

Abstract
Increases in reactive oxygen species (ROS) and tissue evidence of oxidative injury are common in patients with inflammatory processes or tissue injury. This has led to many clinical attempts to scavenge ROS and reduce oxidative injury. However, we live in an oxygen rich environment and ROS and their chemical reactions are part of the basic chemical processes of normal metabolism. Accordingly, organisms have evolved sophisticated mechanisms to control these reactive molecules. Recently, it has become increasingly evident that ROS also play a role in the regulation of many intracellular signaling pathways that are important for normal cell growth and inflammatory responses that are essential for host defense. Thus, simply trying to scavenge ROS is likely not possible and potentially harmful. The 'normal' level of ROS will also likely vary in different tissues and even in different parts of cells. In this paper, the terminology and basic chemistry of reactive species are reviewed. Examples and mechanisms of tissue injury by ROS as well as their positive role as signaling molecules are discussed. Hopefully, a better understanding of the nature of ROS will lead to better planned therapeutic attempts to manipulate the concentrations of these important molecules. We need to regulate ROS, not eradicate them.
Introduction
Production of reactive oxygen species (ROS) and oxidative stress are associated with tissue injury and many pathological processes, including septic shock [1,2]. This has prompted clinical attempts to regulate oxygen radical production and oxidative stress [3-7]. Signs of oxidative stress often have been reduced, at least in blood, but by and large these clinical trials have had little beneficial outcome, although a reduction of mortality was observed in one trial [8] and a reduction in multi-organ system failure in another [9]. An underlying assumption has been that ROS randomly and indiscriminately attack important chemical pathways and, thereby, cause cell injury or death, but more recently it has become evident that ROS can act as important signaling molecules under physiological and pathophysiological conditions [10-14]. Thus, to understand the potential benefits and limitations of therapeutic approaches aimed at increasing ROS scavenging, one must understand the 'meaning' of oxidation and ROS. It will then become evident that although ROS are potentially very toxic, they are also essential factors in normal metabolism.
Oxygen is now the most prevalent element in the earth's crust [15]. It exists in air as a diatomic molecule, O2. Except for a small number of anaerobic bacteria, all living organisms use O2 for energy production and it is thus essential for life as we know it. Energy production from food material by organisms requires 'oxidation', which means the loss of electrons. In anaerobic organisms, electrons are taken up by hydrogen, but in aerobic organisms, the loss of electrons occurs much more efficiently through the use of electron carriers such as nicotinamide adenine dinucleotide (NAD+) and flavins, which are 'reduced' in the process by gaining electrons from target molecules and are re-oxidized by donating electrons to O2 through oxidative phosphorylation. The potential for O2 to oxidize other molecules also makes it toxic. Oxidation is the basic process in combustion; fires do not burn without O2. It is also the cause of rust. Oxidation can inactivate important enzymes and anaerobes that do not have anti-oxidant mechanisms do not survive in an O2 environment. Thus, for organisms to have evolved in an O2 world there has had to be evolution of potent mechanisms to control oxidative processes.
Terminology
Before continuing with a discussion of potential beneficial and harmful aspects of ROS, we need to review the terms involved [15]. Oxidation is the gain of oxygen by a substance or a loss of an electron. A useful reminder is 'LEO', which stands for 'lose electron oxidized'. Reduction is the loss of oxygen by a substance, the gain of an electron or the gain of hydrogen; a useful reminder is 'GER', which stands for 'gain electron reduced'. An oxidizing agent takes an electron or hydrogen from another chemical or adds oxygen. A reducing agent supplies electrons or hydrogen to another chemical, or removes oxygen. An important chemical principle is that because of their spin, electrons are most stable when they are paired in their orbits. Unpaired electrons are attracted to magnetic fields, which makes them more reactive. Substances that have unpaired electrons and are capable of independent existence are called free radicals. By this definition, atomic hydrogen is a free radical because it only has one electron. O2 is a radical because it has two unpaired electrons in its outer orbitals, and this gives O2 its reactivity. However, the two unpaired electrons on O2 have parallel spins, which means that O2 can only oxidize another molecule by accepting a pair of electrons that have antiparallel spin so as to fit into the two vacant spaces of O2. This tends to make O2 only accept one electron at a time and thus react sluggishly with non-radicals. Thus O2 is the most stable state of oxygen. Superoxide (O2•-) has one more electron than O2. Since only one electron is unpaired in O2•-, it is simpler for it to accept an electron and is thus more reactive than O2. However, O2•- is still not a very reactive radical; in the presence of H+ or HO2• it can reduce O2•- to H2O2 or be oxidized to O2.
Another term that is often used is 'reactive oxygen species' (ROS). This term includes radicals as well as chemicals that can take part in radical type reactions (i.e. gain or loose electrons), but are not true radicals in that they do not have unpaired electrons. Examples of non-radical ROS include hydrogen peroxide (H2O2), hypochlorous acid (HOCI), ozone (O3) and singlet oxygen (1ΔgO2). An important product of the two radicals O2•- and NO is peroxynitrite (ONOO-); this reaction occurs at a diffusion limited rate [16,17]. Although not a radical itself, ONOO- can result in cytotoxic processes, including lipid peroxidation, the formation of nitrotyrosine residues that can inactivate enzymes, depletion of glutathione, and DNA injury. Besides oxygen-based radicals, there are also reactive nitrogen species such as nitric oxide (NO) and nitrogen dioxide (NO2), sulfur based molecules such as thinyl (RS) and perthinyl (RSS), as well as carbon centered molecules such as trichloromethyl (CCI3•), which is a product of metabolism of carbon tetrachloride (CCI4) [15].
Sources of O2•- and ROS
Under the conditions of normal metabolism the most important source of O2•- is the mitochondrial electron transport chain, which leaks a few electrons directly onto O2 as part of normal metabolism. It is estimated that 1 % to 3% of O2 reduced in mitochondria is in the form of O2•- [18]. This comes from two sites, complex 1 (NADH dehydrogenase) and complex III (ubiquinone-cytochrome c reductase), with the latter being the major source under normal conditions [11].
Several enzymes also contribute to O2•- production. One of the best characterized is xanthine oxidase, which is present in the cytosol of many tissues but also can be found in circulating blood and bound to glycosaminoglyan sites in the arterial wall [19]. Normally the enzyme acts as a dehydrogenase and transfers electrons to NAD+ rather than O2, but in ischemia reperfusion [20,21] or in sepsis [21,22] the active site of the enzyme is oxidized and the enzyme acts as an oxidase and produces O2•-.
In phagocytic cells the major source of O2•- is a multi-component oxidase called NAD(P)H oxidase [23,24]. In response to membrane signals this complex produces a burst of O2•- that is important for killing invading microorganisms. Genetic mutations in components of the complex result in chronic granulomatous disease, which is characterized by repeated infections. There are at least five components to the complex. Two, p22Phox (phox stands for phagocyte associated oxidase) and gp91Phox (subsequently called NOX2) are found in membranes [25,26]. NOX2 is the component that produces O2•-. The complex is activated when the cytosolic component p67Phox is transported to the membrane complex by the transporter molecule p47Phox [27]. The attachment of p67phox to the membrane complex results in a conformational change in p22Phox that exposes the active site on NOX2. The small g-protein Rac also contributes to the activity of the enzyme and transmits membrane signals to the complex. Recently, a family of non-phagocytic NOXs with the same basic components as the phagocytic type have been identified in numerous types of cells, including vascular smooth muscle, endothelial, skeletal muscle, fibroblast, and mesangial cells [28-30]. The non-phagocytic form produces much lower amounts of O2•- compared to the phagocytic form but is constitutively active.
O2•- is also produced by a number of metabolically active enzymes as part of their normal function or when there is inadequate substrate. For example, cytochrome P450 enzymes can produce O2•- as a side reaction when they breakdown target molecules [15]. Nitric oxide synthases, the family of enzymes that produce NO, produce O2•- when the substrates L-arginine or co-factor tetrahydropteridines are insufficient [21,31,32].
O2•- can also be produced by cyclooxygenase as part of arachidonic acid metabolism. O2•- even can be produced through auto-oxidation of molecules such as gylceraldehyde, FMNH2, FADH2, adrenalin, noradrenalin, dopamine and thiol containing molecules such as cysteine in the presence of O2 [1,15]. Since we live in an oxygen rich environment, and ROS are byproducts of normal metabolism, potent protective mechanisms have evolved to allow life to continue. One of the most fundamental antioxidant enzymes is superoxide dismutase (SOD), which catalyzes the reaction of two O2•- and two H+ to H2O2 (reduced) and O2 (oxidized) [33]. There are three forms to this enzyme: SOD1, a copper/zinc (Cu/Zn) isoform present in the cytosol; SOD2, a manganese (Mn) isoform present in mitochondria; and SOD3, a Cu/Zn isoform present in the extracellular space. Knockout of SOD2 in mice is lethal in the first week of life [34,35] whereas deficiencies of SOD1 and SOD3 are not lethal but result in less tolerance of neuronal injury [36] or hyperoxia, respectively [37]. H2O2 itself is not a radical but is a ROS and may actually account for most of the O2•- reactions. What makes H2O2 so important is that it is more stable than O2•- and can diffuse across membranes. In the presence of iron in the ferrous form (Fe2+), H2O2 can be reduced to the highly reactive OH• radical. It is thus important that H2O2 also be reduced in a controlled manner and this is achieved by catalase or glutathione peroxidase. Other antioxidants include cysteine, glutathione itself, ascorbic acid (vitamin C) and α-tocopherol (vitamin E), which can also scavenge peroxynitrite.
Production of injury by ROS
One of the major toxic effects of excessive ROS is damage to cellular membranes by the process of lipid peroxidation. Species such as OH•, HO2•-, and OONO-, but not O2•-, can extract an H from methylene (-CH2-), which creates the carbon radical -•CH-. This carbon radical then attacks other -CH2- groups in lipid molecules, and creates a chain reaction that alters the fluidity and shape of the membrane. This is the same process that makes oil rancid. A consequence of the change to the cell membrane is disruption of calcium handling, which is essential for intracellular signaling. Lipid peroxides can also damage DNA and proteins.
Attack of DNA by ROS results in DNA strand breaks. As with lipids, O2•- and H2O2 do not do this by themselves but do so in the presence of hypochlorous acid (HOCI). It is also possible that oxidative stress results in the release of bound intracellular iron and copper ions that can then generate the highly toxic OH• through what is known as the Fenton reaction [15]. The potential for this to produce mutations and to alter normal transcriptional and translational processes is obvious. Besides these direct effects of oxidative injury, there can be indirect injury because the nicks and breaks in DNA strands can trigger activation of Poly(ADP) polymerase (PARP), which alters gene expression, DNA replication and may trigger apoptosis. It can also deplete NAD+, which leads to cellular ATP depletion [38].
Proteins, too, can be targets of oxidative alterations. Protein oxidation disrupts receptors, enzyme function and signal transduction pathways. The amino acid tyrosine is particularly prone to attack by ROS, especially reactive nitrogen species such as OONO- [39]. The product of OONO- and tyrosine is 3-nitrotyrosine and antibodies against it are used as a 'footprint' of protein oxidation [40]. Oxidation of proteins also can lead to products with carbonyl groups [41]. The amino acids histidine, arginine, lysine and proline are especially vulnerable. However, just because a protein is oxidized does not mean that it has lost its function and the biological significance of oxidation of a particular protein needs to be confirmed by evidence of an alteration in function. An example of a protein function altered by oxidation is the inactivation of the intra-mitochondrial SOD, SOD2, by peroxynitrite [42]. Because O2•- scavenging is reduced, oxidative processes are accelerated. Potentially important functional sites for oxidation of proteins are the -SH groups because the formation of -S-S- bonds between different protein strands or parts of the same strand can result in conformational changes in the protein that alter its function (see below).
ROS in sepsis
There is evidence from animal studies that an increase in ROS in sepsis is of pathophysiological importance. Oxygen radical scavengers reduce lung injury in animal models [43-48] and improve hemodynamics [48,49]. An interesting and potentially clinically important example of O2•- induced injury is the deactivation of catecholamines in inflammatory reactions [50]. Catecholamines can act as antioxidants because of their ability to interact with ROS, but this process also leads to their deactivation and the formation of adrenochromes, which are toxic themselves. Of interest, in the first identification of SOD, one of the tests of the activity of the enzyme was the prevention of oxidation of catecholamines [33]. The potential clinical importance of the oxidation of catecholamines was demonstrated by Salvemini and coworkers [50] who showed that ROS decrease the activity of catecholamines and oxygen radical scavengers restore cardiovascular responsiveness to catecholamines in an animal model of sepsis.
There is also evidence for a clinically significant role for ROS in humans. Patients with sepsis who are able to achieve a normal antioxidant potential in their plasma have better survival [51] and treatment of septic patients with the antioxidants glutathione and N-acetylcysteine decreases measures of oxidative injury [4]. N-acetylcysteine reduces the respiratory burst from neutrophils of septic patients [52] and patients with lung injury randomized to antioxidant therapy with N-acetylcysteine versus placebo had an improvement in systemic oxygenation and a reduction in the need for ventilatory support [5]. An improvement in hepatic blood flow in septic patients has also been observed [6]. On the other hand, no significant clinical advantage to the administration of N-acetylcysteine was observed in two studies [3,7]. An important limitation of N-acetylcysteine is that it works by increasing the intracellular cysteine concentration, which normally is high relative to the plasma concentration [53]. Thus, potentially toxic plasma levels are needed to reach the necessary intracellular levels. N-acetylcysteine also has a low Km for the removal of O2•-, which is why it has to be present at high concentrations. Augmenting oxygen radical scavenging activity in patients with septic shock by combining N-acetylcysteine and glutathione produced a trend towards less organ damage [4] but results were not conclusive. To date there is no clear evidence that antioxidant therapy alters outcome in septic patients [54], although as noted in the introduction, supplementation of feeds with vitamins was shown to reduce mortality of a general group of severely ill patients [8] and reduce multiorgan dysfunction in a group of critically ill patients who were primarily trauma victims [9].
ROS and cell signalling
Perhaps the failure to find a clinical role for therapies aimed at the reduction of ROS is that they are based on the limited paradigm that ROS only cause injury. An alternative view is that although ROS are potentially highly toxic, redox reactions are also part of the basic chemical processes of life [10,11]. Since organisms have had to develop efficient regulatory mechanisms to keep the production of ROS under control, these same mechanisms could be used to regulate other intracellular processes [12-14,53,55]. A parallel might be seen with that of Ca2+ handling. The intracellular Ca2+ concentration is kept at less than 1/10,000 of extracellular Ca2+ so as to avoid the interaction of Ca2+ and phosphate and bone formation. Because of the large transmembrane gradient of Ca2+, the leak of small amounts of Ca2+ across cell membranes through specialized channels can provide one of the cell's basic signalling mechanisms. Similarly, regulation of extracellular and intracellular levels of O2•- and H2O2 could provide potential for signalling of extracelluar to intracellular mechanisms. In this paradigm, ROS are not just random destructive species but regulators of metabolic processes and part of the chemistry of life [10,11]. Furthermore, evidence of oxidative injury may be the end result of the inflammatory process rather than the major cause of injury, in which case the use of antioxidants may be too late. Another analogy might be helpful. Consider walking along a beach and observing a rusted old ship lying on the shore. You conclude that the reason why the ship was abandoned is because it is so rusted (oxidized) until you walk past the ship and notice a large hole in the hull. You then realize that the ship was abandoned because of the hole and rusted when it was no longer cared for. Signs of oxidative changes may simply indicate that molecules or cells have been abandoned by the organism and are not themselves the major cause of the disease process.
Although there is a lot of evidence indicating that ROS and the redox state have a signaling role in bacteria and plants, there was less evidence in mammalian cells until recently. For example, in bacteria the transcription factor OxyR is redox sensitive [13]. There is now an increasing number of examples in animals of ROS-based signaling, including protein tyrosine phosphatase 1B (PTP-1B) [56], thioredoxin [57], SERCA2 [58] and Ras [59]. A well-characterized radical that has a major role in normal physiological function is nitric oxide (NO•). This radical has a central role in the regulation of vascular tone, nerve function and immune regulation. Even the potentially toxic by-product of NO• and O2•-, OONO-, has recently been shown to play a role in the regulation of vascular tone [58]. Cohen and coworkers found that NO• induced dilatation occurs by the production of low concentrations of OONO-, which directly stimulates the sarco/endoplasmic reticulum calcium (Ca2+) ATPase (SERCA) to decrease intracellular Ca2+ and thereby produce vasodilatation. This occurs by reversible S-glutathiolation of the thiol of a cysteine molecule on SERCA. Thus, by removing O2•- and preventing the formation of OONO-, superoxide scavengers actually blocked NO-induced vascular relaxation. However, high levels of oxidative stress, including high concentrations of OONO-, resulted in irreversible oxidation of key thiols and prevented normal NO-induced relaxation. An important lesson may be learnt from the NO system. Endothelial and neuronal cells that use NO for signalling produce NO in small amounts, whereas macrophages and neutrophils that use NO to attack invading organisms produce large amounts. Similarly, the NAD(P)H oxidase in phagocytic cells produces large quantities of O2•-, whereas the NAD(P)H oxidases in non-phagocytic cells produce much smaller amounts of O2•-, consistent with a signalling role.
The role of ROS in the signaling of a number of growth factors has also been well established. An excellent example is the role of ROS in angiotensin signaling as established by Griendling and co-workers [29,60-62]. They showed that exposure of vascular smooth muscle to angiotensin II results in smooth muscle growth that is dependent upon increased production of O2•- by NAD(P)H oxidase and its subsequent dismutation to H2O2. H2O2 then activates downstream prosurvival pathways and, in vivo, this results in vascular hypertrophy. Other growth factors such as platelet derived growth factor have been shown to have similar signaling mechanisms [63].
ROS also play a role in the intracellular signaling of tumor necrosis factor-α [22,64-72] and this too seems to occur through O2•- produced by NAD(P)H oxidase and likey involves regulation of the transcriptional activity of NFκB. Similarly, it has recently been shown that lipopolysaccharide activation of Toll-like receptor 4 increases O2•- production by NAD(P)H oxidase and this too leads to NFκB activation [73].
Various mechanisms have been explored recently that can explain how ROS can signal intracellular events. These generally involve the oxidation of cysteine residues and formation of -S-S- bonds [12,14,53,74,75] (Figs 1 to 3). These bonds can be within a molecule and result in a conformational change (Fig. 1) or between protein strands, in which case they result in dimerization of proteins. The creation of -S-S- bonds can also result in the release of an inhibitory molecule (Fig. 2). Some reactions are irreversible and result in protein instability or irreversible protein cross-linking. However, an interesting reversible process is oxidation of cysteinyl thiols by S-glutathiolation from thiol disulfide exchange reactions involving oxidized glutathione or from direct oxidation of protein cysteinyl thiols followed by reaction with reduced glutathione [75] (Fig. 3). In the case of PTP-1 B, stabilization of an oxidized cysteine occurs through the formation of a mixed disulfide with glutathione (Fig. 3). The formation of the mixed disulfide prevents the irreversible oxidation of the thiol to sulfinic or sulfonic acid and allows for the reactivation of the enzyme by cellular thioreductase, although recently it has become apparent that even sufinic groups can be re-oxidized [76-78].
Figure 1.
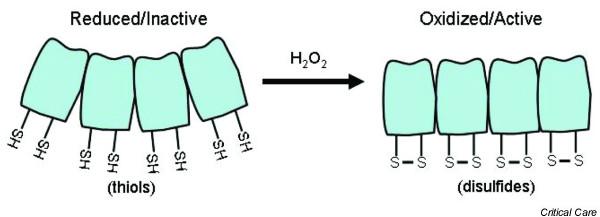
The change from thiols (-SH) to disulfide bonds (-S-S-) can produce a conformational change that may allow better protein-protein or protein-DNA interactions. Adapted from Droge et al. [11].
Figure 3.

Regulation of phosphatase by the redox state. Cysteine molecules have sulfur atoms (S) that are protonated and not reactive in most proteins. However, on some molecules, such as phosphatases, S can form thiolates (S-) at normal pH and these can be reversibly oxidized. The top of the figure shows the balance between phosphatase activity (which dephosphorylates molecules) and kinase activity (which phosphorylates and activates molecules). Phosphatase activity is regulated by the redox state as shown in the cycle below the bracket. Oxidation to sulfenic acid (-S-OH) is reversible. This can occur by glutathiolation (GSH) or by the formation of disulfides. However, excessive oxidation leads to sulfinic acid, which cannot easily be converted back to reduced forms of sulfur.
Figure 2.
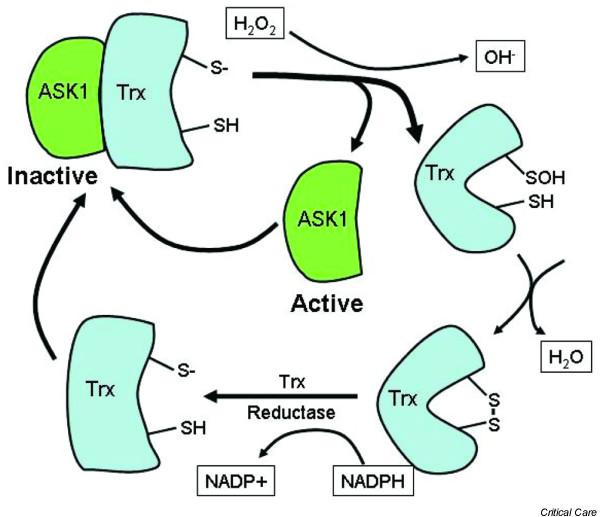
Oxidation of thioredoxin (Trx) by hydrogen peroxide (H2O2) leads to a change in shape of the molecule and the release of the transcriptional factor ASK1. Trx is then reduced again by Trx reductase, which allows it to again bind to ASK1 and inactivate this transcriptional factor. Through this mechanism the redox state of the cell can regulate the activity of the transcriptional factor ASK1.
Implications
ROS are an essential part of many metabolic pathways; they are part of the flame of basic energy producing processes. Organisms have had to evolve elaborate mechanisms to live with these reactive molecules and seem also to have evolved to use the reactive nature of these molecules for intracellular signal transduction. Thus, a key concept in dealing with ROS must be to regulate but not eradicate, for turning off production of ROS is tantamount to turning off the engine that powers us. ROS also seem to have specific roles in different cell types and thus therapeutic strategies for the manipulation of ROS should take into account the source of ROS, the targets of the ROS, specific cell types involved and the specific location of ROS production in these cells, for one needs to know that the potential therapeutic agent actually can get to the site of excess ROS production. A list of things to consider when examining the potential of a therapeutic agent to deal with ROS is given in Table 1. In the management of ROS we will need to be careful to not repeat the mistake that was made with global inhibition of NO production.
Table 1.
A check list for the evaluation of the utility of anti-oxidant therapies
| 1 | What is the reactive oxygen species that is causing the oxidative injury? |
| 2 | What is the target molecule? |
| 3 | What is the source of the ROS? |
| 4 | What types of cells produce the ROS? |
| 5 | Where in the cells are the ROS produced? |
| 6 | What is the potentially useful role of the ROS? |
Abbreviations
PTP-1B = phosphatase 1B; ROS = reactive oxygen species; SERCA = sarco/endoplasmic reticulum calcium ATPase; SOD = superoxide dismutase.
Competing interests
The author(s) declare that they have no competing interests.
References
- Salvemini D, Cuzzocrea S. Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radical Biol Med. 2002;33:1173–1185. doi: 10.1016/S0891-5849(02)00961-9. [DOI] [PubMed] [Google Scholar]
- Halliwell B. The role of oxygen radicals in human disease, with particular reference to the vascular system. Haemostasis. 1993;23:118–126. doi: 10.1159/000216921. [DOI] [PubMed] [Google Scholar]
- Jepsen S, Herlevsen P, Knudsen P, Bud MI, Klausen NO. Antioxidant treatment with N-acetylcysteine during adult respiratory distress syndrome: A prospective, randomized, placebo-controlled study. Crit Care Med. 1992;20:918–923. doi: 10.1097/00003246-199207000-00004. [DOI] [PubMed] [Google Scholar]
- Ortolani O, Conti A, De Gaudio AR, Moraldi E, Cantini Q, Novelli G. The effect of glutathione and N-acetylcysteine on lipoperoxidative damage in patients with early septic shock. Am J Resp Crit Care Med. 2000;161:1907–1911. doi: 10.1164/ajrccm.161.6.9903043. [DOI] [PubMed] [Google Scholar]
- Suter P, Domenighetti G, Schaller M-D, Laverriere M-C, Ritz R, Perret C. N-acetylcysteine enhances recovery from acute lung injury in man. Chest. 1994;105:190–194. doi: 10.1378/chest.105.1.190. [DOI] [PubMed] [Google Scholar]
- Rank N, Michel C, Haertel C, Lenhart A, Welte M, Meier-Hellmann A, Spies C. N-acetylcysteine increases liver blood flow and improves liver function in septic shock patients: Results of a prospective, randomized, double-blind study. Crit Care Med. 2000;28:3799–3807. doi: 10.1097/00003246-200012000-00006. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Wheeler AP, Arons MM, Morris PE, Paz HL, Russell JA, Wright PE. A trial of antioxidants N-acetylcysteine and procysteine in ARDS. Chest. 1997;112:164–172. doi: 10.1378/chest.112.1.164. [DOI] [PubMed] [Google Scholar]
- Crimi E, Liguori A, Condorelli M, Cioffi M, Astuto M, Bontempo P, Pignalosa O, Vietri MT, Molinari AM, Sica V, Della Corte F, Napoli C. The beneficial effects of antioxidant supplementation in enteral feeding in critically ill patients: a prospective, randomized, double-blind, placebo-controlled trial. Anesth Analg. 2004;99:857–863. doi: 10.1213/01.ANE.0000133144.60584.F6. [DOI] [PubMed] [Google Scholar]
- Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg. 2002;236:814–822. doi: 10.1097/00000658-200212000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/S0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Torres M, Fukuto J. Redox signaling. Mol Cell Biochem. 2002;234–235:49–62. doi: 10.1023/A:1015913229650. [DOI] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signalling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3. Oxford: Oxford University Press; 1999. [Google Scholar]
- Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol. 1996;40:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CR, Darley-Usmar V, Berrington WR, McAdams M, Gore JZ, Thompson JA, Parks DA, Tarpey MM, Freeman BA. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci USA. 1996;93:8745–8749. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich V, Bachschmid M. Superoxide as a messenger of endothelial function. Biochem Biophys Res Commun. 2000;278:1–8. doi: 10.1006/bbrc.2000.3733. [DOI] [PubMed] [Google Scholar]
- Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: redox mechanisms in blood vessels. Arterioscler Thromb Vasc Biol. 2005;25:274–278. doi: 10.1161/01.ATV.0000149143.04821.eb. [DOI] [PubMed] [Google Scholar]
- Brandes RP, Koddenberg G, Gwinner W, Kim D, Kruse HJ, Busse R, Mugge A. Role of increased production of superoxide anions by NAD(P)H oxidase and xanthine oxidase in prolonged endotoxemia. Hypertension. 1999;33:1243–1249. doi: 10.1161/01.hyp.33.5.1243. [DOI] [PubMed] [Google Scholar]
- Quinn MT, Gauss KA. Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol. 2004;76:760–781. doi: 10.1189/jlb.0404216. [DOI] [PubMed] [Google Scholar]
- Babior MB. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- Geiszt M, Leto TL. The Nox family of NAD(P)H oxidases: host defense and beyond. J Biol Chem. 2004;279:51715–51718. doi: 10.1074/jbc.R400024200. [DOI] [PubMed] [Google Scholar]
- Nisimoto Y, Motalebi S, Han CH, Lambeth JD. The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b(558) J Biol Chem. 1999;274:22999–23005. doi: 10.1074/jbc.274.33.22999. [DOI] [PubMed] [Google Scholar]
- Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase. Role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc Natl Acad Sci USA. 1996;93:6770–6774. doi: 10.1073/pnas.93.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA. 1997;94:6954–6958. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci USA. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Zingarelli B, O'Connor M, Salzman AL. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxyni-trite. Proc Natl Acad Sci. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-U. [DOI] [PubMed] [Google Scholar]
- Beckman JS. Reactions between nitric oxide, superoxide, and peroxynitrite: Footprints of peroxynitrite in vivo. Adv Pharmacol. 1995;34:17–43. doi: 10.1016/s1054-3589(08)61079-0. [DOI] [PubMed] [Google Scholar]
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determiantion of oxidatively modified proteins. Methods Enzymol. 1994;233:346–363. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- Yamakura F, Taka H, Fujimura T, Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- Olson NC, Anderson DL, Grizzle MK. Dimethylthiourea attenuates endotoxin-induced acute respiratory failure in pigs. J Appl Physiol. 1987;63:2426–2432. doi: 10.1152/jappl.1987.63.6.2426. [DOI] [PubMed] [Google Scholar]
- Seekamp A, Lalonde C, Zhu D, Demling R. Catalase prevents prostanoid release and lung lipid peroxidation after endotoxemia in sheep. J Appl Physiol. 1988;65:1210–1216. doi: 10.1152/jappl.1988.65.3.1210. [DOI] [PubMed] [Google Scholar]
- Olson NC, Grizzle MK, Anderson DL. Effect of polyethylene glycol-superoxide dismutase and catalase on endotoxemia in pigs. J Appl Physiol. 1987;63:1526–1532. doi: 10.1152/jappl.1987.63.4.1526. [DOI] [PubMed] [Google Scholar]
- Petrone WF, English DK, Wong K, McCord JM. Free radicals and inflammation: Superoxide-dependent activation of a neutrophil chemotactic factor in plasma. Medical Sci. 1980;77:1159–1163. doi: 10.1073/pnas.77.2.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard BR, Lucht WD, Niedermeyer ME, Snapper JR, Olgetree LM, Bricham KL. Effect of N-acetylcysteine on the pulmonary response to endotoxin in the awake sheep and upon in vitro granulocyte function. J Clin Invest. 1984;73:1772–1780. doi: 10.1172/JCI111386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez PK, Zhuang J, Doctrow SR, Malfroy B, Benson PF, Menconi MJ, Fink MP. EUK-8, a synthetic superoxide dismutase and catalase mimetic, ameliorates acute lung injury in endotoxemic swine. J Pharmacol Exp Therapeut. 1995;275:798–806. [PubMed] [Google Scholar]
- Saetre T, Hoiby EA, Aspelin T, Lermark G, Egeland T, Lyberg T. Aminoethyl-isothiourea, a nitric oxide synthase inhibitor and oxygen radical scavenger, improves survival and counteracts hemodynamic deterioration in a porcine model of streptococcal. Crit Care Med. 2000;28:2697–2706. doi: 10.1097/00003246-200008000-00003. [DOI] [PubMed] [Google Scholar]
- Macarthur H, Westfall TC, Riley DP, Misko TP, Salvemini D. Inactivation of catecholamines by superoxide gives new insights on the pathogenesis of septic shock. Proc Natl Acad Sci USA. 2000;97:9753–9758. doi: 10.1073/pnas.97.17.9753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley HC, Bacon PJ, Goode HF, Webster NR, Jones JG, Menon DK. Plasma antioxidant potential in severe sepsis: a comparison of survivors and nonsurvivors. Crit Care Med. 1996;24:1179–1183. doi: 10.1097/00003246-199607000-00019. [DOI] [PubMed] [Google Scholar]
- Heller AR, Groth G, Heller SC, Breitkreutz R, Nebe T, Quintel M, Koch T. N-acetylcysteine reduces respiratory burst but augments neutrophil phagocytosis in intensive care unit patients. Crit Care Med. 2001;29:272–276. doi: 10.1097/00003246-200102000-00009. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Greene R. From mucolytic to antioxidant and liver protection: New aspects in the intensive care unit career of N-acetylcys-teine. Crit Care Med. 2000;28:3935–3938. doi: 10.1097/00003246-200012000-00037. [DOI] [PubMed] [Google Scholar]
- Janssen-Heininger YMW, Poynter ME, Baeuerle PA. Recent advances torwards understanding redox mechanisms in the activation of nuclear factor [kappa]b. Free Radic Biol Med. 2000;28:1317–1327. doi: 10.1016/S0891-5849(00)00218-5. [DOI] [PubMed] [Google Scholar]
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Myazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- Adachi T, Pimentel DR, Heibeck T, Hou X, Lele YJ, Jiang B, Ido Y, Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;27:29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW, Akers M, Yin Q, Fujio Y, Walsh K, Griendling KK. Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells. J Biol Chem. 1999;274:22699–22704. doi: 10.1074/jbc.274.32.22699. [DOI] [PubMed] [Google Scholar]
- Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- Sundaresan M, Yu Z-X, Ferrans VJ, Irani K, Fingel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Ziff M. Increased superoxide anion release from human endothelial cells in response to cytokines. J Immunol. 1986;137:3295–3298. [PubMed] [Google Scholar]
- Ferro TJ, Gertzberg N, Selden L, Neumann P, Johnson A. Endothelial barrier dysfunction and p42 oxidation induced by TNF-alpha are mediated by nitric oxide. Am J Physiol Lung Cell Mol Physiol. 1997;272:L979–L988. doi: 10.1152/ajplung.1997.272.5.L979. [DOI] [PubMed] [Google Scholar]
- Rahman A, Kefer J, Bando M, Niles W, Malik AB. E-selectin expression in human endothelial cells by TNF-alpha-induced oxidant generation and MF-kB activation. Am J Physiol Lung Cell Mol Physiol. 1998;275:L533–L544. doi: 10.1152/ajplung.1998.275.3.L533. [DOI] [PubMed] [Google Scholar]
- De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, Griendling KK. Tumour necrosis factor a activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J. 1998;329:653–657. doi: 10.1042/bj3290653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy H, Shayman J, Till GO, Mahrougui M, Owens C, Ryan U, Ward PA. Superoxide responses of endothelial cells to C5a and TNF-alpha: divergent signal transduction pathways. Am J Physiol Lung Cell Mol Physiol. 1992;263:L51–L59. doi: 10.1152/ajplung.1992.263.1.L51. [DOI] [PubMed] [Google Scholar]
- Phelps DT, Ferro TJ, Higgins PJ, Shankar R, Parker DM, Johnson A. TNF-alpha induces peroxynitrite-mediated depletion of lung endothelial glutathione via protein kinase C. Am J Physiol. 1995;269:L551–L559. doi: 10.1152/ajplung.1995.269.4.L551. [DOI] [PubMed] [Google Scholar]
- Goode HF, Webster NR. Free radicals and antioxidants in sepsis. Crit Care Med. 1993;21:1770–1776. doi: 10.1097/00003246-199311000-00029. [DOI] [PubMed] [Google Scholar]
- Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Cell Physiol. 2001;280:C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719. [DOI] [PubMed] [Google Scholar]
- Demling RH, Lalonde C, Jin L-J, Ryan P, Fox R. Endotoxemia causes increased lung tissue lipid peroxidation in unanesthetized sheep. J Appl Physiol. 1986;60:2094–2100. doi: 10.1152/jappl.1986.60.6.2094. [DOI] [PubMed] [Google Scholar]
- Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J Immunol. 2004;173:3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- Xu D, Rovira II, Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Dev Cell. 2002;2:251–252. doi: 10.1016/S1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- Filomeni G, Rotilio G, Ciriolo MR. Cell signalling and the glutathione redox system. Biochem Pharmacol. 2002;64:1057–1064. doi: 10.1016/S0006-2952(02)01176-0. [DOI] [PubMed] [Google Scholar]
- Woo HA, Chae HA, Hwang SC, Yang K-S, Kang SW, Kim K, Rhee SG. Reversing the inactivatino of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Woo HA, Kang SW, Kim HK, Yang K-S, Chae HZ, Rhee SG. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid: Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J Biol Chem. 2000;278:47361–47364. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]