

Abstract
BRCA1 is a breast and ovarian cancer-specific tumor suppressor, with properties of a transcription factor involved in DNA repair. We previously have shown the transactivation of heterologous promoters by the carboxyl terminus of BRCA1. We now describe that BRCA1-mediated transactivation is enhanced by p300/CBP (CREB binding protein) and that this effect was suppressed by the adenovirus E1A oncoprotein. We show a physical association of BRCA1 with the transcriptional coactivators/acetyltransferases p300 and CBP. Endogenous as well as overexpressed BRCA1 and p300 were found to associate in a phosphorylation-independent manner. BRCA1 interacts with the cAMP response element binding protein (CREB) domain of p300/CBP via both its amino and carboxyl termini. Finally, full-length BRCA1 is shown to transcriptionally activate the Rous sarcoma virus-long terminal repeat promoter, which was further stimulated by p300. Immunocolocalization analyses suggest that BRCA1 and p300 associate in a cell cycle-dependent manner. Our results support a role for BRCA1 in transcription.
BRCA1 is the first of the two identified familial breast cancer genes (1). The human BRCA1 gene encodes a 1,863-aa-long, predominantly nuclear phosphoprotein (2–5). At its amino terminus BRCA1 possesses a RING finger domain thought to mediate protein–protein interactions. At its extreme carboxyl terminus BRCA1 has two BRCT repeats that are present in a large number of DNA damage-responsive cell cycle checkpoint proteins found from bacteria to humans (6, 7). Interestingly, BARD1, a protein that physically interacts with the RING finger binding domain of BRCA1 also possesses BRCT repeats as well as a RING finger (8). BRCA1 is essential in mouse embryonic development (9–11). BRCA1 −/− embryos do not die at a specific time point but over a period starting from embryonic day (E) 6.5 to E13 because of a gastrulation failure or defects in neural tube closure depending on the mutation (9–11). However, there is one report of a naturally occurring BRCA1 knockout in a breast cancer patient (12). Tissue-specific conditional knockouts of BRCA1 do indeed induce breast epithelial hyperplasias in mice, albeit at a low frequency after 10–13 months (13). If true, this difference might not be unexpected given the surprisingly poor sequence conservation of 58% identity at the amino acid level between the human and mouse BRCA1 orthologues (14). The levels of BRCA1 fluctuate through the cell cycle, reaching a maximum in S phase and a minimum during G1 (4, 15, 16). This change parallels the phosphorylation state of BRCA1, which becomes hyperphosphorylated predominantly on serine residues (4, 15, 17). BRCA1 is known to associate with RAD51, the mammalian homologue of the bacterial RecA protein (5), suggesting that BRCA1 is involved in the maintenance of genomic integrity and/or in DNA damage-dependent responses. Further evidence pointing in this direction stems from the recent observation that DNA-damaging agents can induce the hyperphosphorylation of BRCA1 on serine residues (17, 18). This phosphorylation is distinct from the cell cycle-dependent phosphorylation in that it produces a specific mobility shift different from that observed in S- or G1-phase cells. In addition, the punctate nuclear staining of BRCA1 in S-phase cells is altered on exposure to DNA damaging agents (18) and gamma irradiation, which relocalizes BRCA1 to Rad50-hMre11-p95 complexes (19).
Although sketchy, other evidence points toward a second function of BRCA1, namely in transcription. The carboxyl terminus of BRCA1, when fused to the heterologous Gal4 DNA binding domain, activates transcription, which is abolished when cancer predisposing mutations are introduced in the BRCA1 component (20, 21). Moreover BRCA1 has been found to be associated in part with the RNA polymerase II holoenzyme (22) and RNA helicase A. BRCA1 overexpression also activates the mdm2 promoter in a p53-dependent fashion (23).
The cAMP response element binding protein (CREB) binding protein (CBP) and p300 are paralogous proteins that initially were identified as a transcriptional coactivator of the CREB and a 300-kDa E1A-associated protein, respectively. Subsequent studies have shown that these two proteins not only bind CREB and E1A but also function as transcriptional coactivators of a multitude of other activated transcription factors and basal factors such as TATA box-binding protein and TFIIB (24). This finding led to the proposal that the mechanistic basis for their transcriptional activation properties was caused by the bridging function between the activated transcription factors and the basal transcriptional machinery. Interestingly, CBP/p300 are themselves histone acetyl-transferases (HATs) (25, 26) and are able to recruit other HATs such as P/CAF and ACTR/SRC-1 (27, 28). Acetylation of the lysines at the amino termini of histones is thought to aid transcriptional activation by decondensing chromatin. Conversely deacetylation of core histones has been shown to be involved in transcriptional repression and silencing (29). Mutations within the CBP gene, which presumably lead to haploinsufficiency, cause the Rubinstein-Taybi syndrome characterized by facial abnormalities, broad thumbs, large toes, mental retardation, and an increased susceptibility to develop tumors. CBP also has been found to be involved in two types of chromosomal translocations present in therapy-related myelodysplasia and acute leukemias. In these translocations CBP is fused to the presumptive transcription factor MLL (30–32) and the MOZ HAT (33). p300 also has been found to be lost in one case of gastric or colon carcinoma (34). All of these facts suggest the possibility that p300 and CBP might be tumor suppressors or oncogenes. In this manuscript we show that CBP/p300 and BRCA1 physically and functionally interact and we discuss the possible significance of this association.
Materials and Methods
Tissue Culture Methods and Transfections.
Human embryonic kidney 293, U2OS human osteosarcoma, and human cervical carcinoma HeLa tissue culture cells were grown in DMEM (Cellgro, Mediatech, Washington, DC) supplemented with 10% FCS and a mixture of penicillin/streptomycin (Sigma). RK13 rabbit kidney cells were grown in Eagle's modified minimal medium with Earl's salts supplemented with 10% FCS and penicillin/streptomycin, and 293 cells were transfected by using the calcium phosphate method as described (35). HeLa cells were transfected by using Superfect (Qiagen, Chatsworth, CA) as described by the manufacturer. The amounts of transfected plasmids for cotransfection assays were as follows: 0.5–1 μg 2× GAL4-luciferase reporter, 4 μg Gal4-BRCA1528–1863, 6 μg p300-hemagglutinin (HA), and 1 μg E1A or E1A-NTD646. Transcriptional activation of the Rous sarcoma virus (RSV)-long terminal repeat (LTR) promoter was assayed by cotransfection assays using 1 μg RSV-LTR LacZ plasmid, 0.5 μg thymidine kinase-luciferase, and 0.5 μg cytomegalovirus-promoter-driven enhanced green fluorescent protein per 6-cm dish. For the BRCA1-dependent activation 8 μg of pCL-MFG-BRCA1 or empty plasmid control was transfected per 6-cm dish. For the p300 dependency 2 μg of pCL-MFG-BRCA1 was used and increasing amounts of p300-HA plasmid DNA (up to 6 μg) were used. (see Fig. 6B). HeLa tTa BRCA1 were transfected with increasing amounts of p300 as indicated in Fig. 6C, an 0.5-μg RSV-LTR LacZ plasmid, and 0.25 μg thymidine kinase-luciferase. Synchronization of HeLa cells was performed as described (4). For immunofluorescence staining, HeLa cells were grown on chamber slides (Falcon) previously coated with poly-l-lysine.
Figure 6.

(A) Transcriptional activation of the RSV-LTR promoter-driven β-galactosidase reporter on cotransfection with BRCA1 over control empty vector. (B) Cotransfection of p300-enhanced BRCA1-dependent transcriptional activation in RK13 cells. Reduced amounts of BRCA1 were transfected in these cases to avoid saturation of the reporter. (C) Transfection of increasing amounts of p300 and constant amounts of a RSV-LTR promoter-driven β-galactosidase reporter into a tetracycline/doxycycline repressible BRCA1 HeLa cell line shows BRCA1-dependent p300 enhancement of RSV-LTR promoter activity.
Immunoprecipitations and Western Blotting.
Cells were washed once in ice-cold PBS before scraping them off at 4°C with 1 ml of PBS. Cells were spun down for 10 min at 400 × g. Cell pellets were frozen in liquid nitrogen and left to thaw on ice. Thawed cells were resuspended in RIPA buffer (20 mM Tris⋅HCl, pH 8.0/100 mM NaCl/0.2% deoxycholate/0.2% Triton X-100/0.2% NP-40/1 mM DTT/10 μg/ml aprotinin/0.1 mM PMSF/16 μg/ml benzamidine/10 μg/ml 1,10-phenanthroline/10 μg/ml leupeptin/10 μg/ml pepstatin A). Cell extract then was passed through a 25-gauge syringe 10 times or sonicated, frozen in liquid nitrogen, thawed on ice, and centrifuged at >21,000 × g for 30–45 min. Supernatants were used as crude extracts for immunoprecipitations. Nonspecific binding was reduced by preincubation of extracts with protein A or protein G Sepharose (Amersham Pharmacia) for 1–2 h while tumbling at 4°C. Pellets were discarded and extracts were incubated with immune sera or controls for 2–4 h. Immunoprecipitations were performed with 5 μl of rabbit affinity-purified BRCA1 antipeptide AbC or AbD antibodies per ml of whole-cell extract (4) or equivalent volumes of monoclonal anti-HA (12CA5) and polyclonal anti-p300 antibodies (C-20, Santa Cruz Biotechnology).
Immunostaining and Microscopy.
Cells were fixed and permeabilized as described (5). BRCA1 was visualized by using mAbs MS110 and MS13 (5) from hybridoma supernatants at a 1:10 dilution. p300 was visualized with a 1:400 dilution of purified rabbit polyclonal antipeptide antibody C-20 (Santa Cruz Biotechnology). Primary antibodies were diluted in 1 mg/ml of BSA in PBS, incubated at 37°C for 20 min, and washed three times with PBS before the fluorophore-conjugated secondary antibody incubation. Secondary antibodies all were derived from donkey. Secondary fluorophore-conjugated antibodies (The Jackson Laboratory) were diluted 1:100 to 1:500 in PBS with 5%–10% donkey serum and incubated for 20 min at 37°C. Cell then were washed five times in PBS, stained with 50 μg/ml of 4′,6-diamidino-2-phenylindole for 5 min on ice, washed five times with PBS, and then mounted and inspected by confocal laser microscopy.
Glutathione S-Transferase (GST) Pull-Downs.
GST-CBP fusion proteins were produced in Escherichia coli and purified as described (36). Binding of proteins to glutathione Sepharose was done in 20 mM Hepes, pH 7.4/50 mM NaCl/1 mM MgCl2/0.2 mM DTT/0.5 mM PMSF/20 μg/ml leupeptin/20 μg/ml aprotinin/0.05% Tween 20.
Phosphorylation-Dependence Study.
Full-length BRCA1 or myc-tagged BRCA1 were overexpressed in 293T cells and subsequently immunoprecipitated from cell lysates as described (4), except that immunoprecipitations were performed in the absence of SDS and without heat denaturation. Proteins were eluted from protein A-Sepharose beads by a 1-min incubation in 50 mM glycine⋅HCl, pH 2.5 buffer, neutralized in bacterial alkaline phosphatase fraction F reaction buffer (4), and treated with or without bacterial alkaline phosphatase for 20 min at 55°C. The proteins then were incubated with a GST-CBP 451–721 bound to glutathione-agarose beads, and GST pull-down assays were performed as described above in the presence of 0.2% Triton X-100. Bound proteins were separated on SDS-polyacrylamide gels and visualized by Western blot analysis.
Cotransfection Assays.
Luciferase and β-galactosidase assays were performed as described in ref. 36 or by the manufacturer (Promega). All luciferase measurements were normalized to β-galactosidase activity driven by the cytomegalovirus promoter, conversely the RSV-LTR-LacZ normalization was done with the cotransfected thymidine kinase-luciferase reporter.
Results
p300/CBP Activates the Gal4-BRCA1 Carboxyl-Terminal Domain Transcription.
Gal4-BRCA11528–1863 previously has been reported to be a transactivator (20, 21). Using a reporter plasmid containing a thymidine kinase minimal promoter-driving luciferase with two upstream Gal4 DNA binding sites, we observed that transfection of p300 in conjunction with Gal4-BRCA11528–1863 in either 293 (Fig. 1A) or RK13 (Fig. 1B) cells showed a 3- to 5-fold increase in luciferase activity compared with Gal4-BRCA11528–1863 alone. This transactivation effect was neither observed with Gal4 DNA binding domain alone nor with p300 cotransfected with the Gal4 DNA binding domain and is thus specific. The adenovirus E1A protein suppresses p300- and CBP-mediated transcriptional activation (27). Consistent with this finding, cotransfection of E1A in either 293 (Fig. 1C) or U2OS cells (Fig. 1D) suppressed the p300-mediated activation of Gal4-BRCA11528–1863, whereas cotransfection of an amino-terminal deletion mutant of E1A (NTD646) that is unable to bind CBP/p300 did not (Fig. 1 C and D).
Figure 1.

Transient cotransfection assays of Gal4-BRCA11528–1863 and p300-HA using a luciferase reporter containing GaL4 DNA binding sites in 293 (A) and RK13 cells (B) showing p300 enhancement of Gal4-BRCA11528–1863 activation of a Gal4 luciferase reporter. Suppression of Gal4-BRCA11528–1863/p300-HA-mediated transcription by E1A and not by E1A-NTD646 in 293 cells (C) and U2OS cells (D).
BRCA1 and p300 Interact in Vivo and in Vitro.
To investigate a possible biochemical association between BRCA1 and the coactivator p300, we expressed BRCA1 and a carboxyl terminally HA-tagged form of p300 either individually or in combination. Lysates were prepared from the transfected 293 cells and immunoprecipitated with either BRCA1 antibody AbD (Fig. 2A) (4) or AbC (Fig. 2B) followed by Western blotting using the 12CA5 anti-HA mAb. p300-HA was immunoprecipitated when BRCA1 and p300-HA were coexpressed (Fig. 2A, lane 4 and B, lane 3). p300-HA occasionally was detected in the BRCA1 immunoprecipitations when it was expressed alone, because of low levels of endogenous BRCA1 in 293 cells (Fig. 2A, lane 3 and C, lane 1). To exclude the possibility that complex formation of p300-HA and BRCA1 was occurring through DNA we performed identical immunoprecipitations in the presence of DNaseI (1,360 units/ml) (Fig. 2C). p300-HA was precipitated by BRCA1 antibodies, indicating that the complex is formed through protein–protein interactions. Identical results were obtained when immunoprecipitations were carried out in the presence of ethidium bromide (0.01 mg/ml) (data not shown). We also checked for endogenous p300/BRCA1 complexes in both HeLa cervical carcinoma (Fig. 2D), MCF7 breast cancer (Fig. 2E), and HBL100 normal breast epithelium (Fig. 2F) whole-cell extracts by immunoprecipitation with BRCA1-AbC or BRCA1-AbD antibodies followed by immunoblotting with an p300 rabbit antibody. p300 was detected when either BRCA1 antibody AbC or AbD was used for immunoprecipitation (Fig. 2 D, lane 1, E lanes 3 and 4, and F, lane 4), but not on preincubation with their cognate peptides (Fig. 2 D, lane 2, E, lane 2, and F, lane 2) or with control anti-IκBα or mSin3A/B serum (Fig. 2 D, lane 3, E, lane 5, and F, lane 5). Antibodies against ACTR/AIB1, a coactivator/acetyltransferase known to interact with p300/CBP, were used as a positive control (Fig. 2F, lane 6). Protein A and protein G beads alone did not immunoprecipitate p300-containing complexes.
Figure 2.

(A) Immunoprecipitation with anti-BRCA1-AbD antibody of overexpressed BRCA1 and p300-HA in transiently transfected 293 cells. Transfected plasmids are indicated. Immune complexes were resolved by SDS/PAGE and immunoblotted with the 12CA5 anti-HA mAb. (B) Immunoprecipitation is as in A except that the immunoprecipitating antibody is AbC. The same immunoprecipitations were performed in the presence of 1,360 units/ml of DNaseI (C). Immunoprecipitation of endogenous BRCA1/p300 complexes from HeLa cells (D). Cell extracts from three 15-cm dishes were immunoprecipitated with anti-BRCA1 AbC antibody (lane 1), with the same antibody after preincubation with its cognate competitor “C” peptide (lane 2) or an unrelated rabbit antiserum (anti-IκBα, lane 3). Immunoprecipitated proteins were resolved by SDS/PAGE and immunoblotted with the C-20 anti-p300 antibody (Santa Cruz Biotechnology). Immunoprecipitations of one 15-cm dish per immunoprecipitation of MCF-7 breast cancer cells were done similarly (E). Immune complexes were precipitated by using either anti-BRCA1 AbC (lane 3) or AbD (lane 4). Negative controls were either precipitates with beads alone (lane 1), the immunoprecipitating antibody with prior incubation with its cognate peptide (lane 2), or the use of an unrelated antibody (Sin3B) (lane 5). One confluent 15-cm dish of HBL100 normal breast epithelial cells was used per immunoprecipitation reaction (F). The immunoprecipitating antibody is as noted above each lane. ACTR/AIB1 serves as a positive control (lane 6). The total amount of p300 signal in the extracts is given by the p300 immunoprecipitation reaction (lane 7). Sin3A and Sin3B antibodies were used as negative controls.
Using either of the two BRCA1 mAbs (MS13 and MS110) (5) and the rabbit polyclonal C-20 p300 antibody, we detected substantial colocalization of the p300 and BRCA1 immunofluorescence signals in confluent HeLa cells. Subnuclear regions of strong BRCA1 and p300 staining coincide at the nuclear rim and other speckled structures within the nucleus (Fig. 3A). This pattern of staining corresponded to that of DNA as seen in 4′,6-diamidino-2-phenylindole staining in these cells (not shown), suggesting that both proteins are in close physical proximity with a DNA-associated structure. No significant colocalization of p300 and BRCA1 could be seen in S-phase cells. (Fig. 3B). p300 showed diffuse staining in the nucleus whereas BRCA1 formed a large number of nuclear dots as described (5).
Figure 3.
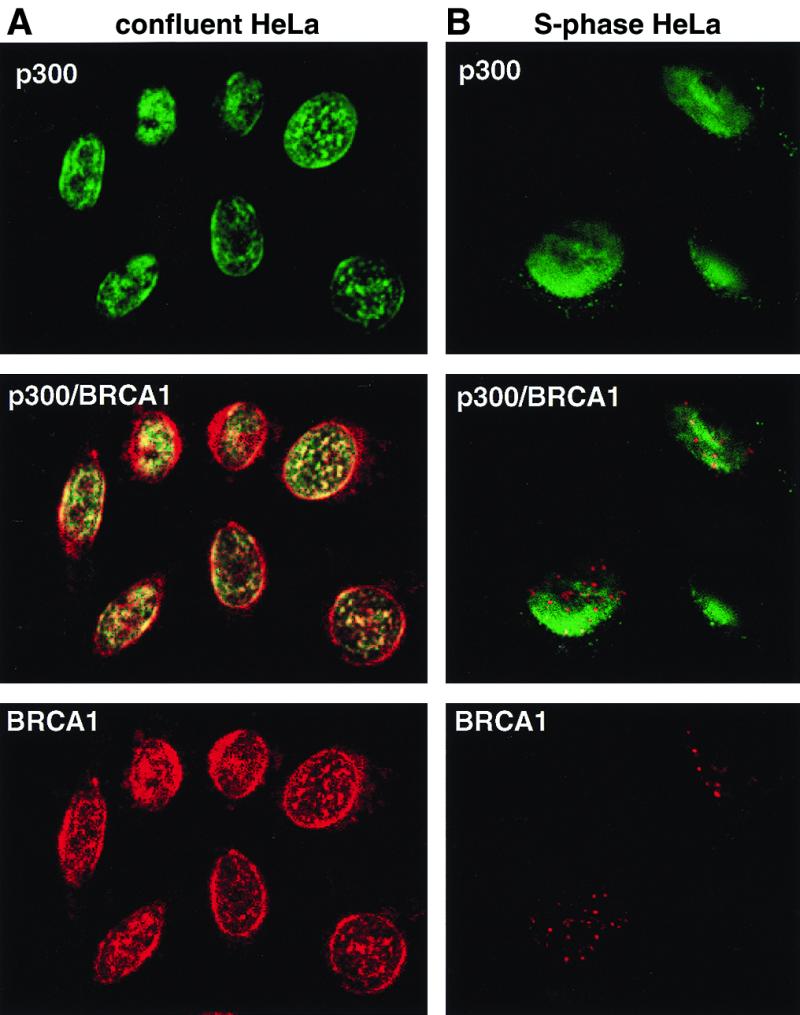
Colocalization of p300 and BRCA1 by immunofluorescence staining. BRCA1 is stained in red (Texas red) and p300 in green (FITC), while coincident signals are seen in yellow in the merged picture obtained by confocal laser microscopy. (A) A field of confluent HeLa cells. (B) S-phase HeLa cells 4 hr after release from aphidicolin-mediated arrest. Magnifications: ×63.
Interaction Domain Mapping.
To map the interaction domain of BRCA1 on CBP/p300, GST pull-downs with six distinct GST-CBP fusions spanning the whole molecule and extracts of 293 cells transfected with BRCA1 were performed. Western blotting of the bound proteins using the BRCA1-AbD antibody showed that only the fragment corresponding to residues 451–721 of CBP interacted with BRCA1 (Fig. 4A, lane 2). Similarly in vitro-translated 35S-Met-labeled full-length BRCA1 interacted specifically with CBP fragment 451–721 (Fig. 4B). Thus the interaction between CBP and BRCA1 is likely to be direct and occurs through the CREB binding domain of CBP (Fig. 4C).
Figure 4.

Mapping of the BRCA1 binding domain on CBP. (A) Pull-downs with the indicated CBP amino acids fused to GST and an extract from 293 cells overexpressing full-length BRCA1. Approximately 10% of the input is shown in lane 7 of the anti-BRCA1-AbD Western blot. The arrow points to full-length BRCA1. (B) GST pull-down of in vitro-translated 35S-labeled BRCA1 using GST or the GST-CBP 451–721 fragment. Pulled-down complexes were resolved by SDS/PAGE and visualized by autoradiography. (C) CBP/p300 domain structure with some of the known interaction partners.
To localize the CBP/p300 interaction domain within BRCA1, we generated four amino-terminal myc epitope-tagged fragments of BRCA1 that spanned the entire protein. Whole-cell extracts of 293T cells overexpressing the individual myc-tagged fragments of BRCA1 were used in pull-down with GST-CBP 451–721. Both the BRCA1:1–303 and BRCA1:1314–1863 fragments interacted strongly with CBP (Fig. 5A, lanes 5 and 8). The BRCA1 fragment spanning residues 303–772 exhibited weak binding (Fig. 5A, lane 6) and residues 772-1314 did not bind at all. To demonstrate the existence of the complexes in vivo, we coexpressed p300-HA and the myc-tagged BRCA1 fragments in 293T cells and immunoprecipitated the complexes by using the 12CA5 anti-HA antibody. Immunoblotting with anti-myc antibodies revealed only binding by the 1–303 and 1314–1863 fragments (Fig. 5B). No binding by the 303–772 fragment occurred, suggesting that the weak binding observed in the in vitro interaction is either insignificant or some other protein binds 303–772 more strongly in vivo.
Figure 5.

(A) Mapping of the CBP interacting region on BRCA1 using GST-CBP 451–721 and myc-tagged BRCA1 fragments. Lanes 1–4 show anti-myc Western blots of the input cell extracts, and lanes 5–8 show the respective GST pulldowns. (B) Mapping by coimmunoprecipitation of cotransfected myc-tagged BRCA1 fragments and p300-HA using anti-HA antibody followed by anti-myc Western blotting. Input is shown in lanes 1–4 and immunoprecipitations in lanes 5–8. (C) Schematic structures of the overexpressed myc-tagged BRCA1 proteins. (D) Binding of BRCA1 to CBP is not affected by phosphorylation. The CBP/p300 interacting fragment of BRCA1 as well as full-length BRCA1 were overexpressed by transient transfection in 293T cells. The overexpressed proteins were immunoprecipitated with anti-BRCA1-AbD antibody and eluted off the antibodies. The proteins then were selectively dephosphorylated with bacterial alkaline phosphatase (BAP). Dephosphorylated and nondephosphorylated fragments then were assayed for binding to GST or GST-CBP 451–721. Bound proteins were resolved by SDS/PAGE and visualized by immunoblotting with either anti-myc or anti-BRCA1 AbD.
The BRCA1/CBP Interaction Is Phosphorylation Independent.
Because phosphorylation of BRCA1 is modulated during the cell cycle and in response to DNA damage (2, 4), we tested the dependency of the BRCA1:CBP interaction on phosphorylation. We expressed full-length as well as myc-tagged BRCA1:1314–1863, which encompasses the transactivation domain of BRCA1, in 293T cells. These two proteins were immunopurified, dephosphorylated, and then tested for binding in a pull-down assay using GST-CBP 451–721. As shown in Fig. 5D, both proteins bound specifically to the GST-CBP 451–721 fragment regardless of treatment with bacterial alkaline phosphatase, suggesting that the BRCA1/CBP interaction is phosphorylation independent.
Full-Length BRCA1 and p300 Activate the RSV LTR.
To extend the paradigm of BRCA1 as a transcriptional activator we sought promoters responsive to full-length BRCA1. We fortuitously found that overexpression of full-length BRCA1 could activate transcription of the RSV-LTR promoter in cotransfection assays in RK13 (Fig. 6A) and HeLa cells (Fig. 6C). This transcriptional activation was further enhanced by cotransfection of p300 when using suboptimal amounts of BRCA1 (Fig. 6B). This effect also is observed on derepression of a tetracycline-repressible BRCA1 cell line (H.R., unpublished results) on cotransfection of p300 along with the RSV-LTR β-galactosidase reporter (Fig. 6C). Thus BRCA1 can mediate transcriptional activation in cooperation with p300 of RSV-LTR, a novel BRCA1 responsive promoter.
Discussion
A growing body of evidence points toward a function of p300/CBP in growth regulatory pathways. In addition to their interaction with tumor suppressors and protooncoproteins such as p53, c-Myb, c-Jun, or c-Fos (24, 37) or involvement in chromosomal translocations with MOZ- (33) and MLL- (31, 32, 38, 39) causing leukemia, p300/CBP are targets of the viral oncoproteins E1A or simian virus 40 large T, which antagonize the p300/CBP function in transcription (27, 40). Thus, the oncogenic potential of E1A could be in part caused by an obstruction of BRCA1:p300/CBP cooperation resulting in the loss of the tumor-suppressing function of BRCA1. In addition to enhancing transcriptional activation mediated by Gal4-BRCA11528–1863, p300/CBP promoted the BRCA1-mediated up-regulation of the RSV LTR. We conclude that BRCA1 and p300/CBP not only physically, but also functionally interact. In contrast to the only two other known promoters, namely MDM2 and P21 (23, 41, 42) stimulated by BRCA1, the RSV LTR does not contain p53-binding sites. Interestingly, the cooperation of p300 with Gal4-BRCA11528–1863 also could be observed with a reporter devoid of Gal4 DNA binding sites in RK13, but not 293 cells (data not shown), suggesting that BRCA1 is a general transcription factor. This transactivation could be mediated by RNA polymerase II through RNA helicase A, to which both BRCA1 and CBP bind, (43, 44), suggesting a multicomponent complex promoting transcription. Although the exact mechanism of BRCA1-mediated transcriptional activation remains unclear, it is probably not caused by the increased histone acetylation, because BRCA1 exhibits neither intrinsic HAT activity nor does it modulate the activity of p300 toward free histones (G.M.P. and R.J., unpublished observations). It is worth noting that BRCA1 also has been found in association with components of the histone deacetylase (45) and CtBP (46–48) complexes that mediate transcriptional repression. These associations are probably not contradictory, as they could be analogous in function to nuclear receptors, which can act as activators or repressors depending on their cofactors. BRCA1 and p300 colocalize substantially in structures around the nuclear rim and speckles within the nucleus in a cell cycle-dependent manner. This pattern is consistent with that of DNA in resting cells, thereby suggesting that BRCA1 and p300 collaborate in the maintenance of the quiescent state. Proteins thought to mediate chromatin remodeling such as MLL and bmi-1 have been reported to have a similar staining pattern (49). Thus, while BRCA1 may act mainly as a transcription factor in conjunction with p300/CBP during the G phases of the cell cycle, it may exert a different function on DNA repair/replication on dissociation from p300/CBP in S phase.
Acknowledgments
We thank Theo Palmer and Dan Petersen for help with confocal microscopy, Ralph Scully and David Livingston for the MS13 and MS110 antibodies, and Hong-Wu Chen from the Ron Evans lab for antibodies against ACTR/AIB1. We thank Milton H. Saier for comments on the manuscript and the members of the Verma and Hunter labs for suggestions and comments. R.J. was supported by a fellowship from the Deutsche Forschungsgemeinschaft, and H.R. was supported by a fellowship of the Schweizerische Nationalfonds für wissenschaftliche Forschung. I.M.V. was supported by grants from the National Institutes of Health and the American Cancer Society, Wayne and Gladys Valley Foundation, and H.N. and Frances C. Berger Foundation. T.H. was supported by National Institutes of Health Grants CA39780 and CA54418. T.H. and I.M.V. are American Cancer Society research professors of molecular biology.
Abbreviations
- CREB
cAMP response element binding protein
- CBP
CREB binding protein
- HAT
histone acetyl-transferase
- RSV
Rous sarcoma virus
- LTR
long terminal repeat
- HA
hemagglutinin
- GST
glutathione S-transferase
References
- 1.Bertwistle D, Ashworth A. Curr Opin Genet Dev. 1998;8:14–20. doi: 10.1016/s0959-437x(98)80056-7. [DOI] [PubMed] [Google Scholar]
- 2.Thomas J E, Smith M, Rubinfeld B, Gutowski M, Beckmann R P, Polakis P. J Biol Chem. 1996;271:28630–28635. doi: 10.1074/jbc.271.45.28630. [DOI] [PubMed] [Google Scholar]
- 3.Wilson C A, Payton M N, Pekar S K, Zhang K, Pacifici R E, Gudas J L, Thukral S, Calzone F J, Reese D M, Slamon D I. Nat Genet. 1996;13:264–265. doi: 10.1038/ng0796-264. [DOI] [PubMed] [Google Scholar]
- 4.Ruffner H, Verma I M. Proc Natl Acad Sci USA. 1997;94:7138–7143. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston D M. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 6.Bork P, Hofmann K, Bucher P, Neuwald A F, Altschul S F, Koonin E V. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- 7.Koonin E V, Altschul S F, Bork P. Nat Genet. 1996;13:266–268. doi: 10.1038/ng0796-266. [DOI] [PubMed] [Google Scholar]
- 8.Wu L C, Wang Z W, Tsan J T, Spillman M A, Phung A, Xu X L, Yang M C, Hwang L Y, Bowcock A M, Baer R. Nat Genet. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 9.Gowen L C, Avrutskaya A V, Latour A M, Koller B H, Leadon S A. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- 10.Hakem R, de la Pompa J L, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 11.Ludwig T, Chapman D L, Papaioannou V E, Efstratiadis A. Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 12.Boyd M, Harris F, McFarlane R, Davidson H R, Black D M. Nature (London) 1995;375:541–542. doi: 10.1038/375541b0. [DOI] [PubMed] [Google Scholar]
- 13.Xu X, Wagner K U, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng C X. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 14.Szabo C I, Wagner L A, Francisco L V, Roach J C, Argonza R, King M C, Ostrander E A. Hum Mol Genet. 1996;5:1289–1298. doi: 10.1093/hmg/5.9.1289. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, Farmer A A, Chen C F, Jones D C, Chen P L, Lee W H. Cancer Res. 1996;56:3168–3172. [PubMed] [Google Scholar]
- 16.Vaughn J P, Davis P L, Jarboe M D, Huper G, Evans A C, Wiseman R W, Berchuck A, Iglehart J D, Futreal P A, Marks J R. Cell Growth Differ. 1996;7:711–715. [PubMed] [Google Scholar]
- 17.Thomas J E, Smith M, Tonkinson J L, Rubinfeld B, Polakis P. Cell Growth Differ. 1997;8:801–809. [PubMed] [Google Scholar]
- 18.Scully R, Chen J, Ochs R L, Keegan K, Hoekstra M, Feunteun J, Livingston D M. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 19.Zhong Q, Chen C F, Li S, Chen Y, Wang C C, Xiao J, Chen P L, Sharp Z D, Lee W H. Science. 1999;285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 20.Chapman M S, Verma I M. Nature (London) 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- 21.Monteiro A N, August A, Hanafusa H. Proc Natl Acad Sci USA. 1996;93:13595–13599. doi: 10.1073/pnas.93.24.13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scully R, Anderson S F, Chao D M, Wei W, Ye L, Young R A, Livingston D M, Parvin J D. Proc Natl Acad Sci USA. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouchi T, Monteiro A N, August A, Aaronson S A, Hanafusa H. Proc Natl Acad Sci USA. 1998;95:2302–2306. doi: 10.1073/pnas.95.5.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janknecht R, Hunter T. Nature (London) 1996;383:22–23. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- 25.Ogryzko V V, Schiltz R L, Russanova V, Howard B H, Nakatani Y. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 26.Bannister A J, Kouzarides T. Nature (London) 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 27.Yang X J, Ogryzko V V, Nishikawa J, Howard B H, Nakatani Y. Nature (London) 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Lin R J, Schiltz R L, Chakravarti D, Nash A, Nagy L, Privalsky M L, Nakatani Y, Evans R M. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 29.Pazin M J, Kadonaga J T. Cell. 1997;89:325–328. doi: 10.1016/s0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- 30.Taki T, Sako M, Tsuchida M, Hayashi Y. Blood. 1997;89:3945–3950. [PubMed] [Google Scholar]
- 31.Sobulo O M, Borrow J, Tomek R, Reshmi S, Harden A, Schlegelberger B, Housman D, Doggett N A, Rowley J D, Zeleznik-Le N J. Proc Natl Acad Sci USA. 1997;94:8732–8737. doi: 10.1073/pnas.94.16.8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rowley J D, Reshmi S, Sobulo O, Musvee T, Anastasi J, Raimondi S, Schneider N R, Barredo J C, Cantu E S, Schlegelberger B, et al. Blood. 1997;90:535–541. [PubMed] [Google Scholar]
- 33.Borrow J, Stanton V P, Jr, Andresen J M, Becher R, Behm F G, Chaganti R S, Civin C I, Disteche C, Dube I, Frischauf A M, et al. Nat Genet. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 34.Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, Chong J M, Iwama T, Miyaki M. Oncogene. 1996;12:1565–1569. [PubMed] [Google Scholar]
- 35.Janknecht R, Hunter T. EMBO J. 1997;16:1620–1627. doi: 10.1093/emboj/16.7.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janknecht R, Nordheim A. Oncogene. 1996;12:1961–1969. [PubMed] [Google Scholar]
- 37.Shikama N, Lyon J, LaThangue N B. Trends Cell Biol. 1997;7:230–236. doi: 10.1016/S0962-8924(97)01048-9. [DOI] [PubMed] [Google Scholar]
- 38.Satake N, Ishida Y, Otoh Y, Hinohara S, Kobayashi H, Sakashita A, Maseki N, Kaneko Y. Genes Chromosomes Cancer. 1997;20:60–63. [PubMed] [Google Scholar]
- 39.Ida K, Kitabayashi I, Taki T, Taniwaki M, Noro K, Yamamoto M, Ohki M, Hayashi Y. Blood. 1997;90:4699–4704. [PubMed] [Google Scholar]
- 40.Eckner R, Ludlow J W, Lill N L, Oldread E, Arany Z, Modjtahedi N, DeCaprio J A, Livingston D M, Morgan J A. Mol Cell Biol. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Somasundaram K, Peng Y, Tian H, Bi D, Weber B L, El-Deiry W S. Oncogene. 1998;16:1713–1721. doi: 10.1038/sj.onc.1201932. [DOI] [PubMed] [Google Scholar]
- 42.Somasundaram K, Zhang H, Zeng Y X, Houvras Y, Peng Y, Wu G S, Licht J D, Weber B L, El-Deiry W S. Nature (London) 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 43.Anderson S F, Schlegel B P, Nakajima T, Wolpin E S, Parvin J D. Nat Genet. 1998;19:254–256. doi: 10.1038/930. [DOI] [PubMed] [Google Scholar]
- 44.Nakajima T, Uchida C, Anderson S F, Lee C G, Hurwitz J, Parvin J D, Montminy M. Cell. 1997;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 45.Yarden R I, Brody L C. Proc Natl Acad Sci USA. 1999;96:4983–4988. doi: 10.1073/pnas.96.9.4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li S, Chen P L, Subramanian T, Chinnadurai G, Tomlinson G, Osborne C K, Sharp Z D, Lee W H. J Biol Chem. 1999;274:11334–11338. doi: 10.1074/jbc.274.16.11334. [DOI] [PubMed] [Google Scholar]
- 47.Wong A K, Ormonde P A, Pero R, Chen Y, Lian L, Salada G, Berry S, Lawrence Q, Dayananth P, Ha P, et al. Oncogene. 1998;17:2279–2285. doi: 10.1038/sj.onc.1202150. [DOI] [PubMed] [Google Scholar]
- 48.Yu X, Wu L C, Bowcock A M, Aronheim A, Baer R. J Biol Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 49.Ennas M G, Sorio C, Greim R, Nieddu M, Scarpa A, Orlandini S, Croce C M, Fey G H, Marschalek R. Cancer Res. 1997;57:2035–2041. [PubMed] [Google Scholar]