

Abstract
Congenital diaphragmatic hernia (CDH) is a relatively common birth defect with a high mortality. Although little is known about its etiology, there is increasing evidence for a strong genetic contribution. Both numerical and structural chromosomal abnormalities have been described in patients with CDH. Partial trisomy 11q and partial trisomy 22 associated with the common t(11;22) has been reported in several cases of CDH. It has been assumed that the diaphragmatic defect seen in these individuals was primarily due to duplication of material from chromosome 22q11. However, in this report we describe a family with a t(11;12) in which one of two brothers with partial trisomy 11q has a left sided posterolateral CDH. This is the second case of CDH in partial trisomy 11q due to an unbalanced translocation other than t(11;22). Using array-based comparative genomic hybridization and fluorescent in situ hybridization, we mapped the breakpoints in both brothers and their mother who is a balanced translocation carrier. Our results suggest that duplication of one or more genes on a ~19 Mb region of 11q23.3-qter predisposes to the development of CDH. These effects may be the primary cause of CDH in individuals t(11;22) or may be additive to effects from the duplication of chromosome 22 material. We also conclude that the partial trisomy 11q syndrome has a variable phenotype and that CDH should be added to the spectrum of anomalies that can be present in this syndrome.
Keywords: congenital diaphragmatic hernia, partial trisomy 11q syndrome, molecular cytogenetic analysis, comparative genomic hybridization
INTRODUCTION
Congenital diaphragmatic hernia (CDH, [OMIM142340]) is a relatively common anomaly with an incidence of 1 in 3,000 births [Torfs et al., 1992]. CDH is characterized by a variable defect in the diaphragm, lung hypoplasia, and postnatal pulmonary hypertension that is often resistant to therapy [Torfs et al., 1992; Beresford and Shaw, 2000]. It has a high mortality. CDH can occur as an isolated defect, in combination with multiple congenital anomalies or as part of a defined syndrome. There is increasing evidence for a genetic cause of CDH. Trisomy 18 and tetrasomy 12p (Pallister–Killian) are the most common identifiable chromosomal causes of CDH [Pober et al., 2006]. Structural chromosomal abnormalities encompassing almost every chromosome have been described [Lurie, 2003]. Deletions/duplications of some chromosomal regions have been described in multiple individuals with CDH. Copy number alterations of one or more genes in these regions are likely to predispose to the development of CDH. We have recently defined a minimally deleted region for CDH on chromosome 15q26 [Klaassens et al., 2005].
A second structural chromosomal anomaly repeatedly implicated in CDH is the t(11;22). The only viable type of unbalanced segregation of this translocation is 3:1 meiotic segregation resulting in a 47,XX or XY,+der(22)t(11;22)(q23.3;q11.2), karyotype which results in trisomy for a portion of 11q and 22 [Fraccaro et al., 1980; Lurie, 2003]. The diaphragmatic defects associated with this abnormality have been attributed primarily to duplication of material from chromosome 22. This was based on several reports of diaphragmatic hernia in individuals with trisomy 22 and the existence of only a single report of CDH associated with partial duplication 11q due to a t(11;13) resulting in 47,XY,+der(13)t(11;13)(q21;q14) [Park et al., 1993].
The phenotype accompanying t(11;22) was first described as “trisomy 22” but this constellation of findings is now known as “partial trisomy 11q syndrome” since most of the features are thought to be caused by the duplication of genes on 11q [Francke et al., 1977; Pihko et al., 1981]. Translocations of 11q with autosomes other than 22 have also been described [Francke et al., 1977; de France et al., 1984; Vianello et al., 1986; Van Opstal et al., 1993; Zhao et al., 2003]. These translocations have been associated with loss or gain of material from the other homologous autosome. Chromosomal breaks on chromosome 11q occur most often at band q23.3, that contains a fragile site that predisposes to breaks and recombination between chromosomes [Pfeiffer and Schutz, 1993].
In this report, we describe two siblings, born to a mother with a t(11;12). This family came to our attention when the youngest child was diagnosed prenatally with CDH. Both siblings were found to carry a partial duplication of chromosome 11q and a partial deletion of 12q. This is the second time that CDH has been reported in an individual with partial trisomy 11q due to an unbalanced translocation other than t(11;22) [Park et al., 1993]. Using complementary cytogenetic techniques, we mapped the breakpoints in three family members carrying either balanced or unbalanced forms of this translocation. Our results suggest that duplication of one or more genes on an ~19 Mb region of 11q23.3-qter is likely to predispose to the development of CDH. These effects may be the primary cause of CDH in individuals with t(11;22) or may be additive to effects from the duplication of chromosome 22 material. We also conclude that partial trisomy 11q syndrome has a variable phenotype which may include CDH.
CLINICAL REPORTS
Patient 1
Patient 1 (IV:2, Fig. 1) is a male child born at term to a 40-year-old G2P0 mother with a history of a single spontaneous abortion. After birth, multiple congenital anomalies were noted including a severe Pierre–Robin sequence (cleft palate, micrognathia, and glossoptosis) requiring tracheostomy after closure of the cleft palate. There were mild dysmorphic features (high anterior hairline, hypotelorism, metopic ridge, upslanting of the eyes with telecanthic folds, short palpebral fissures, broad, flattened upturned nose, prominent philtrum, open mouth appearance), prominent heels, and a micropenis. Because of these abnormalities and marked delay in psycho- and motor-development, he was referred to the clinical genetics department for evaluation at the age of 1 year and 3 months. Cytogenetic analysis revealed an unbalanced translocation: 46,XY,der(12)t(11;12)(q23;q24).
Fig. 1.
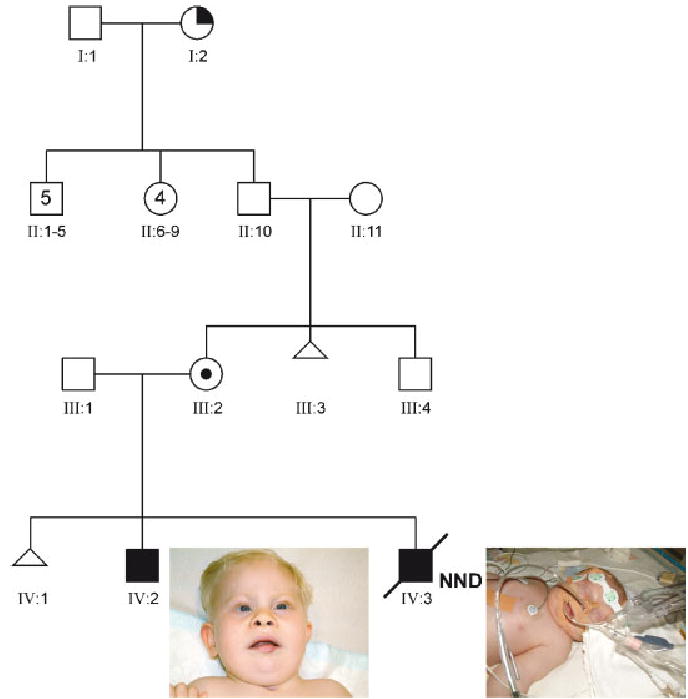
Pedigree of family with t(11;12). III:2—Healthy carrier mother; IV:1—spontaneous abortion; IV:2—Patient 1, note the dysmorphic features such as short nose, micrognathia, and prominent upper lip; IV:3—Patient 2, dysmorphic features difficult to see due to presence of tubes and facial edema. Pictures of Patient 1 (IV:3) and 2 (IV:2) are shown with parental permission. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Re-evaluation of Patient 1 at 24 months of age revealed marked delay in his psychomotordevelopment and hypotonia. His upper airway compromise had diminished and he no longer required his tracheostomy. The dysmorphic features were still present.
Patient 2
At the time of diagnosis, Patient 1’s mother was already pregnant with Patient 2 (IV:3, Fig. 1). Prenatal ultrasound examination during this pregnancy revealed a male fetus with a left-sided CDH, unilateral mild hydronephrosis, prominent heels, and a micropenis. Amniocentesis was performed and a G-banded chromosome analysis revealed a 46,XY,der(12)t(11;12)(q23;q24)mat chromosomal complement.
Patient 2 was delivered at 40 weeks of gestation and had a birth weight of ~3,000 g (~50th centile). Accordingly to standardized treatment for CDH, he was intubated at birth and standard ventilator care for CDH was initiated. Physical and radiological examination confirmed the presence of the left-sided diaphragmatic hernia with severe pulmonary hypoplasia, seen on prenatal ultrasound. Additional anomalies identified at the time of birth included mild dysmorphic features (low-set, slightly dysplastic ears, and a short, broad neck), remarkably loose skin and subcutaneous tissue, hypoplastic toenails, prominent heels, and a micropenis. Cardiac ultrasound revealed a ventricular septal defect. No other signs of Fryns syndrome, besides hypoplastic nails, were present.
Patient 2’s perinatal course was complicated by severe, therapy-resistant, pulmonary hypertension, and recurrent pneumothorax. The patient was not considered a candidate for surgical repair because of instability of the clinical situation. Extra-corporeal membrane oxygenation (ECMO) therapy was not initiated because of his unbalanced chromosomal abnormality. The patient died at the age of 7 days as the result of therapy-resistant pulmonary hypertension and circulatory failure. Although parents did not grant permission for autopsy, consent was obtained for further clinical and cytogenetic analysis.
CYTOGENETIC ANALYSIS
G-banded chromosome analysis of peripheral blood lymphocytes from Patient 1 initially showed no chromosomal abnormalities. A repeat investigation at the age of 15 months revealed a chromosomal translocation with a 46,XY,der(12)t(11;12)(q23.3; q24.3)mat chromosome complement resulting in partial trisomy for 11q and partial monosomy for 12q. Prenatal chromosome analysis of Patient 2 revealed the same unbalanced translocation. This was confirmed on peripheral blood lymphocytes by GTG banding, by whole chromosome painting of chromosomes 11 and 12 (Figure 2), and by Multiplex ligation-dependent probe amplification (MLPA, results not shown).
Fig. 2.
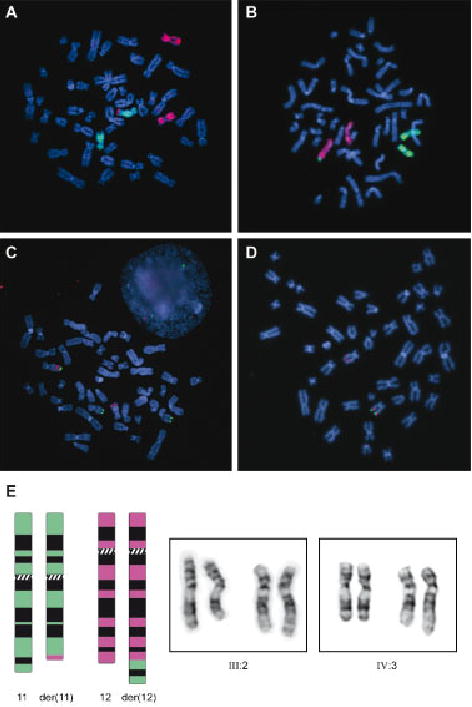
A–D: Fluorescence in situ hybridization experiments: WCP11 (red) & WCP12 (green) paint of chromosomes from Patient 1 showing additional chromosome 11 material on the derivative chromosome 12 (A), WCP11 (green) & WCP12 (red) paint of maternal chromosomes showing balanced translocation of chromosome 11 material to chromosome 12. B: RP11-19F21 (red signal) & RP1-26N8 (green signal) showing the presence of three copies of the terminal region of 11q in Patient 1 (C), RP11-19F21 (red signal) & RP1-221K18 (green signal) showing only a single signal from the terminal region of 12q in Patient 2 (D). E: Ideograms and partial karyotypes of mother (III:2) and Patient 2 (IV:3). On the left a schematic representation of the balanced translocation is shown. Chromosome 11 (green) and chromosome 12 (pink) are shown. On the right pictures of chromosomes 11 and 12 of both III:2 (balanced translocation carrier) and IV:3 (Patient 2) are shown. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Conventional G-banded chromosome analysis performed on parental peripheral blood lymphocytes revealed a maternal reciprocal translocation resulting in a 46,XX,t(11;12)(q23;q24.3) karyotype (Fig. 2). Paternal chromosomes were normal. No biological material was available from the mother’s first spontaneous abortion. All other family members of the mother are living abroad and were unavailable for investigation.
To delineate the exact size of the deletion and duplication present in Patient 2, array-based comparative genomic hybridization (array-CGH) was performed using a whole-genome tiling BAC array with a resolution of ~300 kb (Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX) [Li et al., 2003]. This analysis revealed an ~19 Mb duplication of chromosome 11q23.3-qter, but no abnormality of chromosome 12 was detected (Table I).
TABLE I.
Results Cytogenetic Analysis
| Probe name | Location | Patient 1 (FISH) | Patient 2 (array-CGH) | Patient 2 (FISH) | Mother (FISH) |
|---|---|---|---|---|---|
| Chromosome 11 | |||||
| RP1-44H16 | 11pter | N | N | N | |
| RP11-42L18 | 11q22 | N | |||
| RP11-235P12 | 11q23.3 | N | N | ||
| RP11-114K7 | 11q23.3 | N | N | ||
| RP11-840K20 | 11q23.3 | N | N | N | |
| RP11-1058J6 | 11q23.3 | N | N | N | |
| RP11-728G20 | 11q23.3 | N/Dupa | Dup | N/Dupa | N/Mb |
| RP11-1147E8 | 11q23.3 | N/Dupa | Dup | N/Dupa | N/Mb |
| RP11-2O20 | 11q23.3 | Dup | Dup | Dup | M |
| RP11-890O20 | 11q23.3 | Dup | Dup | M | |
| RP11-786F19 | 11q23.3 | Dup | Dup | M | |
| RP11-664F8 | 11q23.3 | Dup | Dup | Dup | M |
| RP11-778O17 | 11q23.3 | Dup | Dup | ||
| RP11-158K18 | 11q23.3 | Dup | Dup | Dup | |
| RP11-77K9 | 11q24.1 | Dup | Dup | ||
| RP11-102M23 | 11q25 | Dup | |||
| RP1-26N8 | 11qter | Dup | Dup | M | |
| Chromosome 12 | |||||
| CTB-124K20 | 12pter | N | N | N | |
| RP11-29G23 | 12q12 | N | |||
| RP11-269C10 | 12q24 | N | N | ||
| RP11-813G9 | 12q24 | N | |||
| RP11-91M21 | 12q24 | N | N | N | |
| RP11-19F21 | 12q24.21 | N | N | ||
| RP11-641K13 | 12q24.32 | N | N | ||
| RP11-1023E10 | 12q24.32 | N | N | N | |
| RP11-503G7 | 12q24.32 | N | N | N | N |
| RP11-1038O14 | 12q24.33 | N | N | N | N |
| RP11-452D11 | 12q24.33 | Del | N | Del | M |
| RP11-962N24 | 12q24.33 | Del | Del | M | |
| RP1-221K18 | 12qter | Del | Del | M | |
N, normal signal; Dup, duplication; Del, deletion; M, moved to derivative chromosome; blank space, not tested.
Lower signal intensity of duplicated signal on derivative chromosome.
Lower signal intensity of signal on derivative chromosome 11 and 12 (“split-spot”).
To validate results found by array-CGH and to map the breakpoints more precisely, BAC clones were selected from the University of California Santa Cruz (UCSC) and Ensembl genome browsers and ordered from BACPAC Resources, or obtained from the Department of Molecular and Human Genetics at Baylor College of Medicine. We used these BAC clones for FISH on metaphase chromosomes (Fig. 2). These results confirmed the ~19 Mb duplication of 11q23.3-qter and mapped the breakpoint to a region located within BAC clones RP11-728G20 and RP11-1147E8 (partially overlapping clones). Using FISH, a terminal deletion of chromosome 12q24.33 was found, proximally bordered by BAC clone RP11-1038O14 and extending towards the telomere. Identical results were found for Patient 1.
A whole chromosome paint of chromosomes 11 and 12 performed on maternal lymphocytes revealed a translocation of chromosome 11 material to chromosome 12. No translocation of chromosome 12 material could be detected using this technique. FISH analysis, however, revealed the same breakpoint locations identified in Patient 1 and 2 with more distally located clones on both chromosomes translocating to the corresponding derivative chromosome, confirming the reciprocal translocation.
DISCUSSION
We describe two siblings with the same unbalanced translocation of chromosomes 11 and 12. Although these siblings are concordant for most of the features and share a phenotype that is consistent with partial trisomy 11q syndrome, they are discordant for several anomalies including CDH (Table II). This leads us to conclude that partial trisomy 11q syndrome has a variable phenotype and that CDH should be added to the spectrum of anomalies that can be present in this syndrome.
TABLE II.
Clinical Features
| Features of “partial trisomy 11q syndrome” | Patient 1 | Patient 2 |
|---|---|---|
| Mental retardation | + | ? |
| Motor retardation | + | ? |
| IUGR | − | − |
| Postnatal growth deficiency | − | − |
| Hypotonia | + | + |
| Microcephaly | − | − |
| Retro/micrognathia | +a | + |
| Craniofacial asymmetry | − | − |
| Abnormal ears | − | + |
| Short nose | + | + |
| Prominent upper lip | + | − |
| Retracted lower lip | − | − |
| Cleft palate/high arched palate/bifid uvula | + | + |
| Glossoptosis | + | − |
| Cardiac abnormality | − | + |
| CDH | − | + |
| Renal abnormality | − | + |
| Small/hypoplastic nails | − | + |
| Dislocated hip joints | − | ? |
| Clavicle defect | − | − |
| Cutis laxa | − | + |
| Short/broad neck | − | + |
| Other abnormalities | − | − |
IUGR, intra-uterine growth retardation, CDH, congenital diaphragmatic hernia; +, present, −, not present; ?, no data available.
Micrognathia more pronounced at birth. No longer obvious as the age of 2 8/12 years.
By means of complementary cytogenetic techniques, we mapped the breakpoints in both patients and their mother who carries the corresponding reciprocal translocation. The unbalanced chromosomal anomaly was detected by conventional G-banded chromosome analysis. The duplication of the ~19 Mb distal portion of 11q was easily detected by array-CGH and confirmed by FISH. The smaller ~0.5 Mb deletion of 12qter was not detected by array-CGH but was confirmed by FISH studies. The failure of the array to detect this deletion is most likely due to low representation of material distal to the breakpoint (Table I). This region contains a cluster of zinc finger genes and a repeat cluster. RP11-452D11, the only BAC representing this region on the array partially overlapped this repeat cluster.
Due to the unbalanced translocation, there is a duplication of part of the long arm of chromosome 11, an abnormality that has been described several times in the literature as “partial trisomy 11q syndrome” or “duplication 11(q21/q23 → qter) syndrome” [Francke et al., 1977; Pihko et al., 1981]. The clinical features of this syndrome and the phenotype of Patients 1 and 2 are summarized in Table II.
CDH has been described in several patients with partial duplication of 11q due to a t(11;22) [Biederman et al., 1980; Kadir et al., 1997; Borys and Taxy, 2004]. To our knowledge, CDH has only been described once in a child with partial duplication 11q due to an unbalanced translocation with another chromosome than 22 [Park et al., 1993]. Patient 2 represents the second case of an unbalanced translocation other than the t(11;22) resulting in partial duplication 11q associated with CDH, in this case associated with a small partial monosomy 12q. Although we cannot exclude a potential role for the genes deleted on 12q or that the CDH in Patient 2 only occurred by coincidence, we believe that it is more likely that one or more genes in the duplicated region on 11q predispose to the development of CDH. There have been two case-reports on monosomy 12q24, but in none of these CDH has been described [Sathya et al., 1999; Plotner et al., 2003]. Ultimately, environmental factors and the genetic background determine whether an individual eventually develops CDH. The discordance in these two siblings might be the result of epigenetic differences between the two brothers, such as different methylation status of genes in the duplicated region.
The duplicated region on 11q23-qter contains almost a hundred known genes and multiple unknown transcipts. Although none of the genes in this region have been implicated in the etiology of CDH, there are several genes that could play a role in the development of CDH based on their known functions. ROBO3 (roundabout, axon guidance receptor, homolog 3 [OMIM 607630]), located at 11q24.2, regulates axon guidance across the midline in the brain [Sabatier et al., 2004] and might be involved in non-neuronal morphogenesis [Anselmo et al., 2003]. ROBO genes encode the receptors for the Slit-family of genes in both Drosophila and vertebrates, and Slit-3 (OMIM 603745) binds to ROBO3 in vertebrates. Although it is unclear at this time whether central- and posterolateral-type CDHs are caused by related genes, it is interesting to note that Slit3 knockout mice display a septum transversum CDH associated with kidney agenesis and cardiac defects [Liu et al., 2003]. Another member of the roundabout-gene-family, ROBO4 (roundabout, axond guidance receptor, homolog 4 [OMIM 607528]), also located on 11q24.2, is also duplicated in our patients. This gene is mainly expressed at sites of active angiogenesis, especially under hypoxic conditions [Huminiecki et al., 2002]. A third duplicated gene is CDON (Cell adhesion molecule-related/downregulated by oncogenes [OMIM 608707]), located at 11q24. In complex with BOC (Brother of CDON [OMIM 608708]), this gene regulates myogenic differentiation [Kang et al., 2002]. Further delineation of breakpoints in patients with smaller partial duplications of 11q and CDH, functional studies, and transgenic animal experiments may help focus attention on one or more of these genes.
Recently, Zhao et al. [2003] described four patients with partial trisomy 11q and severe upper airway malformations. They attributed the upper airway malformations in these patients to duplication of the 11q21–q23.2 region. This conclusion was based on the proximal location of the breakpoints in patients with the upper airway anomalies, which started at q21, in comparison to patients without upper airway malformations, in which the breakpoints were located more distally at q23.2. The phenotype of Patient 1, who presented with severe Pierre–Robin sequence, would suggest that the 11q23.2-qter region is involved in upper airway formation and that incomplete penetrance may account for those individuals with 11q23-qter duplications who lack this phenotype.
In conclusion, the “partial trisomy 11q syndrome” has a highly variable phenotype. Our findings implicate that CDH should be added to the spectrum of abnormalities that can be present in this syndrome.
ELECTRONIC-DATABASE INFORMATION
Accession number and URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensemb.org/Homo_Sapiens/Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?.db=OMIM Unigene, http://www.ncbi.nlm.nih.gov/Genomes/index.html University of California Santa Cruz (UCSC) Genome Browser, http://genome.cse.ucsc.edu/.
Acknowledgments
We are grateful to family members for their cooperation. The authors would also like to thank Tom de Vries Lentsch for help in preparing the figures, Hakima el Idrissi for performing some of the FISH experiments, Gracia Mancini for translating some publications, Carla van de Sijs for providing some of the clinical data, and the Baylor Human Genome Sequencing Center for providing BAC clones used in this study. This research was funded in part by the Sophia Foundation for Scientific Research, Rotterdam, The Netherlands (SSWO project 441), NIEHS P01 ES11253 (B.L.), the Baylor MRDDRC NIH HD024064, and NICHD K08 HD050583.
Footnotes
Grant sponsor: Sophia Foundation for Scientific Research, Rotterdam, The Netherlands (SSWO project 441); Grant sponsor: NIEHS; Grant number: P01 ES11253; Grant sponsor: Baylor MRDDRC NIH; Grant number: HD024064; Grant sponsor: NICHD; Grant number: K08 HD050583.
References
- Anselmo MA, Dalvin S, Prodhan P, Komatsuzaki K, Aidlen JT, Schnitzer JJ, Wu JY, Kinane TB. Slit and robo: Expression patterns in lung development. Gene Expr Patterns. 2003;3:13–19. doi: 10.1016/s1567-133x(02)00095-9. [DOI] [PubMed] [Google Scholar]
- Beresford MW, Shaw NJ. Outcome of congenital diaphragmatic hernia. Pediatr Pulmonol. 2000;30:249–256. doi: 10.1002/1099-0496(200009)30:3<249::aid-ppul9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Biederman BM, Lin CC, Lowry RB, Somerville R. Tertiary trisomy (22q11q),47,+der(22),t(11;22) Hum Genet. 1980;53:173–177. doi: 10.1007/BF00273491. [DOI] [PubMed] [Google Scholar]
- Borys D, Taxy JB. Congenital diaphragmatic hernia and chromosomal anomalies: Autopsy study. Pediatr Dev Pathol. 2004;7:35–38. doi: 10.1007/s10024-003-2133-7. [DOI] [PubMed] [Google Scholar]
- de France HF, Beemer FA, Senders RC, Gerards LJ, Cats BP. Partial trisomy 11q due to paternal t(11q;18p); further delineation of the clinical picture. Clin Genet. 1984;25:295–299. doi: 10.1111/j.1399-0004.1984.tb01992.x. [DOI] [PubMed] [Google Scholar]
- Fraccaro M, Lindsten J, Ford CE, Iselius L. The 11q;22q translocation: A European collaborative analysis of 43 cases. Hum Genet. 1980;56:21–51. doi: 10.1007/BF00281567. [DOI] [PubMed] [Google Scholar]
- Francke U, Weber F, Sparkes RS, Mattson PD, Mann J. Duplication 11 (q21 to 23 leads to qter) syndrome. Birth Defects Orig Artic Ser. 1977;13:167–186. [PubMed] [Google Scholar]
- Huminiecki L, Gorn M, Suchting S, Poulsom R, Bicknell R. Magic roundabout is a new member of the roundabout receptor family that is endothelial specific and expressed at sites of active angiogenesis. Genomics. 2002;79:547–552. doi: 10.1006/geno.2002.6745. [DOI] [PubMed] [Google Scholar]
- Kadir RA, Hastings R, Economides DL. Prenatal diagnosis of supernumerary chromosome derivative (22) due to maternal balanced translocation in association with diaphragmatic hernia: A case report. Prenat Diagn. 1997;17:761–764. [PubMed] [Google Scholar]
- Kang JS, Mulieri PJ, Hu Y, Taliana L, Krauss RS. BOC, an Ig superfamily member, associates with CDO to positively regulate myogenic differentiation. EMBO J. 2002;21:114–124. doi: 10.1093/emboj/21.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital diaphragmatic hernia and chromosome 15q26: Determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jiang T, Bejjani B, Rajcan-Separovic E, Cai WW. High-resolution human genome scanning using whole-genome BAC arrays. Cold Spring Harb Symp Quant Biol. 2003;68:323–329. doi: 10.1101/sqb.2003.68.323. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhang L, Wang D, Shen H, Jiang M, Mei P, Hayden PS, Sedor JR, Hu H. Congenital diaphragmatic hernia, kidney agenesis and cardiac defects associated with Slit3-deficiency in mice. Mech Dev. 2003;120:1059–1070. doi: 10.1016/s0925-4773(03)00161-8. [DOI] [PubMed] [Google Scholar]
- Lurie IW. Where to look for the genes related to diaphragmatic hernia? Genet Couns. 2003;14:75–93. [PubMed] [Google Scholar]
- Park JP, McDermet MK, Doody AM, Marin-Padilla JM, Moeschler JB, Wurster-Hill DH. Familial t(11;13)(q21;q14) and the duplication 11q, 13q phenotype. Am J Med Genet. 1993;45:46–48. doi: 10.1002/ajmg.1320450113. [DOI] [PubMed] [Google Scholar]
- Pfeiffer RA, Schutz C. Tandem duplication 11q23-ter in the dysmorphic child of a retarded mother mosaic for the same anomaly with no apparent abnormalities. Ann Genet. 1993;36:163–166. [PubMed] [Google Scholar]
- Pihko H, Therman E, Uchida IA. Partial 11q trisomy syndrome. Hum Genet. 1981;58:129–134. doi: 10.1007/BF00278696. [DOI] [PubMed] [Google Scholar]
- Plotner PL, Smith JL, Northrup H. Deletion 12q: A second patient with 12q24.31q24.32 deletion. Am J Med Genet Part A. 2003;118A:350–352. doi: 10.1002/ajmg.a.10232. [DOI] [PubMed] [Google Scholar]
- Pober BR, Russell M, Ackerman KG. 2006. Congenital Diaphragmatic Hernia Overview. GeneReviews (in press).
- Sabatier C, Plump AS, Le M, Brose K, Tamada A, Murakami F, Lee EY, Tessier-Lavigne M. The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell. 2004;117:157–169. doi: 10.1016/s0092-8674(04)00303-4. [DOI] [PubMed] [Google Scholar]
- Sathya P, Tomkins DJ, Freeman V, Paes B, Nowaczyk MJ. De novo deletion 12q: Report of a patient with 12q24.31q24.33 deletion. Am J Med Genet. 1999;84:116–119. doi: 10.1002/(sici)1096-8628(19990521)84:2<116::aid-ajmg6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Torfs CP, Curry CJ, Bateson TF, Honore LH. A population-based study of congenital diaphragmatic hernia. Teratology. 1992;46:555–565. doi: 10.1002/tera.1420460605. [DOI] [PubMed] [Google Scholar]
- Van Opstal D, Eussen HJ, Van Hemel JO, Sachs ES. Application of fluorescent in situ hybridization for ‘de novo’ anomalies in prenatal diagnosis. Prenat Diagn. 1993;13:825–832. doi: 10.1002/pd.1970130906. [DOI] [PubMed] [Google Scholar]
- Vianello MG, Chiossi F, Fasce L, Besio B, Chiossi M. [Trisomy 11qter(11q21 and 11q23—qter) syndrome. Presentation of a case with familial translocation t(11;12) (q23,1—qter;q24,3)] Minerva Pediatr. 1986;38:183–192. [PubMed] [Google Scholar]
- Zhao HQ, Rope AF, Saal HM, Blough-Pfau RI, Hopkin RJ. Upper airway malformation associated with partial trisomy 11q. Am J Med Genet Part A. 2003;120A:331–337. doi: 10.1002/ajmg.a.20134. [DOI] [PubMed] [Google Scholar]