1997;272:26693–26701. doi: 10.1074/jbc.272.42.26693. [DOI] [PubMed] [Google Scholar]
- 18.Renaud J P, Rochel N, Ruff M, Vivat V, Chambon P, Gronemeyer H, Moras D. Nature (London) 1995;378:681–689. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 19.Totrov M, Abagyan R. Nat Struct Biol. 1994;1:259–263. doi: 10.1038/nsb0494-259. [DOI] [PubMed] [Google Scholar]
- 20.Strynadka N C, Eisenstein M, Katchalski-Katzir E, Shoichet B K, Kuntz I D, Abagyan R, Totrov M, Janin J, Cherfils J, Zimmerman F, et al. Nat Struct Biol. 1996;3:233–239. doi: 10.1038/nsb0396-233. [DOI] [PubMed] [Google Scholar]
- 21.Nolte R T, Wisely G B, Westin S, Cobb J E, Lambert M H, Kurokawa R, Rosenfeld M G, Willson T M, Glass C K, Milburn M V. Nature (London) 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 22.Abagyan R, Totrov M. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 23.MolSoft. icm 2.7 Program Manual. San Diego: MolSoft; 1998. [Google Scholar]
- 24.Totrov M, Abagyan R. Proteins Suppl. 1997;1:215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 25.Totrov M, Abagyan R. Proceedings of the Third Annual International Conference on Computational Molecular Biology, April 1999, Lyon, France. New York: ACM Press; 1999. pp. 37–38. [Google Scholar]
- 26.Qi J S, Desai-Yajnik V, Greene M E, Raaka B M, Samuels H H. Mol Cell Biol. 1995;15:1817–1825. doi: 10.1128/mcb.15.3.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li D, Desai-Yajnik V, Lo E, Schapira M, Abagyan R, Samuels H H. Mol Cell Biol. 1999;19:7191–7202. doi: 10.1128/mcb.19.10.7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toma S, Isnardi L, Raffo P, Riccardi L, Dastoli G, Apfel C, LeMotte P, Bollag W. Int J Cancer. 1998;78:86–94. doi: 10.1002/(sici)1097-0215(19980925)78:1<86::aid-ijc14>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 29.Chandraratna R A. J Am Acad Dermatol. 1998;39:S149–S152. doi: 10.1016/s0190-9622(98)70313-5. [DOI] [PubMed] [Google Scholar]
- 30.Lipinski C A, Lombard F, Dominy B W, Feeney P J. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 31.Paige L A, Christensen D J, Gron H, Norris J D, Gottlin E B, Padilla K M, Chang C Y, Ballas L M, Hamilton P T, McDonnell D P, Fowlkes D M. Proc Natl Acad Sci USA. 1999;96:3999–4004. doi: 10.1073/pnas.96.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Nature (London) 1995;375:377–382. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 33.Teng M, Duong T T, Klein E S, Pino M E, Chandraratna R A. J Med Chem. 1996;39:3035–3038. doi: 10.1021/jm9603532. [DOI] [PubMed] [Google Scholar]
- 34.Sun S Y, Yue P, Dawson M I, Shroot B, Michel S, Lamph W W, Heyman R A, Teng M, Chandraratna R A, Shudo K, et al. Cancer Res. 1997;57:4931–4939. [PubMed] [Google Scholar]
- 35.Tremblay A, Tremblay G B, Labrie C, Labrie F, Giguere V. Endocrinology. 1998;139:111–118. doi: 10.1210/endo.139.1.5702. [DOI] [PubMed] [Google Scholar]