

Abstract
In disseminated intravascular coagulation (DIC) there is extensive crosstalk between activation of inflammation and coagulation. Endogenous anticoagulatory pathways are downregulated by inflammation, thus decreasing the natural anti-inflammatory mechanisms that these pathways possess. Supportive strategies aimed at inhibiting activation of coagulation and inflammation may theoretically be justified and have been found to be beneficial in experimental and initial clinical studies. This review assembles the available experimental and clinical data on biological mechanisms of antithrombin in inflammatory coagulation activation. Preclinical research has demonstrated partial interference of heparin – administered even at low doses – with the therapeutic effects of antithrombin, and has confirmed – at the level of cellular mechanisms – a regulatory role for antithrombin in DIC. Against this biological background, re-analyses of data from randomized controlled trials of antithrombin in sepsis suggest that antithrombin has the potential to be developed further as a therapeutic agent in the treatment of DIC. Even though there is a lack of studies employing satisfactory methodology, the results of investigations conducted thus far into the mechanisms of action of antithrombin allow one to infer that there is biological plausibility in the value of this agent. Final assessment of the drug's effectiveness, however, must await the availability of positive, prospective, randomized and placebo-controlled studies.
Introduction
Antithrombin is a vitamin K independent glycoprotein and an essential inhibitor of thrombin and other serine proteases such as factors Xa and IXa [1]. Acquired antithrombin deficiency is more frequent than congenital deficiency and develops primarily through increased consumption or loss of antithrombin. The reduced plasma level in acquired deficiency is a strong predictor of a severe disease course, particularly in patients who have suffered trauma or severe sepsis. Increased consumption of antithrombin occurs primarily in disseminated intravascular coagulation (DIC) [2-8]. A diagnosis of DIC is made after taking into consideration the triggering disease, clinical picture, and unambiguous and striking hemostatic findings such as a drop in thrombocytes, loss of antithrombin activity, increased concentrations of D-dimers or fibrin monomers [7].
Based on the assumed inhibition by antithrombin of activated clotting factors in the circulating vascular system, concentrated human plasmatic antithrombin was administered in the setting of DIC in isolated cases [2,4-6,8], in animal experiments [6,9-13], and in clinical studies [14-18] with the aim of interrupting the complex cycle of DIC and preventing multiple organ failure. In these studies the duration of DIC was significantly shortened and organ functions were improved, but no significant reduction in mortality in the antithrombin groups was achieved. Proof of reduced lethality from DIC with administration of concentrated human plasmatic antithrombin in prospective controlled clinical studies has not yet been reported.
The largest number of patients with acquired antithrombin deficiency treated with human plasmatic antithrombin in a prospective controlled clinical study can be found in the KyberSept trial [19]. Patients with severe sepsis were recruited for this study independent of the cause or duration of sepsis and irrespective of the presence or absence of DIC. Therapeutic doses of heparin were not permitted because of the interaction of this agent with antithrombin and its anticoagulatory effects. In prophylactic doses, however, administration of heparin was permitted. Compared with placebo, treatment with high-dose antithrombin over 4 days had no significant effect on mortality at 28 days. Failure to demonstrate effectiveness of antithrombin may be due to the heterogeneity of the patient population investigated. Interaction between antithrombin and heparin was considered a possible reason, because antithrombin achieved a significant reduction in mortality at 90 days in a subgroup of patients who did not simultaneously receive prophylactic doses of heparin.
This review addresses the potential relevance of new knowledge on the mechanisms of action of antithrombin to the practising clinician.
Characteristics of antithrombin
The two basic endogenous anticoagulation principles that have an inhibitory effect on the coagulation cascade are the heparin–antithrombin and the protein C thrombomodulating mechanisms. Both of these inhibit serine proteases and cofactors/activated coagulation factors [1].
Antithrombin is a 58 kDa glycoprotein that is composed of a single amino acid chain with a plasma concentration of about 150–150 μg/ml (2–3 μmol/l). The total 432 amino acids carry four N glycosylated oligosaccharide chains and three disulfide bridges. In the presence of heparin, antithrombin strongly inhibits not only thrombin and factor Xa but also factor IXa, and it weakly inhibits factors XIa and XIIa, trypsin, plasmin, kallikrein and factor VIIa [15-17,20,21]. This broad spectrum of inhibition makes antithrombin the key regulator of the coagulation system. In addition, antithrombin appears to have anti-inflammatory properties that appear to be distinct in their mechanism from this effect on coagulation that is dependent on enzyme inhibition.
Two isoforms of antithrombin circulate in the plasma, namely the α and the β forms, which do not differ from each other in their thrombin inhibiting effect, but they differ in their affinity for heparin. Under normal conditions, 85–95% of circulating antithrombins are glycosylated at four of their asparagine molecules; these are of the α isoform. The remaining 5–15% of circulating antithrombins, of the β isoform, lack glycosylation at asparagine 135, which results in their 3- to 10-fold greater affinity for heparin [22].
Anticoagulation
Heparin exerts an anticoagulatory effect by binding to antithrombin and thereby alters its conformation in such a manner that it inhibits thrombin 1000-fold more strongly [23]. It builds up a stoichiometric complex with thrombin, at a 1:1 ratio, over a reactive arginine-containing loop and a catalytic serine-containing interaction site. As soon as a thrombin–antithrombin (TAT) complex is formed, both molecules are inactivated (Fig. 1). The TAT complex has a short plasma half-life of only 5 min and is disposed of by its binding to the so-called low-density lipoprotein receptor-related protein of the liver [24,25].
Figure 1.
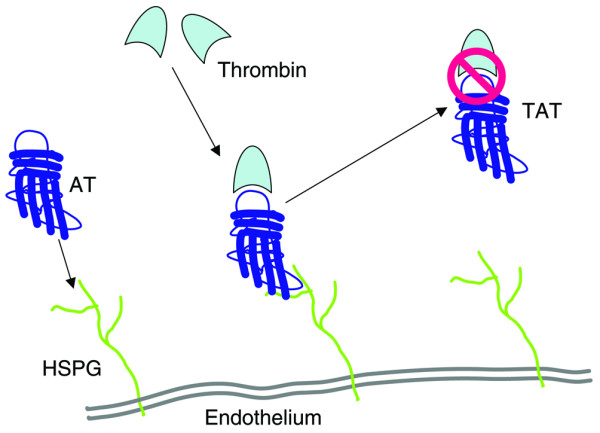
Interaction of antithrombin with endothelium. The affinity of antithrombin (AT) to thrombin and its enzymatic inhibition is increased by binding of AT with the heparan-binding site to cellular heparin sulfate proteoglycans (HSPGs). TAT, thrombin–antithrombin.
In order for thrombin to be inactivated by antithrombin, not only must heparin be bound to antithrombin but it must also accumulate on thrombin. Nonfractionated, high-molecular-weight heparins, which have several pentasaccharide sequences, can thus promote the antithrombin-dependent inactivation of thrombin. This bivalent interaction does not require the presence of antithrombin to inhibit other serine proteases, such as factor Xa or the contact factors. This explains why, compared with the other factors, thrombin is inhibited more weakly by low-molecular-weight heparins.
Proteoglycans, such as heparan sulfate proteoglycans (HSPGs), are expressed on the surface of vessel endothelium. It has been estimated that there are about 500,000 binding sites for antithrombin on each single endothelial cell on the inner surface of the blood vessels [26]. If the heparin-binding capacity of circulating antithrombin is not blocked or exhausted by soluble heparin or by HSPG detached from the endothelium, the deposition of antithrombin on surface heparins of the endothelium can help antagonize the activated thrombin also deposited at the same sites of the endothelium in vessel injury [26]. This mechanism might be of significance in the maintenance of microcirculation.
Anti-inflammation
The anticoagulatory properties of antithrombin have been investigated relatively exhaustively and have been known for some time for their inhibitory effect on serine proteases. Since the late 1980s, additional results from several laboratories have shown that antithrombin also has strong anti-inflammatory properties that are independent of its anticoagulatory characteristics. These effects were first postulated by Taylor and coworkers [27,28] based on their experiments on DIC in a monkey model. Infusions of antithrombin significantly reduced mortality in monkeys previously treated intravenously with lethal doses of Escherichia coli. The therapeutic effects on systemic blood pressure and organ failure, however, could not be explained on the basis of its direct effect on thrombin. These observations were supported by the fact that DEGR–factor Xa, a catalytically inactive factor Xa derivative that selectively blocks production of thrombin, also had no influence on organ failure in this model.
An early explanation for this anti-inflammatory effect of antithrombin presented itself when it was shown that antithrombin promotes the release of prostacyclin (prostaglandin I2) from the endothelium, which – both in vitro and in vivo – can interact with heparin-like glycosaminoglycans of the endothelium surface [29-31] (Fig. 2).
Figure 2.
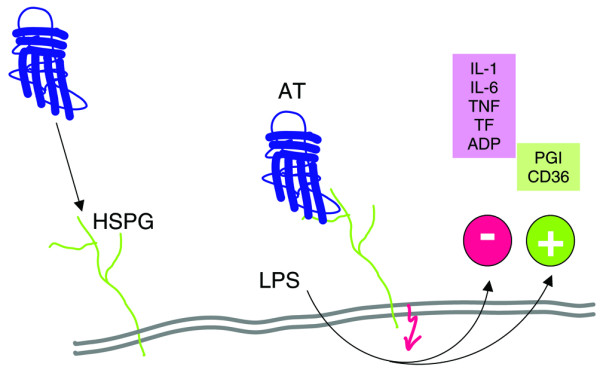
Inflammation-modulating effect of antithrombin on the endothelium. Ligation of heparan sulfate proteoglycans (HSPGs) of endothelium with antithrombin (AT) induces cellular signalling events that alter the cell's biochemical and functional responses to inflammatory stimuli (e.g. bacterial lipopolysaccharide [LPS]). Changes include reduced release of inflammatory and procoagulatory mediators (e.g. interleukin [IL]-1, IL-6, tumour necrosis factor-α[TNF]), tissue factor (TF), adenosine diphosphate (ADP) and cellular adhesion molecules (not shown), as well as increased release of anticoagulatory prostacyclin (prostaglandin [PG]I) or CD39/ATPDase. In neuronal tissue, protective mechanisms may by mediated via the release of calcitonin gene-related peptide and nitric oxide with the potential to affect prostacyclin release [55].
Endotoxin-induced damage to pulmonary blood vessels [32] as well as hepatic ischaemia/reperfusion lesions [31] can be decreased with antithrombin via induction of endothelial prostaglandin production. The protective effect could not be achieved with Trp49–antithrombin (a modified antithrombin) because, lacking affinity to heparin, it was unable to induce release of endothelial prostacyclin. In independent investigations conducted in a mesenteric ischaemia/reperfusion feline model, Ostrovsky and coworkers [33] showed that antithrombin exerts its protective effect by inhibiting rolling and adhesion of leucocytes on the endothelium. Hoffmann and coworkers [34] and Neviere and colleagues [35] later confirmed this effect in a rat model by intravital videomicroscopy of damage to the small intestine in endotoxinaemia. In both of these studies, the effect of antithrombin on the leucocyte–endothelium interaction could be abolished by indomethacin, which is an inhibitor of prostacyclin production. HSPGs (heparin-like glycosaminoglycans) are involved as mediators in the effect of antithrombin on the leucocyte–endothelium interaction. There are indications that syndecan-4 (an HSPG member of the syndecan family) mediates, as one of the antithrombin receptors on endothelial cells, these direct effects of antithrombin [36-38]. Antithrombin reduced the ischaemia/reperfusion induced renal or hepatic injury by inhibiting leucocyte activation, suggesting that therapeutic effects of antithrombin might be mainly mediated by prostacyclin released from endothelial cells through interaction of antithrombin with cell surface glycosaminoglycans [39,40]. Organ systems protected by antithrombin in ischaemia/ reperfusion via such mechanisms may include those of the central nervous system [41,42].
Syndecan-4 is expressed by leucocytes, and moreover antithrombin can regulate the migratory behaviour on the cell surface of neutrophils, monocytes, lymphocytes and eosinophiles [36,38,43]. In its highly conserved heparin sulfate ectodomains, syndecan-4 possesses pentasaccharide arrays that are recognized by the heparin-binding site of the antithrombin molecule [30,44]. With reagents such as heparinase or natrium chlorate that degrade heparin sulfate, the effect of antithrombin on leucocyte migration could be abolished [36-38,43]. In agreement with this is the observation that syndecan-4 deficiency in knockout mice increases mortality in endotoxinaemia [45]. Even the smallest doses of heparin such as those used in thrombosis prophy-laxis can have a negative impact on the beneficial effect of antithrombin on microcirculatory disturbances in endotoxinaemia [46]. This might be due to the fact that heparin competitively inhibits the binding of antithrombin to syndecan-4 or other HSPGs. It has been reported in human umbilical cord endothelial cells that, under static conditions, antithrombin inhibits endotoxin or cytokine induced signaling and adherence of neutrophils [47] (Fig. 2). Coincubation of antithrombin with a synthetic exogenous pentasaccharide or pretreatment of endothelial cells with heparinase or chondroitinase abolished the effect of antithrombin on neutrophil adherence. Based on these observations it was concluded that antithrombin has the capacity to block endothelial cell activation via activation of syndecan-4-dependent signal transduction.
In their investigations into the intracellular mechanisms that underlie the anti-inflammatory effects of antithrombin, Oelschläger and coworkers [48] confirmed that antithrombin inhibits activation of nuclear factor-κB (NF-κB) in cultured human monocytes and endothelial cells [49]. The transcription factor NF-κB activates various genes that encode immune factors in reactions to inflammatory stimuli [50]. That this mechanism is inhibited with antithrombin represents a plausible explanation for the anti-inflammatory effects described. This inhibition of NF-κB was observed in an in vitro concentration of antithrombin corresponding to its level in the plasma, a concentration required for therapeutic anti-inflammation in vivo [51] or for other effects described in vitro [52].
Additional in vitro findings on inhibition of β2 integrin expression in neutrophils and monocytes by antithrombin [53] suggest that direct molecular mechanisms underlie inhibition of leucocyte adhesion to the endothelium. Antithrombin inhibited the tumour necrosis factor (TNF)-α induced increase in E-selectin expression in endothelial cells by inhibiting the interaction of NF-κB with CBP/p300 through cAMP-dependent protein kinase A-induced CREB activation [54]. This inhibitory activity of antithrombin probably also depends on its binding to heparin-like substances on the endothelial cell.
Because of the widespread expression of HSPGs, sites of action for antithrombin may not be restricted to endothelium and leucocytes but may well include additional targets. Evidence for this came from studies in ischaemic/reperfusion injury of the liver; antithrombin might increase hepatic tissue levels of prostacyclin via enhancement of hepatic injury induced activation of capsaicin-sensitive sensory neurones, thereby reducing liver injury in rats. In this process, calcitonin gene-related peptide-induced activation of both endothelial nitric oxide synthase and cyclo-oxygenase-1 might be critically involved [55]. Because in such a scenario the site of action of antithrombin is within the interstitium, prior extravasation of circulating antithrombin would have to be a prerequisite for anti-inflammatory activity to be exerted. Of course, this also has implications for estimating dose–response relationships because it will be difficult to determine extravascular concentrations of antithrombin.
Interaction between antithrombin and heparin
It is clear that antithrombin has anti-inflammatory properties in addition to its anticoagulation ones. Because the anti-inflammatory effects are exerted via interaction between antithrombin and its heparin-binding domains and the heparin-like receptors on the surfaces of endothelial cells and leucocytes such as HSPG syndecan-4, it is not surprising that the antithrombin concentrations required for this differ from those for anticoagulation and lie in 'supraphysiological' region. In vitro and in animal models, low doses of unfractionated or low-molecular-weight heparin antagonized this HSPG-mediated effect, because the binding of antithrombin to the cellular HSPG was blocked by exogenous heparin [46] (Fig. 3). It was previously shown antithrombin–heparin treatment significantly attenuated the endotoxin-induced consumption of fibrinogen and completely prevented the increase in soluble fibrin in plasma; however, no significant effect of antithrombin III-heparin was observed on endotoxin-induced mortality and dysfunction in pulmonary gas exchange. We therefore conclude that a purified antithrombin–heparin complex inhibits thrombin effects and prevents development of DIC, but it fails to influence clinical outcome significantly in endotoxic shock in pig [56].
Figure 3.
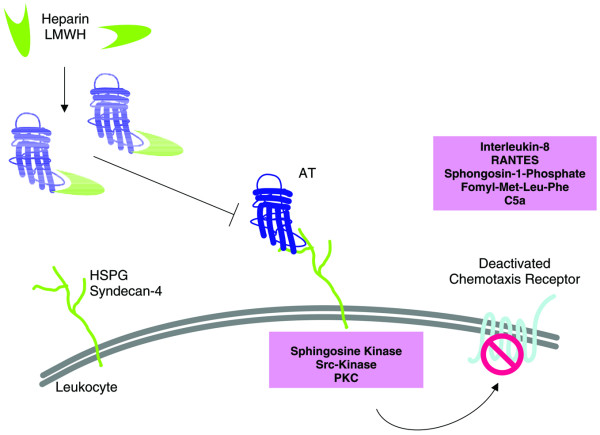
Molecular mechanism underlying the interaction between antithrombin and heparin. Signalling events triggered by antithrombin (AT) affect chemotaxis receptor function. Mediator release and adhesion molecule expression is altered by inhibiting the interaction of nuclear factor-κB (NF-κB) with CBP/p300 through cAMP-dependent protein kinase A-induced CREB activation [54]. Exogenous unfractionated heparin or low-molecular-weight heparin (LMWH) may prevent AT from interacting with heparin sulfate proteoglycans (HSPGs) by competitive ligand binding at the heparin binding site of AT. PK, protein kinase; RANTES, regulated upon activation, normal T-cell expressed and secreted.
Therapy for severe sepsis, which in its initial phase is a proinflammatory, life-threatening disease, with antithrombin was evaluated in a randomized, controlled study (KyberSept) conducted in 2314 patients [19]. Total mortality on day 28 after the start of therapy in patients in the intention-to-treat group (who received antithrombin) was no different from that in the placebo group (38.9% for antithrombin versus 38.7% for placebo). This finding was counter to the raised expectations stimulated by phase II trials [14-18,21]. Concomitant – with the study medication, one subgroup of patients was given heparin for thrombosis prophylaxis. Because, according to subgroup analysis, antithrombin reduced 90-day mortality only in the group of patients who did not receive heparin at the same time, an interaction between antithrombin and heparin was postulated that probably offset the beneficial anti-inflammatory properties of the former [19,57,58].
Although prophylactic and therapeutic use of heparin in venous thromobosis and pulmonary embolism has been extensively investigated during the past 50 years, its employment in patients with sepsis has not yet been evaluated in prospective studies. In small studies a beneficial effect of heparin on survival of patients with sepsis could not be demonstrated [59]. In the KyberSept study, administration of prophylactic doses of heparin was permitted by the protocol [19,57]. Because the possibility of an interaction with antithrombin had not been excluded, the subgroup analysis was determined a priori, before the start of the study, although there was no stratification for heparin. Analyses of heparin were carried out post hoc in Kybersept, which distinguishes it from other retrospective calculations that were justifiably the subject of criticism [60].
The post hoc analyses of the effectiveness of antithrombin in – severe sepsis without concomitant administration of heparin [57] supported the hypothesis that prophylactic administration of heparin interferes with the therapeutic effect of antithrombin. Although definitive statistical proof is lacking, it is quite likely that the increasing loss of efficacy of antithrombin over the time course of the KyberSept trial is associated with increasing concomitant administration of heparin [57]. In addition, the combination of antithrombin and heparin led to a significant increase in bleeding complications, even though no increase in mortality was shown to result from them. Conversely, antithrombin therapy without heparin did not result in an increase in the number of severe – thromboembolic events, which would suggest that withholding prophylactic heparin during antithrombin therapy does not present any new safety problems [57].
Further, rather less direct confirmation of the role of heparin as an unfavourable interaction partner with antithrombin in severe sepsis comes from another predefined subgroup analysis of the KyberSept trial. It was shown that, in KyberSept, those patients who failed to derive any benefit from antithrombin either were too ill to survive sepsis despite optimal therapy (predicted mortality >60%) or were too well to die in adequate numbers for the study conditions (predicted mortality <30%). In the group of patients with the 'right' degree of illness severity (predicted mortality between 30% and 60%), a statistically significant improvement in survival to day 90 of the observation period was seen in the antithrombin group, irrespective of whether the patients in this group concomitantly received heparin; if those patients who received heparin were excluded from the analysis, the therapeutic effect of antithrombin was even stronger [61].
Results of preclinical research carried out after the KyberSept study suggested the possibility of an undesirable drug interaction between antithrombin and heparin, and this – together with previous knowledge on this subject – provided the justification for re-analyses of the findings. The data support the assumption that if administration of heparin concomitantly with antithrombin is withheld, then the anti-inflammatory potential of the latter can be improved.
Disseminated intravascular coagulation and antithrombin-dependent inhibition of inflammation
DIC is a complex event both from diagnostic and from therapeutic perspectives. DIC can be triggered by numerous diseases that are quite independent from each other. It is not a disease entity in its own right but manifests as a consequence or complication of some severe underlying disease. Activation of coagulation within the context of such an underlying disease induces excessive intravascular coagulation that can manifest in various ways. It can lead to the formation and deposition of fibrin so that microthromboses develop in multiple organs (Fig. 4). The extensive and persisting activation of the coagulation system may also result in reduced levels of coagulation factors. Consumption of these factors is promoted by reduced synthesis in the liver, and their shortened half-life is dependent on activation of proteases. Consumption of coagulation factors and the subsequent thrombocytopenia can lead to the development of microthromboses and severe vascular complications.
Figure 4.
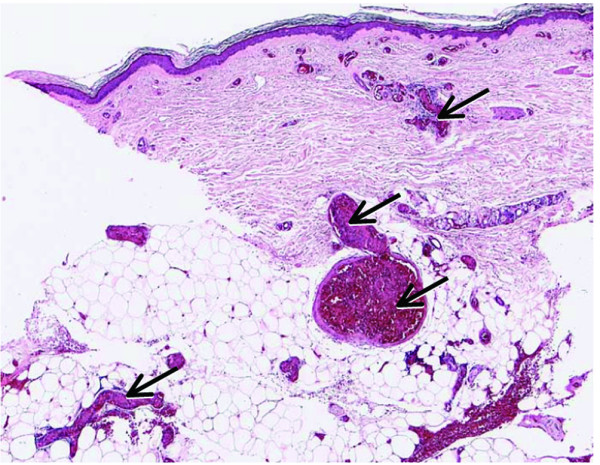
Disseminated intravascular coagulation. Arrows indicate intravascular fibrin deposition.
DIC is among the most frequent complications of sepsis, which is almost always associated with hemostatic changes. Patients with septic shock who develop DIC exhibit higher mortality rates than do those without signs of DIC. With its ubiquitous microthrombosis, DIC plays an essential role in the development of organ dysfunction in septic shock, such that DIC can be considered a primary pathogenetic factor in multi-organ failure. In sepsis, laboratory data demonstrate not only changes in the number of leucocytes but also a reduction in the number of thrombocytes and antithrombin concentration.
Inflammation-dependent activation of coagulation
Increased levels of circulating cytokines follow a marked increase in the markers for thrombin formation (prothrombin fragments F1 and F2, and TAT complex) and for the conversion of fibrinogen to fibrin (fibrinopeptide A and fibrin monomers) [62]. A similar activation of the coagulation system can be observed when TNF is injected into individuals [63]. Concomitant administration of pentoxifyllin and endotoxin in chimpanzees blocked endotoxin-induced TNF expression, and also inhibited activation of coagulation [64]. Thus, TNF is not only a central mediator in the cytokine cascade but also must be viewed as a decisive mediator of endotoxin-induced activation of the coagulation system.
Initial activation of the coagulation system in sepsis occurs primarily via the extrinsic system; that is, it is dependent on tissue factor (TF). In primates, infusions of high doses of E. coli were followed by strong expression of TF [65]. Furthermore, in vitro studies showed that endotoxin and cytokine induced TF expression in monocytes and endothelial cells [66]. TF binds to factor VIIa, resulting in a TF–factor VIIa complex, which is responsible for the conversion of factor X to factor Xa. In children with meningococcal sepsis, monocytes exhibit increased TF expression [67]. One can assume that the TF-dependent system plays a predominant role in activation of coagulation in sepsis.
The coagulation system comprises several inhibitory systems that are of great importance, with antithrombin and protein C–protein S systems in the foreground. In sepsis, however, reduced activity of inhibitory systems has been measured in general [68]. This further contributes to the development of a procoagulatory condition.
As a rule, the number of thrombocytes is reduced within the context of a septic event. This reduction is explained by their being used up in the region of fibrin deposits, by their adhesion to altered endothelial cells, and by pulmonary and hepatic sequestration. Generally speaking, there is a close correlation between lowering of thrombocyte numbers and mortality in patients. Disturbances in platelet function not only result from sepsis but can also arise in the context of antibiotic therapy, or they may be associated with sepsis-dependent uraemia.
Effects of antithrombin on monocytes and thrombocytes
Antithrombin binds to syndecan-4 in monocytes and thrombocytes, activates various signal transduction pathways, and thereby influences the functional behaviour of both of these blood cell types, which are so important in DIC [36,37,53,69]. Signal transduction enzymes that are involved in monocytes in vitro include protein kinase C [37], Rho-GTP associated kinases [38], src tyrosine kinases [69] and sphingosine kinase [37].
Antithrombin enhances Tyr416 and inhibits Tyr527 phosphorylation of src tyrosine kinases; in combination with the chemokine RANTES (regulated upon activation, normal T-cell expressed and secreted); however, it weakens the RANTES-enhanced phosphorylation of Tyr416 [69]. These effects of antithrombin correspond to a large extent with the well known syndecan-4 induced signalling pathways [70]. It can be assumed that antithrombin is also able to exert specific anticoagulatory and anti-inflammatory effects in thrombocytes via analogous biochemical mechanisms because it is known that syndecan-4 is also expressed by thrombocytes and that antisyndecan-4 antibodies can influence thrombocyte aggregation [71].
Antithrombin inhibits chemotaxis [36,37] and expression of TF and cytokines [52] in monocytes. In activated thrombocytes, antithrombin inhibits CD40 ligand expression, syndecan-4 shedding, and release of adenosine diphosphate and adenosine triphosphate, together with inhibition of thrombocyte-regulated release of free radicals from neutrophils [71]. Because all of these mechanisms play a role in activating thrombocytes in DIC patients, it can be assumed that antithrombin deficiency also contributes to the development of DIC via such direct cellular mechanisms, under the stipulation that the in vitro observations of functional regulation of thrombocytes are confirmed in vivo. The same applies for the possible relevance of direct antithrombin effects on monocytes.
Antithrombin therapy in acquired disseminated intravascular coagulation in sepsis
Post hoc analyses of the effectiveness of antithrombin in severe sepsis without concomitant administration of heparin supported the hypothesis of the KyberSept study that prophylactic heparin interferes with the therapeutic effects of antithrombin [57,61]. Because antithrombin therapy without concomitant treatment with heparin was effective in the KyberSept trial, it appeared reasonable to investigate the same cohort of patients to find out whether the effectiveness of such therapy was dependent on the presence of DIC. Such an investigation was reported by Kienast and coworkers [72]. A total of 563 patients who did not receive heparin concomitantly with antithrombin were evaluated (n = 277 for placebo; n = 286 for antithrombin). At the beginning of therapy, 40.7% of the patients (229/563) had DIC. In the placebo group DIC signified enhanced mortality due to sepsis (28-day mortality: 40.0% versus 22.2%; P < 0.01).
DIC patients treated with antithrombin exhibited an absolute reduction (14.6%) in 28-day mortality in comparison with placebo-treated patients (P = 0.02); in contrast, no such effects were noted among patients without DIC (0.1% reduction in mortality; P = 1.0). These analyses in a randomized study population confirmed that antithrombin therapy can have a favourable influence not only on the morbidity associated with severe sepsis but also on mortality if, in addition to withholding concomitant administration of heparin, substitution therapy is given, particularly to those patients with acquired antithrombin deficiency in whom acute DIC has been diagnosed [72].
Limitations
Experimental studies have shown that high doses of antithrombin (>500 UI/kg body weight) elicit anti-inflammatory effects and most experimental studies used a high-dose pretreatment protocol, which is of little clinical relevance [2,10,11,13,27]. However, supranormal levels have not been achieved in human trials because the highest dose regimens could only induce 200–250% antithrombin activity. It is therefore unknown whether anti-inflammatory effects of high-dose antithrombin indeed play a clinical role in humans.
Timing of antithrombin administration in experimental and clinical studies may be another critical determinant of efficacy. Most experimental studies used a high-dose pretreatment protocol, which is of little clinical relevance. On the other hand, very early suppression of inflammation may exert adverse effects on the initial response to injury. In experimental acute lung injury induced by inhalation of Gram-negative bacteria, simultaneous intravenous administration of recombinant human antithrombin caused histologic damage, injury to the alveolar capillary barrier and increased permeability in rodents [73]. These effects of inhibition of thrombin activation by antithrombin led to the hypothesis that there is a beneficial role of early coagulation activation in acute lung injury, and that inhibition of initial procoagulation is potentially dangerous.
Furthermore, biologically, the effect of antithrombin may be modified directly by bacterial endotoxins because porins (nonspecific channels of the bacterial outer membrane that allow transmembrane passage of nutrient molecules that are usually small and hydrophilic) not only enhanced thrombin activity but also inhibited the enhanced antithrombin activity resulting from antithrombin administration [74]. The mechanisms of such inhibitory interaction of porins with antithrombin remain to be detailed; however, antithrombin's heparin-binding site may well be among the potential targets because affinity of porins to heparin-binding domains of matrix proteins was recently reported [75].
Finally, antithrombin may interfere with the cytotoxic effects of lymphocytes via its serine protease-inhibitory (SERPIN) action on granzymes, which are death-inducing proteases stored in the granules of cytotoxic lymphocytes that allow the immune system to eliminate intracellular pathogens and transformed cells rapidly [76]. In severe sepsis, elevation of granzyme plasma levels correlates with disease severity [77]. Whether this SERPIN activity of antithrombin is beneficial or deleterious is unknown.
Conclusion
On one hand, antithrombin research conducted during the past two decades has attempted to establish the effectiveness and safety of antithrombin substitution in cases of severe sepsis in accordance with the criteria of evidence-based medicine. On the other hand, plausible mechanisms of action of this drug were investigated in preclinical research to assess its therapeutic use. KyberSept [19], the first large-scale study conducted within the context of intensive care medicine, investigated use of high-dose antithrombin in severe sepsis, but it was unable to establish its effectiveness because of the highly heterogeneous nature of the patient population and a lack of clearly defined standard therapy [78]. Results of preclinical research carried out simultaneously, however, demonstrated partial interference of heparin – administered even at low doses – with the therapeutic effects of antithrombin. They also confirmed, at the level of cellular mechanisms, the important role of antithrombin in DIC. This knowledge provides a biological background against which re-analyses of the results of the KyberSept study appear to be justified; such analyses suggest that antithrombin has the potential to be developed further as a therapeutic agent in the treatment of DIC. Even though there is a lack of studies employing satisfactory methodology, the results of investigations conducted thus far into the mechanisms of action of antithrombin allow one to infer that there is biological plausibility in the value of this agent [79,80]. Even though several new aspects regarding the biological role of antithrombin and it pharmacodynamcis have recently been discovered, knowledge is still incomplete. Final assessment of the drug's effectiveness must await the availability of positive, prospective, randomized and placebo-controlled studies.
Abbreviations
DIC = disseminated intravascular coagulation; HSPG = heparan sulfate proteoglycan; NF-κB = nuclear factor-κB; TAT = thrombin–antithrombin; TF = tissue factor; TNF = tumour necrosis factor.
Competing interests
CW has received fees for speaking and funding of a research grant at the Medical University of Innsbruck, Austria (PI-Wiedermann) from ZLB Behring.
References
- Rosenberg RD, Bauer KA. Thrombosis in inherited deficiencies of antithrombin, protein C, and protein S. Hum Pathol. 1987;18:253–262. doi: 10.1016/s0046-8177(87)80008-4. [DOI] [PubMed] [Google Scholar]
- Hauptman JG, Hassouna HI, Bell TG, Penner JA, Emerson TE. Efficacy of antithrombin III in endotoxin-induced disseminated intravascular coagulation. Circ Shock. 1988;25:111–122. [PubMed] [Google Scholar]
- Hiller E, Pihusch R. Thrombophilia caused by congenital disorders of blood coagulation. Fortschr Med. 1998;116:26–32. [PubMed] [Google Scholar]
- Mammen EF. The role of antithrombin III in DIC. Biol Clin Haematol. 1987. pp. 69–73.
- de Jonge E, Levi M, Stoutenbeek CP, van Deventer SJ. Current drug treatment strategies for disseminated intravascular coagulation. Drugs. 1998;55:767–777. doi: 10.2165/00003495-199855060-00004. [DOI] [PubMed] [Google Scholar]
- Mammen EF, Miyakawa T, Phillips TF, Assarian GS, Brown JM, Murano G. Human antithrombin concentrates and experimental disseminated intravascular coagulation. Semin Thromb Hemost. 1985;11:373–383. doi: 10.1055/s-2007-1004397. [DOI] [PubMed] [Google Scholar]
- Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341:586–592. doi: 10.1056/NEJM199908193410807. [DOI] [PubMed] [Google Scholar]
- Schregel W, Straub H, Wolk G, Schneider R. Successful therapy of consumption coagulopathy in EPH gestosis with multiple organ failure [in German] Anasth Intensivther Not-fallmed. 1984;19:201–203. [PubMed] [Google Scholar]
- Dickneite G, Paques EP. Reduction of mortality with antithrombin III in septicemic rats: a study of Klebsiella pneumoniae-induced sepsis. Thromb Haemost. 1993;69:98–102. [PubMed] [Google Scholar]
- Emerson TE, Jr, Fournel MA, Leach WJ, Redens TB. Protection against disseminated intravascular coagulation and death by antithrombin-III in the Escherichia coli endotoxemic rat. Circ Shock. 1987;21:1–13. [PubMed] [Google Scholar]
- Redens TB, Leach WJ, Bogdanoff DA, Emerson TE., Jr Synergistic protection from lung damage by combining antithrombin-III and alpha-1-proteinase inhibitor in the E. coli endotoxemic sheep pulmonary dysfunction model. Circ Shock. 1988;26:15–26. [PubMed] [Google Scholar]
- Ronneberger H, Hein B. Effect of antithrombin III on experimental hepatotoxin poisoning in dogs [in German] Arzneimittelforschung. 1984;34:277–279. [PubMed] [Google Scholar]
- Taylor FB, Jr, Emerson TE, Jr, Jordan R, Chang AK, Blick KE. Antithrombin-III prevents the lethal effects of Escherichia coli infusion in baboons. Circ Shock. 1988;26:227–235. [PubMed] [Google Scholar]
- Baudo F, Caimi TM, de Cataldo F, Ravizza A, Arlati S, Casella G, Carugo D, Palareti G, Legnani C, Ridolfi L, et al. Antithrombin III (ATIII) replacement therapy in patients with sepsis and/or postsurgical complications: a controlled double-blind, randomized, multicenter study. Intensive Care Med. 1998;24:336–342. doi: 10.1007/s001340050576. [DOI] [PubMed] [Google Scholar]
- Eisele B, Lamy M, Thijs LG, Keinecke HO, Schuster HP, Matthias FR, Fourrier F, Heinrichs H, Delvos U. Antithrombin III in patients with severe sepsis. A randomized, placebo-controlled, double-blind multicenter trial plus a meta-analysis on all randomized, placebo-controlled, double-blind trials with antithrombin III in severe sepsis. Intensive Care Med. 1998;24:663–672. doi: 10.1007/s001340050642. [DOI] [PubMed] [Google Scholar]
- Fourrier F, Chopin C, Huart JJ, Runge I, Caron C. Double-blind, placebo-controlled trial of antithrombin III concentrates in septic shock with disseminated intravascular coagulation. Chest. 1993;104:882–888. doi: 10.1378/chest.104.3.882. [DOI] [PubMed] [Google Scholar]
- Inthorn D, Hoffmann N, Hartl WH, Mühlbayer D, Jochum M. Effect of antithrombin III supplementation on inflammatory response in patients with severe sepsis. Shock. 1998;10:90–96. doi: 10.1097/00024382-199808000-00002. [DOI] [PubMed] [Google Scholar]
- Vinazzer H. Therapeutic use of antithrombin III in shock and disseminated intravascular coagulation. Semin Thromb Hemost. 1989;15:347–352. doi: 10.1055/s-2007-1002727. [DOI] [PubMed] [Google Scholar]
- Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Penzes I, Kubler A, KyberSept Trial Study Group et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- Abildgaard U. Binding of thrombin to antithrombin III. Scand J Clin Lab Invest. 1969;24:23–27. doi: 10.3109/00365516909080127. [DOI] [PubMed] [Google Scholar]
- Schuster HP, Eisele B, Keinecke HO. S-antithrombin III study: antithrombin III in patients with sepsis. Intensive Care Med. 1998. p. 76. [DOI] [PubMed]
- Turk B, Brieditis I, Bock SC, Olson ST, Bjork I. The oligosaccharide side chain on Asn-135 of alpha-antithrombin, absent in beta-antithrombin, decreases the heparin affinity of the inhibitor by affecting the heparin-induced conformational change. Biochemistry. 1997;36:6682–6691. doi: 10.1021/bi9702492. [DOI] [PubMed] [Google Scholar]
- Andersson LO, Engman L, Henningsson E. Crossed immuno-electrophoresis as applied to studies on complex formation. The binding of heparin to antithrombin III and the antithrombin III-thrombin complex. J Immunol Methods. 1977;14:271–281. doi: 10.1016/0022-1759(77)90138-7. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH, Glover GI, Rivetna M, Schasteen CS, Fallon RJ. Identification of a serpin-enzyme complex receptor on human hepatoma cells and human monocytes. Proc Natl Acad Sci USA. 1990;87:3753–3757. doi: 10.1073/pnas.87.10.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo SV. Serpin receptor 1: a hepatic receptor that mediates the clearance of antithrombin III-proteinase complexes. Am J Med. 1989;87:10S–14S. doi: 10.1016/0002-9343(89)80524-8. [DOI] [PubMed] [Google Scholar]
- Bombeli T, Mueller M, Haeberli A. Anticoagulant properties of the vascular endothelium. Thromb Haemost. 1997;77:408–423. [PubMed] [Google Scholar]
- Taylor FB, Jr, Emerson TE, Jr, Jordan R, Chang AK, Blick KE. Antithrombin-III prevents the lethal effects of Escherichia coli infusion in baboons. Circ Shock. 1988;26:227–235. [PubMed] [Google Scholar]
- Taylor FB, Jr, Chang AC, Peer GT, Mather T, Blick K, Catlett R, Lockhart MS, Esmon CT. DEGR-factor Xa blocks disseminated intravascular coagulation initiated by Escherichia coli without preventing shock or organ damage. Blood. 1991;78:364–368. [PubMed] [Google Scholar]
- Yamauchi T, Umeda F, Inoguchi T, Nawata H. Antithrombin III stimulates prostacyclin production by cultured aortic endothelial cells. Biochem Biophys Res Commun. 1989;163:1404–1411. doi: 10.1016/0006-291X(89)91135-2. [DOI] [PubMed] [Google Scholar]
- Horie S, Ishii H, Kazama M. Heparin-like glycosaminoglycan is a receptor for antithrombin III-dependent but not for thrombin-dependent prostacyclin production in human endothelial cells. Thromb Res. 1990;59:895–904. doi: 10.1016/0049-3848(90)90113-Q. [DOI] [PubMed] [Google Scholar]
- Harada N, Okajima K, Kushimoto S, Isobe H, Tanaka K. Antithrombin reduces ischemia/reperfusion injury of rat liver by increasing the hepatic level of prostacyclin. Blood. 1999;93:157–164. [PubMed] [Google Scholar]
- Uchiba M, Okajima K, Murakami K, Okabe H, Takatsuki K. Attenuation of endotoxin-induced pulmonary vascular injury by antithrombin III. Am J Physiol. 1996;270:L921–L930. doi: 10.1152/ajplung.1996.270.6.L921. [DOI] [PubMed] [Google Scholar]
- Ostrovsky L, Woodman RC, Payne D, Teoh D, Kubes P. Antithrombin III prevents and rapidly reverses leukocyte recruitment in ischemia/reperfusion. Circulation. 1997;96:2302–2310. doi: 10.1161/01.cir.96.7.2302. [DOI] [PubMed] [Google Scholar]
- Hoffmann JN, Vollmar B, Inthorn D, Schildberg FW, Menger MD. Antithrombin reduces leukocyte adhesion during chronic endotoxemia by modulation of the cyclooxygenase pathway. Am J Physiol Cell Physiol. 2000;279:C98–C107. doi: 10.1152/ajpcell.2000.279.1.C98. [DOI] [PubMed] [Google Scholar]
- Neviere R, Tournoys A, Mordon S, Marechal X, Song FL, Jourdain M, Fourrier F. Antithrombin reduces mesenteric venular leukocyte interactions and small intestine injury in endotoxemic rats. Shock. 2001;15:220–225. doi: 10.1097/00024382-200115030-00010. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Reinisch CM, Dunzendorfer S, Romisch J, Wieder-mann CJ. Syndecan-4 mediates antithrombin-induced chemotaxis of human peripheral blood lymphocytes and monocytes. J Cell Sci. 2002;115:227–236. doi: 10.1242/jcs.115.1.227. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Egger P, Dunzendorfer S, Wiedermann CJ. Syndecan-4 as antithrombin receptor of human neutrophils. Biochem Biophys Res Commun. 2001;287:424–426. doi: 10.1006/bbrc.2001.5534. [DOI] [PubMed] [Google Scholar]
- Dunzendorfer S, Kaneider N, Rabensteiner A, Meierhofer C, Reinisch C, Romisch J, Wiedermann CJ. Cell-surface heparan sulfate proteoglycan-mediated regulation of human neu-trophil migration by the serpin antithrombin III. Blood. 2001;97:1079–1085. doi: 10.1182/blood.V97.4.1079. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Okajima K, Uchiba M, Isobe H, Harada N, Mizutani S, Noguchi T. Antithrombin reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation through promotion of prostacyclin production. Blood. 2003;101:3029–3036. doi: 10.1182/blood-2002-08-2406. [DOI] [PubMed] [Google Scholar]
- Harada N, Okajima K, Uchiba M, Kushimoto S, Isobe H. Antithrombin reduces ischemia/reperfusion-induced liver injury in rats by activation of cyclooxygenase-1. Thromb Haemost. 2004;92:550–558. doi: 10.1160/TH03-07-0460. [DOI] [PubMed] [Google Scholar]
- Hirose K, Okajima K, Uchiba M, Nakano KY, Utoh J, Kitamura N. Antithrombin reduces the ischemia/reperfusion-induced spinal cord injury in rats by attenuating inflammatory responses. Thromb Haemost. 2004;91:162–170. doi: 10.1160/TH03-06-0385. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K, Uchiba M. Antithrombin reduces compression-induced spinal cord injury in rats. J Neurotrauma. 2004;21:1818–1830. doi: 10.1089/neu.2004.21.1818. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Kaneider NC, Sturn DH, Wiedermann CJ. Syndecan-4-dependent migration of human eosinophils. Clin Exp Allergy. 2004;34:696–703. doi: 10.1111/j.1365-2222.2004.1853.x. [DOI] [PubMed] [Google Scholar]
- Olson ST, Bjork I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J Biol Chem. 1992;267:12528–12538. [PubMed] [Google Scholar]
- Ishiguro K, Kojima T, Muramatsu T. Syndecan-4 as a molecule involved in defense mechanisms. Glycoconj J. 2002;19:315–318. doi: 10.1023/A:1025308702966. [DOI] [PubMed] [Google Scholar]
- Hoffmann JN, Vollmar B, Laschke MW, Inthorn D, Kaneider NC, Dunzendorfer S, Wiedermann CJ, Romisch J, Schildberg FW, Menger MD. Adverse effect of heparin on antithrombin action during endotoxemia: microhemodynamic and cellular mechanisms. Thromb Haemost. 2002;88:242–252. [PubMed] [Google Scholar]
- Kaneider NC, Forster E, Mosheimer B, Sturn DH, Wiedermann CJ. Syndecan-4-dependent signaling in the inhibition of endotoxin-induced endothelial adherence of neutrophils by antithrombin. Thromb Haemost. 2003;90:1150–1157. doi: 10.1160/TH03-03-0184. [DOI] [PubMed] [Google Scholar]
- Oelschlager C, Romisch J, Staubitz A, Stauss H, Leithauser B, Tillmanns H, Holschermann H. Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood. 2002;99:4015–4020. doi: 10.1182/blood.V99.11.4015. [DOI] [PubMed] [Google Scholar]
- Mansell A, Reinicke A, Worrall DM, O'Neill LA. The serine protease inhibitor antithrombin III inhibits LPS-mediated NF-kappaB activation by TLR-4. FEBS Lett. 2001;508:313–317. doi: 10.1016/S0014-5793(01)03077-0. [DOI] [PubMed] [Google Scholar]
- Hatada EN, Krappmann D, Scheidereit C. NF-kappaB and the innate immune response. Curr Opin Immunol. 2000;12:52–58. doi: 10.1016/S0952-7915(99)00050-3. [DOI] [PubMed] [Google Scholar]
- Minnema MC, Chang AC, Jansen PM, Lubbers YT, Pratt BM, Whittaker BG, Taylor FB, Hack CE, Friedman B. Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood. 2000;95:1117–1123. [PubMed] [Google Scholar]
- Souter PJ, Thomas S, Hubbard AR, Poole S, Romisch J, Gray E. Antithrombin inhibits lipopolysaccharide-induced tissue factor and interleukin-6 production by mononuclear cells, human umbilical vein endothelial cells, and whole blood. Crit Care Med. 2001;29:134–139. doi: 10.1097/00003246-200101000-00027. [DOI] [PubMed] [Google Scholar]
- Gritti D, Malinverno A, Gasparetto C, Wiedermann CJ, Ricevuti R. Attenuation of leukocyte beta 2-integrin expression by antithrombin-III. Int J Immunopathol Pharmacol. 2004;17:27–32. doi: 10.1177/039463200401700104. [DOI] [PubMed] [Google Scholar]
- Uchiba M, Okajima K, Kaun C, Wojta J, Binder BR. Inhibition of the endothelial cell activation by antithrombin in vitro. Thromb Haemost. 2004;92:1420–1427. doi: 10.1160/TH04-03-0139. [DOI] [PubMed] [Google Scholar]
- Harada N, Okajima K, Yuksel M, Isobe H. Contribution of capsaicin-sensitive sensory neurons to antithrombin-induced reduction of ischemia/reperfusion-induced liver injury in rats. Thromb Haemost. 2005;93:48–56. doi: 10.1160/TH04-02-0106. [DOI] [PubMed] [Google Scholar]
- Spannagl M, Hoffmann H, Siebeck M, Weipert J, Schwarz HP, Schramm W. A purified antithrombin III-heparin complex as a potent inhibitor of thrombin in porcine endotoxin shock. Thromb Res. 1991;61:1–10. doi: 10.1016/0049-3848(91)90163-Q. [DOI] [PubMed] [Google Scholar]
- Hoffmann J, Wiedermann CJ, Juers M, Ostermann H, Kienast J, Briegel J, Strauss R, Warren BL, Opal SM. Benefit/risk profile of high-dose antithrombin III in the treatment of severe sepsis without concomitant heparin. Thromb Haemost. 2006. [PubMed]
- Opal SM, Kessler CM, Roemisch J, Knaub S. Antithrombin, heparin, and heparan sulfate. Crit Care Med. 2002;(Suppl):S325–S331. doi: 10.1097/00003246-200205001-00024. [DOI] [PubMed] [Google Scholar]
- Corrigan JJ, Jordan CM. Heparin therapy in septicemia with disseminated intravascular coagulation. N Engl J Med. 1970;283:778–782. doi: 10.1056/NEJM197010082831502. [DOI] [PubMed] [Google Scholar]
- Sleight P. Debate: subgroup analyses in clinical trials: fun to look at but don't believe them! Curr Control Trials Cardiovasc Med. 2000;1:25–27. doi: 10.1186/CVM-1-1-025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedermann CJ, Hoffmann JN, Juers M, Ostermann H, Kienast J, Briegel J, Strauss R, Keinecke HO, Warren BL, Opal SM, for the KyberSept investigators High-dose antithrombin III in the treatment of severe sepsis with a high risk of death: efficacy and safety. Crit Care Med. 2006;34:285–292. doi: 10.1097/01.CCM.0000194731.08896.99. [DOI] [PubMed] [Google Scholar]
- van Deventer SJ, Buller HR, ten Cate JW, Aarden LA, Hack CE, Sturk A. Experimental endotoxemia in humans: analysis of cytokine release and coagulation, fibrinolytic, and complement pathways. Blood. 1990;76:2520–2526. [PubMed] [Google Scholar]
- van der Poll T, Buller HR, ten Cate H, Wortel CH, Bauer KA, van Deventer SJ, Hack CE, Sauerwein HP, Rosenberg RD, ten Cate JW. Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med. 1990;322:1622–1627. doi: 10.1056/NEJM199006073222302. [DOI] [PubMed] [Google Scholar]
- Levi M, ten Cate H, Bauer KA, van der Poll T, Edgington TS, Buller HR, van Deventer SJ, Hack CE, ten Cate JW, Rosenberg RD. Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J Clin Invest. 1994;93:114–120. doi: 10.1172/JCI116934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake TA, Cheng J, Chang A, Taylor FB., Jr Expression of tissue factor, thrombomodulin, and E-selectin in baboons with lethal Escherichia coli sepsis. Am J Pathol. 1993;142:1458–1470. [PMC free article] [PubMed] [Google Scholar]
- Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163:740–745. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterud B, Flaegstad T. Increased tissue thromboplastin activity in monocytes of patients with meningococcal infection: related to an unfavourable prognosis. Thromb Haemost. 1983;49:5–7. [PubMed] [Google Scholar]
- Martinez MA, Pena JM, Fernandez A, Jimenez M, Juarez S, Madero R, Vazquez JJ. Time course and prognostic significance of hemostatic changes in sepsis: relation to tumor necrosis factor-alpha. Crit Care Med. 1999;27:1303–1308. doi: 10.1097/00003246-199907000-00017. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Mosheimer BA, Tancevski I, Kaneider NC, Sturn DH, Patsch JR, Wiedermann CJ. Src tyrosine kinase-dependent migratory effects of antithrombin in leukocytes. Exp Cell Res. 2005;305:214–220. doi: 10.1016/j.yexcr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Simons M, Horowitz A. Syndecan-4-mediated signalling. Cell Signal. 2001;13:855–862. doi: 10.1016/S0898-6568(01)00190-5. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Feistritzer C, Gritti D, Mosheimer BA, Ricevuti G, Patsch JR, Wiedermann CJ. Expression and function of syndecan-4 in human platelets. Thromb Haemost. 2005;93:1120–1127. doi: 10.1160/TH04-11-0763. [DOI] [PubMed] [Google Scholar]
- Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Briegel J, Strauss R, Warren BL, Opal SM. Treatment effects of high-dose antithrombin III without concomitant heparin in patients with severe sepsis with and without disseminated intravascular coagulation. J Thromb Haemost. 2006;4:90–97. doi: 10.1111/j.1538-7836.2005.01697.x. [DOI] [PubMed] [Google Scholar]
- Kipnis E, Guery BP, Tournoys A, Leroy X, Robriquet L, Fialdes P, Neviere R, Fourrier F. Massive alveolar thrombin activation in Pseudomonas aeruginosa-induced acute lung injury. Shock. 2004;21:444–451. doi: 10.1097/00024382-200405000-00008. [DOI] [PubMed] [Google Scholar]
- Di Micco B, Di Micco P, Lepretti M, Stiuso P, Donnarumma G, Iovene MR, Capasso R, Tufano MA. Hyperproduction of fibrin and inefficacy of antithrombin III and α2 macroglobulin in the presence of bacterial porins. Int J Exp Pathol. 2005;86:241–245. doi: 10.1111/j.0959-9673.2005.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AM, Jenkinson HF, Woodward MJ, Dymock D. Binding properties and adhesion-mediating regions of the major sheath protein of Treponema denticola ATCC 35405. Infect Immun. 2005;73:2891–2898. doi: 10.1128/IAI.73.5.2891-2898.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson D, Tschopp J. Inhibition of lymphocyte protease granzyme A by antithrombin III. Mol Immunol. 1988;25:1283–1289. doi: 10.1016/0161-5890(88)90043-0. [DOI] [PubMed] [Google Scholar]
- Zeerleder S, Hack CE, Caliezi C, van Mierlo G, Eerenberg-Belmer A, Wolbink A, Wuillenmin WA. Activated cytotoxic T cells and NK cells in severe sepsis and septic shock and their role in multiple organ dysfunction. Clin Immunol. 2005;116:158–165. doi: 10.1016/j.clim.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Wiedermann CJ, Kaneider NC. Comparison of mechanisms after post-hoc analyses of the drotrecogin alfa (activated) and antithrombin III trials in severe sepsis. Ann Med. 2004;36:194–203. doi: 10.1080/07853890410027943. [DOI] [PubMed] [Google Scholar]
- Kountchev J, Bijuklic K, Bellmann R, Wiedermann CJ, Joannidis M. Reduction of D-dimer levels after therapeutic administration of antithrombin in acquired antithrombin deficiency of severe sepsis. Crit Care. 2005;9:R596–R600. doi: 10.1186/cc3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi M. Antithrombin in sepsis revisited. Crit Care. 2005;9:624–625. doi: 10.1186/cc3819. [DOI] [PMC free article] [PubMed] [Google Scholar]