

Abstract
Mesotrypsin, an inhibitor-resistant human trypsin isoform, does not activate or degrade pancreatic protease zymogens at a significant rate. These observations led to the proposal that mesotrypsin is a defective digestive protease on protein substrates. Surprisingly, studies reported here with α1-antitrypsin (α1AT) revealed that even though mesotrypsin was completely resistant to this serpin-type inhibitor, it selectively cleaved the Lys10-Thr11 peptide bond at the N terminus. Analyzing a library of α1AT mutants in which Thr11 was mutated to various amino-acids, we found that mesotrypsin hydrolyzed lysyl peptide-bonds containing Thr or Ser at the P1′ position with relatively high specificity (kcat/KM ~105 M−1 s−1). Compared to Thr or Ser, P1′ Gly or Met inhibited cleavage 13 and 25-fold; respectively, whereas P1′ Asn, Asp, Ile, Phe or Tyr resulted in 100–200-fold diminished rates of proteolysis, and Pro completely abolished cleavage. Consistent with the Ser/Thr P1′ preference, mesotrypsin cleaved the Arg358-Ser359 reactive-site peptide bond of α1AT Pittsburgh and was rapidly inactivated by the serpin mechanism (ka ~106 M−1 s−1). Taken together, the results indicate that mesotrypsin is not a defective protease on polypeptide substrates in general, but exhibits a relatively high specificity for Lys/Arg-Ser/Thr peptide bonds. This restricted, thrombin-like subsite specificity explains why mesotrypsin cannot activate pancreatic zymogens, but might activate certain proteinase-activated receptors. The observations also identify α1AT Pittsburgh as an effective mesotrypsin inhibitor and the serpin mechanism as a viable stratagem to overcome the inhibitor-resistance of mesotrypsin.
Keywords: serpin, α1-antitrypsin, antitrypsin Pittsburgh, trypsin inhibitor, proteinase-activated receptors
The exceptional resistance of human mesotrypsin against polypeptide trypsin inhibitors was first described in 1978, and characterized in more detail in 1984 by Rinderknecht et al. [1, 2]. Subsequently, cloning of the cDNA, analysis of a crystal structure and mutagenesis studies revealed that the unique Arg198 residue (Arg193 in the conventional chymotrypsin numbering, chymo#) is responsible for the inhibitor resistance [3–5]. This position is normally occupied by a conserved Gly residue in the chymotrypsin-like serine proteases. In the crystal structure, the side-chain of Arg198 is in an extended conformation and appears to occupy the S2′ subsite, which should result in a steric clash with the P2′ residues of trypsin inhibitors [4]. As a result, canonical trypsin inhibitors typically bind to mesotrypsin with micromolar affinities, and thus act as weak-binding, competitive inhibitors [4, 5]. An interesting exception is the Kunitz protease inhibitory domain of the amyloid precursor protein, which inhibits mesotrypsin with a Ki of 30 nM [4]. Mesotrypsin exhibits normal affinity towards benzamidine and readily hydrolyzes small chromogenic peptides [2, 4, 5], indicating that the specificity pocket and the catalytic machinery per se are intact. This stands in contrast to reports showing that mutations of Gly193 (chymo#) in thrombin or factor XI resulted in perturbation of the oxyanion hole and impaired catalysis [6, 7]. Recently, we demonstrated that mesotrypsin rapidly cleaved the reactive-site peptide-bond of the Kunitz-type soybean trypsin inhibitor and completely degraded the Kazal-type pancreatic secretory trypsin inhibitor [5]. On the basis of these observations, we proposed that the biological function of mesotrypsin is the digestive degradation of dietary trypsin inhibitors.
The ability of mesotrypsin to cleave protein substrates other than trypsin inhibitors has remained contentious. Early models suggesting that mesotrypsin might play a role either in activation or degradation of pancreatic protease zymogens were proven untenable, because several laboratories showed that mesotrypsin did not activate human cationic or anionic trypsinogens, bovine chymotrypsinogen or human proelastase 2 to any significant extent [2, 5, 8]. Furthermore, degradation of human cationic and anionic trypsinogens by mesotrypsin was 500- and 20-fold slower, respectively, relative to the rate of degradation by cationic trypsin [5]. On the other hand, more recent observations demonstrated that mesotrypsin might act as an agonist for certain protease-activated receptors (PARs). The two studies published to date disagree which PAR isoforms are susceptible to activation by mesotrypsin, nonetheless, the findings raise the possibility that mesotrypsin might exhibit a unique substrate specificity, and invite investigations into the identification and characterization of mesotrypsin-specific substrates [9, 10].
In the present paper, we studied the interaction of mesotrypsin with the archetypal serpin α1-antitrypsin (α1AT) and its Pittsburgh variant (Met358→Arg). Serpins inhibit serine proteases by entering the catalytic cycle of the protease, and kinetically stabilizing the covalently linked acyl-enzyme intermediate [for a recent review see ref 11]. As shown in Fig 1, the first step of the serpin inhibitory mechanism is similar to that of canonical trypsin inhibitors and involves formation of the non-covalent Michaelis complex. The protease then cleaves the reactive-site peptide bond of the serpin in a substrate-like fashion, which triggers a significant conformational change, resulting in the distortion and inactivation of the acylated protease. The covalent inhibitory complex can slowly dissociate into free enzyme and inactive serpin. An alternative to the inhibitory pathway is the rapid deacylation of the acyl-enzyme complex, before the conformational change and protease trapping could occur. Thus, in this futile "proteolytic pathway" the serpin is simply cleaved as a substrate and becomes inactivated. Typically, in physiologically important serpin-protease reactions the proteolytic pathway is negligible.
Figure 1.
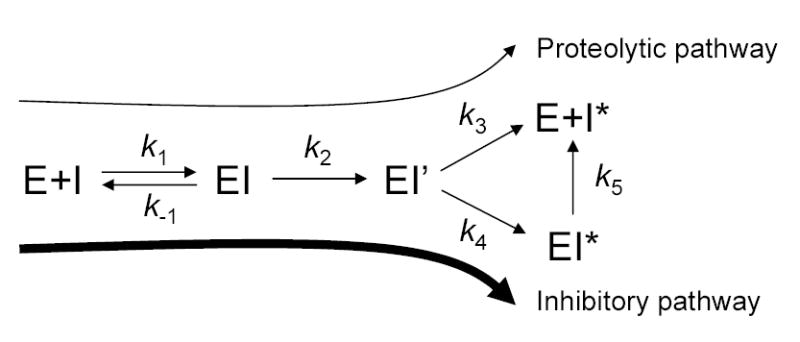
Protease inhibition by the serpin mechanism. I, inhibitor (e.g. α1-antitrypsin); E, enzyme (e.g. trypsin); k1 and k−1 denote the forward and reverse rate constants of the formation of the non-covalent complex EI; k2 is the rate constant of the formation of the acyl-enzyme intermediate EI’; k3 is the rate constant of deacylation, resulting in free enzyme and inactivated, cleaved serpin I*; k4 is the rate constant of the formation of the kinetically trapped, stable covalent complex EI*; k5 is the dissociation rate constant of the covalent complex. Adapted with modifications from [11].
Unexpectedly, we observed that mesotrypsin selectively and rapidly cleaved the Lys10-Thr11 peptide bond at the N terminus of α1AT. Subsequent mutagenesis studies confirmed that mesotrypsin preferentially hydrolyzed lysyl peptide-bonds containing Thr or Ser at the P1′ position. Furthermore, although mesotrypsin was completely resistant to wild-type α1AT, it readily cleaved the Arg358-Ser359 reactive-site peptide bond of α1AT Pittsburgh and was inactivated by the serpin. Taken together, the observations clearly re-define the substrate specificity of mesotrypsin and demonstrate that in addition to the reactive-site peptide-bonds of canonical trypsin inhibitors, mesotrypsin can also efficiently digest Lys/Arg-Thr/Ser peptide bonds in polypeptide substrates.
RESULTS
Mesotrypsin exhibits complete resistance against wild-type α1-antitrypsin
Incubation of 20 nM mesotrypsin with increasing concentrations of wild-type α1AT for 20 min did not result in any detectable inhibition up to 2 μM inhibitor concentration, whereas human cationic and anionic trypsins were fully inhibited (Fig 2A). When the time-course of incubation was extended up to 2.5 h, and 2 μM mesotrypsin was incubated with 5 μM wild-type α1AT, no measurable inhibition of mesotrypsin activity was observed either. Again, under these conditions human cationic and anionic trypsins were inhibited rapidly (Fig 2B). Essentially identical results were obtained with native α1AT purified from human serum or recombinant α1AT expressed in E. coli. The results confirm the early observations of Rinderknecht et al., who in their seminal paper list α1AT as one of the proteinaceous inhibitors that are inactive against mesotrypsin (see Table 5 in ref [2]). Sequence alignments, crystallographic data and mutagenesis experiments showed that the unique Arg198 residue is responsible for the resistance of mesotrypsin against canonical trypsin inhibitors [3–5]. To determine the role of Arg198 in the resistance of mesotrypsin against α1AT, Arg198 was substituted with Gly, the residue characteristically found at this position in the chymotrypsin-like serine proteases. The R198G mutant mesotrypsin was inhibited by wild-type α1AT in a manner that was comparable to inhibition of cationic and anionic trypsins, demonstrating that Arg198 is the critical determinant of resistance against α1AT (Fig 2AB).
Figure 2.
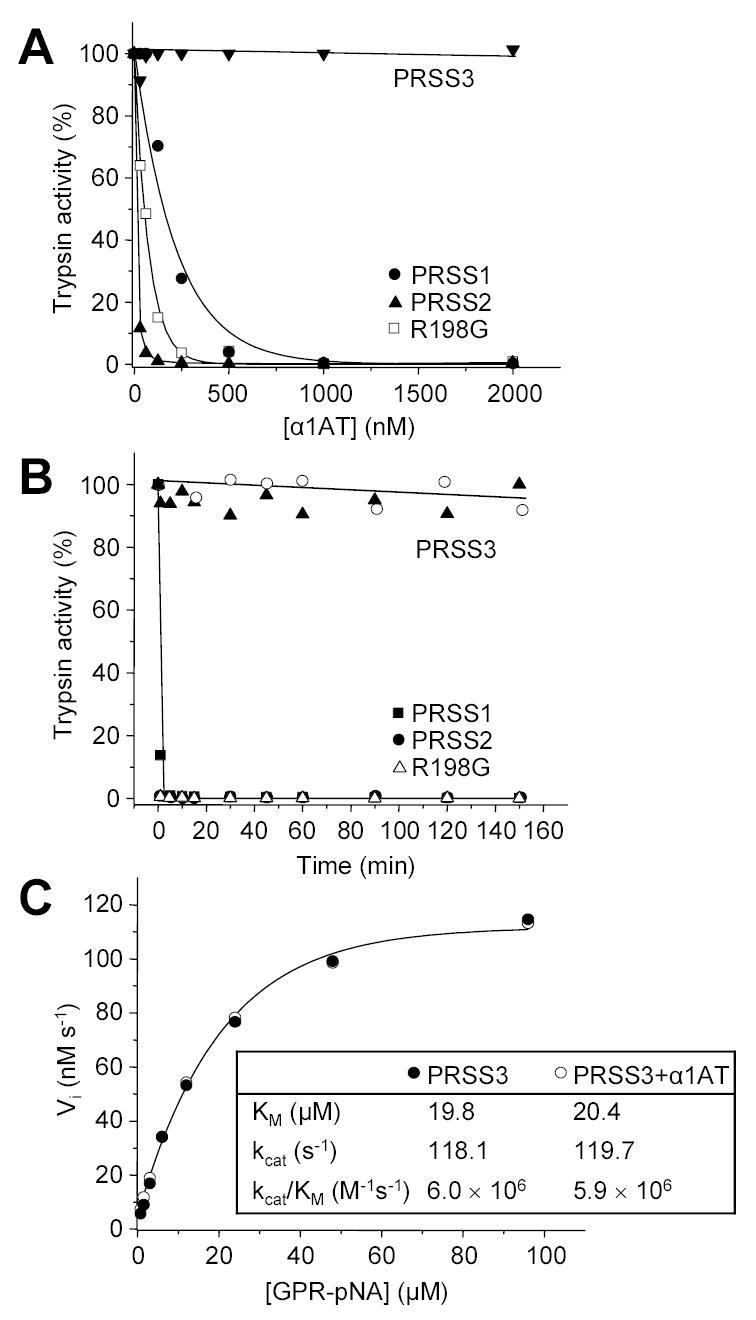
Inhibition of human trypsins by α1-antitrypsin (α1AT). A. Cationic trypsin (PRSS1), anionic trypsin (PRSS2), mesotrypsin (PRSS3) and the R198G mesotrypsin mutant were incubated at 20 nM concentration with the indicated concentrations of α1AT in 100 μL final volume of 0.1 M Tris-HCl (pH 8.0) and 1 mM CaCl2, at room temperature for 20 min. Trypsin activity was then assayed with 0.1 mM N-CBZ-Gly-Pro-Arg-p-nitroanilide (final concentration), and expressed as percentage of initial activity (without inhibition). B. Trypsins (2 μM concentration) were incubated with 5 μM α1AT (final concentration) in 0.1 M Tris-HCl (pH 8.0), 2 mg/mL BSA, and 1 mM CaCl2 at 37 C°. At indicated time-points 2 μL aliquots were withdrawn and trypsin activity was measured. Recombinant α1AT was used to inhibit trypsins in these experiments, with the exception of mesotrypsin (PRSS3), which was incubated with recombinant (solid triangles) and native α1AT purified from human serum (open circles). C. Competitive inhibition of mesotrypsin by α1AT. The initial rate (Vi) of substrate hydrolysis by 1 nM mesotrypsin (final concentration) was measured at the indicated N-CBZ-Gly-Pro-Arg-p-nitroanilide (GPR-pNA) concentrations, in the presence (○) or absence (●) of 7.5 μM α1AT (final concentration), in 0.1 M Tris-HCl (pH 8.0) and 1 mM CaCl2 at room temperature. The KM and kcat parameters were determined from hyperbolic fits.
Fig 2A also indicates that the apparent stoichiometry of inhibition (SI) for the different trypsin isoforms varies between 1 and 40. However, these values do not represent the true SI, because the reactions have not reached completion under the experimental conditions used. Instead, the observed differences in apparent SI suggest different rates of association. Indeed, the measured second-order rate constants (ka) indicate that cationic trypsin associates with wild-type α1AT almost 20-fold more slowly than anionic trypsin (Table 1). Similar ka values were reported previously by Vercaigne-Marko et al. [12]. When the incubation times were extended up to 4 h to allow complete association between α1AT and trypsins, the determined SI values for cationic and anionic trypsins and R198G-mesotrypsin all approached unity (not shown).
Table 1.
Observed association rate constants (kobs) between human trypsins and wild-type or Pittsburgh mutant α1-antitrypsin (α1AT). PRSS1, cationic trypsin; PRSS2, anionic trypsin; PRSS3, mesotrypsin; R198G, mesotrypsin mutant Arg198→Gly. Rate constants were determined from three independent measurements, using a discontinuous or continuous assay, as described in Experimental Procedures. The errors of curve fits are indicated. To obtain the true second order association rate constants, the kobs values need to be multiplied with the stoichiometry of inhibition (SI). With the exception of mesotrypsin, the SI was approximately unity, therefore kobs equals ka. Mesotrypsin associates with α1AT Pittsburgh with an SI of 2, and the calculated ka is 1.1 × 106 M−1 s−1. N. D., not determined. Using purified pancreatic cationic and anionic trypsins and native wild-type α1AT, Vercaigne-Marko et al. (1989) reported association rate constants of 1.35 x 104 M−1 s−1and 1.8 x 105 M−1 s−1, respectively [12].
|
kobs M−1 s−1 |
||
|---|---|---|
| α1AT | α1AT Pittsburgh | |
| PRSS1 | 8.7 ± 0.2 × 103 | 2.3 ± 0.1 × 106 |
| PRSS2 | 1.6 ± 0.1 × 105 | 2.0 ± 0.1 × 106 |
| PRSS3 | N.D. | 5.4 ± 0.3 × 105 |
| R198G | 3.3 ± 0.2 × 104 | 2.2 ± 0.1 × 106 |
Previous studies demonstrated that canonical trypsin inhibitors do not form tight inhibitory complexes with mesotrypsin, however, they still can act as weak, competitive inhibitors [4, 5]. The weak inhibitory effect is not necessarily evident in the typical inhibition assays when the pre-incubated enzyme-inhibitor mixture is diluted into a high concentration of substrate solution. Under these conditions the loosely associated complexes rapidly dissociate and no inhibition is observed. To detect competitive inhibition, kinetic parameters (KM, kcat) for the hydrolysis of the trypsin substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide by mesotrypsin were determined in the absence and presence of 7.5 μM α1AT (Fig 2C). Clearly, wild-type α1AT had no effect whatsoever on mesotrypsin activity, ruling out the possibility of competitive inhibition, at least at the concentration studied.
To visualize the interaction between human trypsins and wild-type α1AT, inhibitory complexes were electrophoresed on 13 % gels and stained with Coomassie blue (Fig 3). Because the serpin mechanism traps the acyl-enzyme intermediate, the covalently linked serpin-protease complexes can be resolved from the reactants by SDS-PAGE. As expected from the functional assays, mesotrypsin did not associate with wild-type α1AT, whereas the R198G mesotrypsin mutant, cationic trypsin and anionic trypsin formed complexes. Partial proteolysis of the complexes was also observed, which resulted in bands migrating between the free α1AT and the intact serpin-protease complex. Mutating Arg122 to Ala (R122A) in cationic and anionic trypsins abolished the major proteolytic bands, confirming that complexes are mostly cleaved at the Arg122-Val123 peptide bond, a well-known autolysis site in trypsin.
Figure 3.
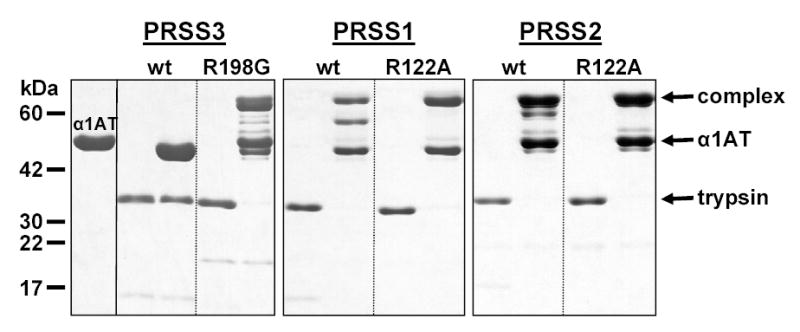
Covalent complex formation between wild-type α1-antitrypsin (α1AT) and human trypsins. Mesotrypsin (PRSS3), the R198G mesotrypsin mutant, cationic trypsin (PRSS1), the R122A cationic trypsin mutant, anionic trypsin (PRSS2), and the R122A anionic trypsin mutant were incubated at 1 μM concentration with or without 3 μM α1AT in 0.1 M Tris-HCl (pH 8.0), and 10 mM CaCl2, at 37 C° for 30 min. The 100 μL incubation mixes were precipitated with 10 % final concentration of trichloroacetic acid and subjected to reducing SDS-PAGE and Coomassie blue staining. The positions of the bands representing the covalent complex, the free α1AT and the free trypsins are indicated. See text for details on the bands migrating between the complex and free α1AT.
N-terminal processing of α1-antitrypsin at the Lys10-Thr11 peptide bond by mesotrypsin
We also observed that incubation of mesotrypsin with wild-type α1AT resulted in a small anodal shift in the position of the free inhibitor band on the gels (Figs 3, 4). Western-blot analysis using an antibody against the N-terminal 6-His epitope of recombinant α1AT revealed that mesotrypsin cleaved off a peptide from the N terminus. Removal of the N terminus was also observed with native α1AT and N-terminal protein sequencing determined that the cleavage occurred at the Lys10-Thr11 peptide bond (Fig 4). N-terminal processing of free and complexed forms of α1AT was also evident after incubation with the slowly-associating cationic trypsin, whereas only partial cleavage occurred during the reaction with anionic trypsin and R198G-mesotrypsin (Fig 3). The inhibitory activity of the N-terminally truncated α1AT remained unaffected when tested on human or bovine trypsins or human neutrophil elastase (not shown).
Figure 4.
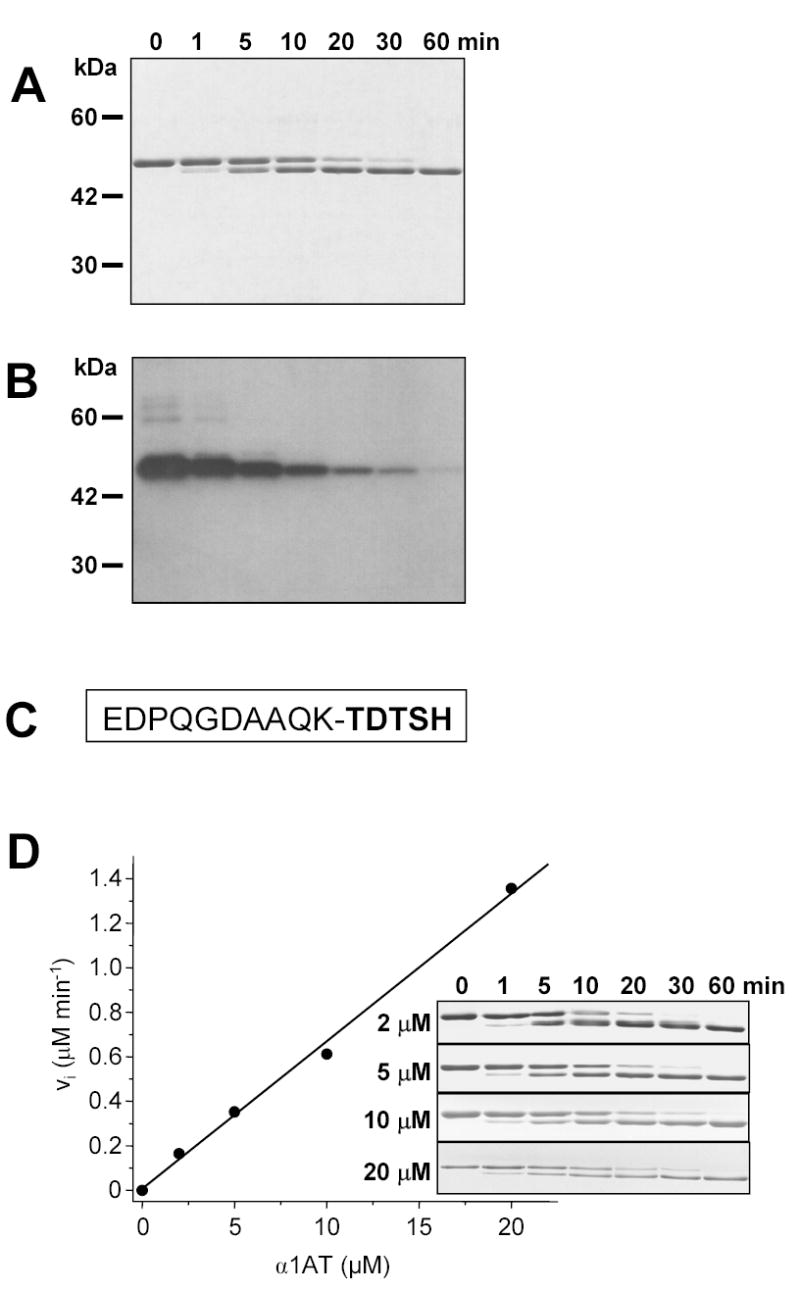
N-terminal processing of α1-antitrypsin (α1AT) by mesotrypsin. A. 5 μM α1AT and 15 nM mesotrypsin (final concentrations) were incubated in 0.1 M Tris-HCl, and 1 mM CaCl2 at 37 C°. Aliquots (20 μL) were precipitated with 10 % final concentration of trichloroacetic acid at the indicated times and resolved on 13 % SDS-polyacrylamide gels followed by Coomassie-blue staining. B. Aliquots were also analyzed by Western blotting. Detection of the N-terminal 6-His-tag in α1AT was carried out with the Tetra-His primary antibody (Qiagen) at 1:1000 dilution, followed by HRP-conjugated anti-mouse IgG diluted at 1:10,000, and SuperSignal West Pico chemiluminescent substrate (Pierce). C. N-terminal sequence of native human α1AT. The cleaved Lys10-Thr11 peptide bond is indicated. The emboldened sequence was determined by Edman degradation. D. Kinetic analysis of the digestion reaction. Mesotrypsin (15 nM concentration) was incubated with the indicated concentrations of α1AT in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 at 37 C°, and the digestions were analyzed by SDS-PAGE (inset) and densitometry. The initial rate (vi) of the reactions was plotted as a function of α1AT concentration.
To determine the kinetic parameters of the reaction, the rate of cleavage was measured at α1AT concentrations ranging from 2 to 20 μM, using gel-electrophoresis and densitometry (Fig 4). The reaction rate showed an apparently linear dependence on the substrate concentration over the range studied, indicating that the KM value must be higher than 20 μM. Using progress curve analysis, the second-order specificity constant kcat/KM was calculated and found to be approximately 105 M−1 s−1.
The Lys10-Ile11-Val12 α1-antitrypsin mutant is not processed by mesotrypsin
The observation that mesotrypsin cleaves the Lys10-Thr11 peptide bond in α1AT with high efficiency is surprising as it stands in contrast with the proposed inability of mesotrypsin to cleave protein substrates other than trypsin inhibitors [2, 5, 8]. The results suggest that mesotrypsin exhibits a uniquely restricted substrate specificity governed by either the conformational properties of the polypeptide substrate or the amino-acid sequences flanking the lysyl/arginyl scissile bonds. To investigate the latter, we introduced the P1′-P2′ amino-acids of the trypsinogen activation site into α1AT by changing the Thr11-Asp12 residues to Ile11-Val12. Rates of cleavage were determined for mesotrypsin, cationic trypsin, anionic trypsin and the R198G-mesotrypsin mutant (Table 2). To eliminate the inhibitory activity, α1AT was first inactivated by digesting the reactive-center loop with the Staphylococcus aureus V8 protease [13, 14]. Rates of N-terminal processing by mesotrypsin were identical before and after V8-protease mediated inactivation of α1AT, indicating that V8 protease does not alter the properties of the N-terminal region. As shown in Table 2, mesotrypsin cleaved the Lys10-Thr11 peptide-bond in α1AT 5-fold better than cationic trypsin or R198G-mesotrypsin, and almost 2-fold better than anionic trypsin. Compared to these cleavage rates, mesotrypsin digested the Lys10-Ile11 peptide bond in the mutant α1AT construct almost 250-fold slower, whereas digestion was enhanced 30-fold by cationic trypsin, and 8-fold by anionic trypsin and R198G-mesotrypsin. Overall, the engineered trypsinogen activation site motif Lys10-Ile11-Val12 in α1AT was hydrolyzed by mesotrypsin 400–1400-fold less efficiently than by other trypsins, which is in perfect agreement with previous observations indicating a 500–1000-fold difference in activation of pancreatic protease zymogens [5]. Clearly, the presence of Arg198 restricts the substrate specificity of mesotrypsin, but does not inhibit digestion of all polypeptide substrates as previously thought.
Table 2.
N-terminal processing of wild-type α1-antitrypsin (Lys10-Thr-Asp) and a mutant with the P1′-P2′ residues of the trypsinogen activation site (Lys10-Ile-Val). PRSS1, cationic trypsin; PRSS2, anionic trypsin; PRSS3, mesotrypsin; R198G, mesotrypsin mutant Arg198→Gly. Second-order rate constants (kobs) were obtained from progress curve analysis of digestion reactions followed by SDS-PAGE and densitometry, as described in Experimental Procedures. Two or more independent experiments were evaluated in a single fitting, and the error of the fit is indicated. Digestion reactions contained 5 μM V8-protease inactivated α1-antitrypsin and 10 nM trypsin (final concentrations), with the exception of PRSS3, which was used at 1 μM concentration to digest the trypsinogen activation site motif (Lys10-Ile-Val).
|
kobs M−1 s−1 |
||
|---|---|---|
| Lys10-Thr-Asp | Lys10-Ile-Val | |
| PRSS1 | 1.9 ± 0.1 × 104 | 6.4 ± 0.7 × 105 |
| PRSS2 | 6.2 ± 0.5 × 104 | 4.9 ± 0.7 × 105 |
| PRSS3 | 1.1 ± 0.1 × 105 | 4.5 ± 1.3 × 102 |
| R198G | 2.2 ± 0.1 × 104 | 1.8 ± 0.1 × 105 |
Mesotrypsin exhibits restricted S1′ subsite specificity
Because Arg198 appears to occupy the S2′ subsite in mesotrypsin [4], we speculated that the positively charged guanidino group might interact with the P2′ Asp12 residue and thus enhance cleavage of the Lys10-Thr11 peptide-bond in α1AT. However, a mutant in which Asp12 was changed to Val was processed by mesotrypsin at a rate that appeared to be only 5-fold decreased (not shown). Due to poor expression, we were unable to study this mutant in more detail. On the other hand, changing Thr11 to Ile resulted in drastic inhibition of cleavage, suggesting that the P1′ position is the critical determinant of mesotrypsin’s specificity. To confirm the significance of the P1′ position, we have replaced Thr11 with 9 different amino-acids of various sizes and physico-chemical properties (in addition to Ile; Asn, Asp, Gly, Met, Phe, Pro, Ser and Tyr). The α1AT mutants were purified and rates of cleavage by mesotrypsin were determined on 13 % SDS-polyacrylamide gels (Fig 5). Surprisingly, in addition to Thr, only Ser allowed rapid cleavage after Lys10, with a kcat/KM value that approached 105 M−1 s−1. Hydrolysis of the Lys10-Gly11 and Lys10-Met11 peptide-bonds was 13-fold and 25-fold slower, respectively, whereas P1′ residues of Asn, Asp, Ile, Phe, or Tyr, resulted in 100–200-fold lower cleavage rates. The Lys10-Pro11 peptide bond was not cleaved to any detectable extent. The results demonstrate that mesotrypsin exhibits an unusually restricted S1′ subsite specificity and accommodates only small, hydrophilic side-chains.
Figure 5.
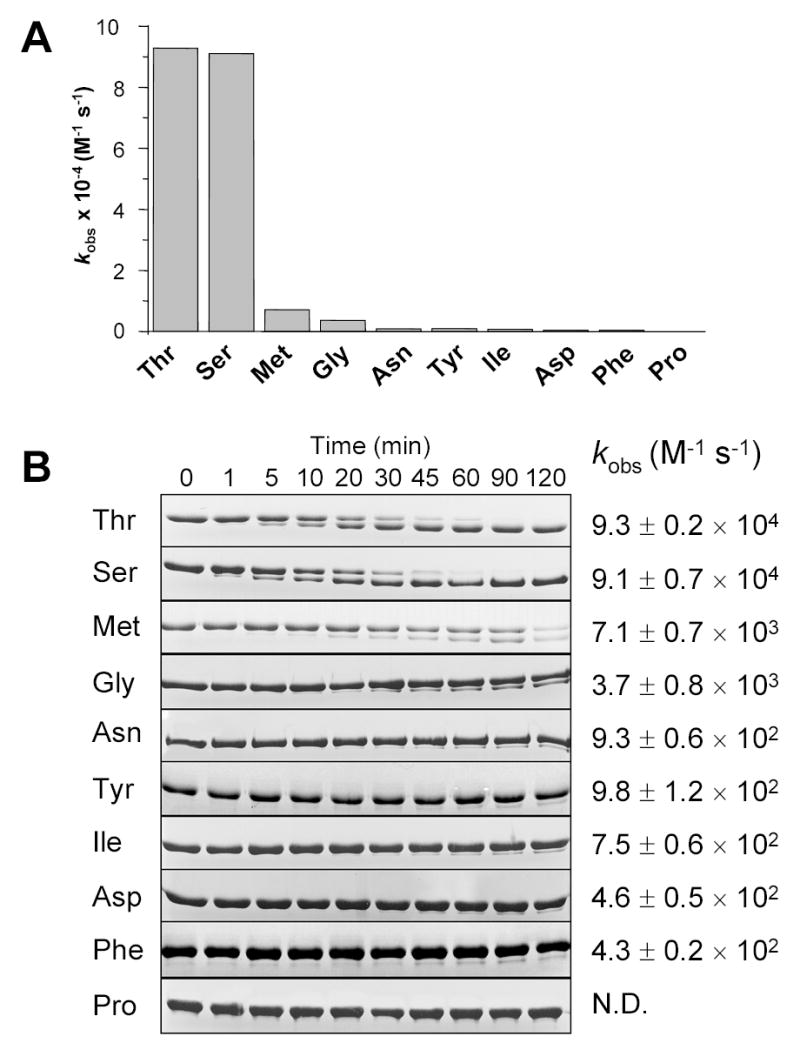
S1′ subsite specificity of mesotrypsin. Wild-type α1-antitrypsin (α1AT) and 9 mutants in which Thr11 was changed to the indicated amino-acids were digested with mesotrypsin in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2 at 37 C°, and the digestion reactions were analyzed by SDS-PAGE and densitometry. Second-order rate constants (kobs) were calculated with progress curve analysis, as described in Experimental Procedures. Two or more independent experiments were evaluated together with a single fit, and the error of the fit is indicated. A. Bar graph representation of the kobs values. B. Coomassie-blue stained gels of digestion reactions with 5 μM wild-type or mutant α1AT and 10 nM mesotrypsin (final concentrations). The gels are shown to illustrate the significant differences in digestion rates. To calculate the kobs values indicated next to the gels, reactions were also performed with mesotrypsin concentrations up to 1 μM to achieve measurable rates of digestion (not shown). N. D., not determined, the Thr11→Pro mutant was not digested to any detectable extent with mesotrypsin.
Mesotrypsin is inactivated by α1-antitrypsin Pittsburgh
The natural Pittsburgh variant of α1AT contains an Arg residue in place of the P1 Met358 in the reactive-site peptide bond [15, 16]. Because of this change, the α1AT Pittsburgh mutant exhibits different specificity than wild-type α1AT. It inhibits thrombin and trypsin-like enzymes significantly better, whereas inhibition of elastases is compromised [17]. Incubation of 20 nM mesotrypsin (final concentration) for 10 min with increasing concentrations of α1AT Pittsburgh resulted in complete inactivation of the protease, with an apparent stoichiometry of inhibition (SI) of 2 (Fig 6A). This value remained the same with increased incubation times, indicating that it corresponds to the true SI between mesotrypsin and α1AT Pittsburgh. Human cationic trypsin, anionic trypsin and the R198G mesotrypsin mutant were also inactivated, with an SI of unity.
Figure 6.
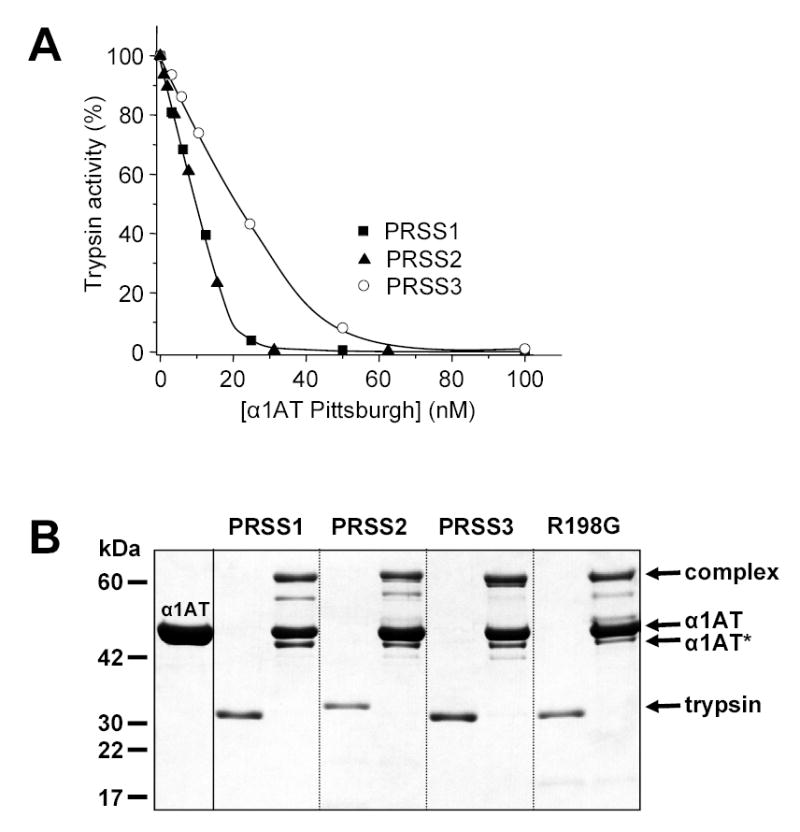
Inhibition of human trypsins by α1-antitrypsin (α1AT) Pittsburgh. A. Cationic trypsin (PRSS1), anionic trypsin (PRSS2) and mesotrypsin (PRSS3) were incubated at 20 nM concentration with the indicated concentrations of α1AT Pittsburgh at room temperature in 100 μL 0.1 M Tris-HCl (pH 8.0), 2 mg/mL BSA, and 1 mM CaCl2 for 10 min. Trypsin activity was then assayed with 0.1 mM N-CBZ-Gly-Pro-Arg-p-nitroanilide (final concentration) and expressed as percentage of initial activity (without inhibition). B. Covalent complex formation between α1-antitrypsin Pittsburgh and human trypsins. Trypsins were incubated at 1 μM concentration with or without 5 μM α1AT Pittsburgh in 0.1 M Tris-HCl (pH 8.0), and 10 mM CaCl2, at 37 C° for 30 min. The incubation mixtures (100 μL) were precipitated with 10 % final concentration of trichloroacetic acid and subjected to reducing SDS-PAGE and Coomassie blue staining. The positions of the bands representing the covalent complex, the free α1AT and the free trypsins are indicated. α1AT* indicates the cleaved, inactive α1AT Pittsburgh. See text for further details.
The second order rate constants for complex association indicated that, after correction for SI, mesotrypsin associated with α1AT Pittsburgh almost as rapidly as cationic or anionic trypsin (Table 1). Notably, association rates for cationic trypsin, anionic trypsin and the R198G mesotrypsin mutant were circa 260-fold, 13-fold and 70-fold higher, respectively, than those with wild-type α1AT.
SDS-PAGE analysis of complex formation confirmed that mesotrypsin covalently associated with α1AT Pittsburgh, in a manner that was essentially identical to inhibition of cationic and anionic trypsins and R198G-mesotrypsin (Fig 6B). Because of the rapid association rates, N-terminal processing of α1AT by free trypsin was not apparent in these experiments. On the other hand, the gels revealed a new α1AT band that migrated somewhat faster than the free α1AT. Western blot analysis showed that the N terminus was intact on this α1AT species, suggesting that this band corresponded to C-terminally truncated, inactive α1AT, cleaved at the Arg358-Ser359 reactive-site peptide-bond. The presence of this band would indicate that some of the covalent complexes rapidly deacylated and thus followed the non-inhibitory proteolytic pathway. Consequently, the stoichiometry of inhibition should be greater than 1, and judging from the band intensities the SI should approach 2. Whereas an increased SI was indeed demonstrated for mesotrypsin (SI ~2), repeated functional assays consistently determined SI values around unity for the other trypsins. Therefore, we must conclude that the C-terminally truncated α1AT band is artefactual, and it is generated from the inhibitory complexes during SDS-denaturation.
The covalent complex between mesotrypsin and α1AT Pittsburgh was stable, however, complexes formed between cationic and anionic trypsins and α1AT Pittsburgh dissociated relatively rapidly, even though these trypsins formed stable complexes with wild-type α1AT (Fig 7). The first-order dissociation rate constants showed that the mesotrypsin - α1AT Pittsburgh complex was 40-fold more stable than complexes with cationic and anionic trypsin. Finally, the R198G mutant mesotrypsin formed equally stable complexes with α1AT Pittsburgh as wild-type mesotrypsin, indicating that increased complex stability is independent of the presence of Arg198 (Table 3).
Figure 7.
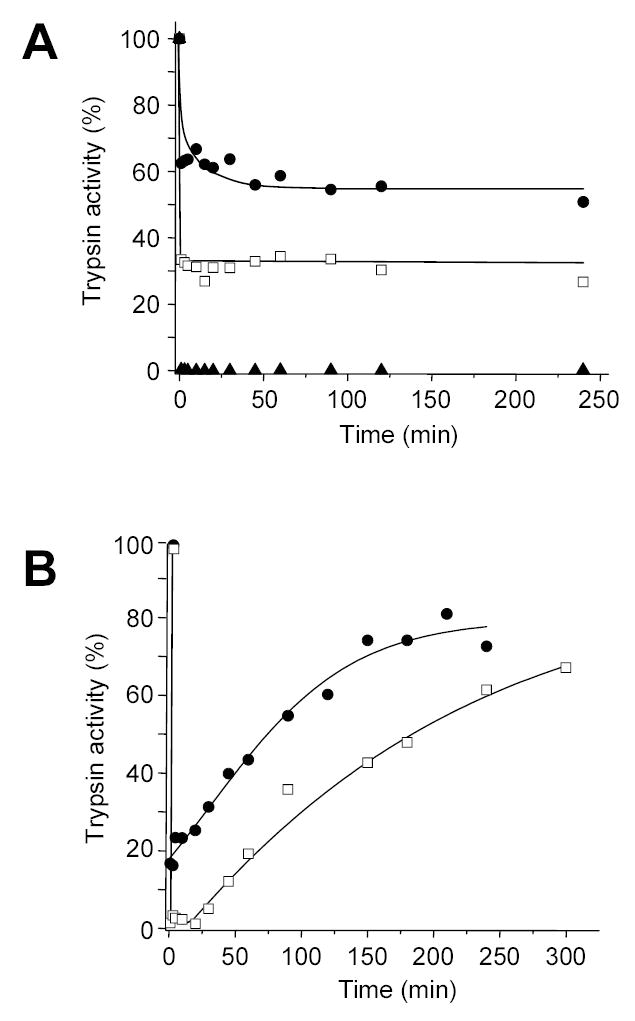
Dissociation of covalent complexes between α1-antitrypsin (α1AT) Pittsburgh and mesotrypsin (A) or cationic trypsin (B). Trypsins were incubated with α1AT Pittsburgh in 0.1 M Tris-HCl (pH 8.0), 10 mM CaCl2, and 2 mg/mL BSA at 37 C°. Aliquots (2 μL) were assayed for trypsin activity at indicated times and trypsin activity was expressed as percentage of initial activity (i.e. before addition of α1AT Pittsburgh). A. Mesotrypsin (1 μM concentration) was incubated with 0.75 μM (●), 1.5 μM (□), or 3 μM (▲) α1AT Pittsburgh. B. Cationic trypsin (1 μM concentration) was incubated with 0.75 μM (●) or 1.5 μM (□) α1AT Pittsburgh.
Table 3.
First-order dissociation rate constants (kdiss) for complexes of human trypsins and wild-type or Pittsburgh variant α1-antitrypsin (α1AT). PRSS1, cationic trypsin; PRSS2, anionic trypsin; PRSS3, mesotrypsin; R198G, mesotrypsin mutant Arg198→Gly. Rates of dissociation were determined at several initial complex concentrations, and kdiss was calculated from linear fits to rate vs. concentration plots, as described in Experimental Procedures. The errors of the linear fits are also indicated. N. D., not determined.
|
kdiss s−1 |
||
|---|---|---|
| α1AT | α1AT Pittsburgh | |
| PRSS1 | 3.8 ± 0.5 × 10−7 | 3.2 ± 0.1 × 10−5 |
| PRSS2 | 3.6 ± 0.3 × 10−6 | 3.3 ± 0.1 × 10−5 |
| PRSS3 | N.D. | 8.0 ± 0.2 × 10−7 |
| R198G | 4.5 ± 0.4 × 10−7 | 4.5 ± 0.3 × 10−7 |
In conclusion, we showed that mesotrypsin cleaved the Arg358-Ser359 reactive-site peptide bond of α1AT Pittsburgh, which resulted in the rapid formation of a covalent inhibitory complex with high kinetic stability. The results provide independent corroboration that a Ser residue at the P1′ position of polypeptide substrates is preferred by mesotrypsin. Furthermore, the serpin mechanism is proven as a feasible strategy to overcome the inhibitor resistance of mesotrypsin, provided the serpin reactive site conforms to the Arg/Lys-Thr/Ser motif.
DISCUSSION
Selective N-terminal processing of α1AT at the Lys10-Thr11 peptide-bond by mesotryspin is the most interesting and unexpected observation of this study. Previously, we proposed that mesotrypsin was a defective digestive protease on polypeptide substrates, because it did not activate pancreatic protease zymogens or degraded trypsinogens [5]. The results presented here clearly negate this notion, and demonstrate that mesotrypsin is a functionally competent digestive enzyme, but it exhibits restricted substrate specificity with a strong preference for Arg/Lys-Thr/Ser peptide-bonds. The data also provide an explanation why mesotrypsin is defective in zymogen activation and degradation. Thus, canonical activation sites of pancreatic protease zymogens contain Ile or Val at the P1′ position. The most sensitive autolysis site in human trypsin(ogen)s, Arg122-Val123, also contains a Val residue at P1′. These peptide bonds are readily cleaved by typical trypsins, which exhibit a broad P1′ specificity, with a moderate preference for hydrophobic amino-acids over Ser or Thr [18–21]. Although Val per se was not tested in our experiments, the presence of the similarly hydrophobic Ile in the P1′ position inhibited cleavage of lysyl peptide bonds by mesotrypsin 120-fold, relative to a P1′ Thr or Ser.
The discovery that mesotrypsin prefers Arg/Lys-Thr/Ser peptide bonds offers supportive evidence that mesotrypsin may indeed act as an agonist for certain PAR receptors. The activating cleavage site is Arg-Ser in PAR-1 and PAR-2, Lys-Thr in PAR-3 and Arg-Gly in PAR-4. Results presented in this study indicate that Arg-Ser or Lys-Thr peptide-bonds are readily cleaved by mesotrypsin, whereas the Arg-Gly bond is hydrolyzed more slowly. Consequently, we could predict that PAR-1, PAR-2 and PAR-3 are good mesotrypsin substrates, whereas PAR-4 should be poorly activated. However, published data are contradictory in this respect. First, PAR-2 and PAR-4 in epithelial cells were identified as mesotrypsin substrates [9]. Later, these findings were disputed, but PAR-1 in the brain was shown to be activated by mesotrypsin [10]. Clearly, beyond the cleavage site per se, other interactions between the protease and the PAR receptor influence whether or not mesotrypsin can activate a given PAR isoform. Furthermore, tissue and species specific glycosylation of PAR can also alter activation properties, which may account for some of the conflicting data published.
The restricted S1′ subsite specificity of mesotrypsin determined here is similar to that of thrombin. Studies using antithrombin-III reactive-site mutants or internally quenched fluorescent substrates revealed that thrombin prefers P1′ Ser, Thr, Gly or Ala residues [22–24]. The majority of thrombin's natural substrates also contain a Ser or Thr residue at the P1′ position (see Table 2 in ref [24]). The structural basis for the restricted S1′ specificity of thrombin is not entirely clear. Crystallographic analysis of thrombin suggested that Lys60f (chymo#) occludes the S1′ subsite and limits its specificity to amino-acids with small side-chains [25]. However, mutagenesis of Lys60f to Ala only partially relieved this restriction, indicating that other determinants are also important [26]. In contrast to thrombin, the S1′ subsite on trypsin is not obstructed and it can accommodate amino-acids of various sizes and polarity [27]. Subsite mapping studies consistently found a modest (10-fold or less) preference for hydrophobic aminoacids over Ser or Thr [18–21], which might be explained by favorable interactions with the hydrophobic side-chain of Lys60 (chymo#) [28]. Superimposition of the crystal structure of human cationic trypsin and mesotrypsin reveals that the structural determinants of the S1′ subsite assume identical conformations in both structures and the S1′ subsite in mesotrypsin does not appear occluded in any way [4, 27]. The Arg198 side-chain in mesotrypsin clearly occupies the S2′ subsite. Evidently, the available structural data offer no explanation for the highly restricted S1′ subsite specificity of mesotrypsin. It appears reasonable to assume that substrate binding leads to conformational changes that mitigate the conflict with Arg198 at the S2′ site and result in the partial obstruction of the S1′ site.
The original objective of the present study was to test the notion whether the serpin inhibitory mechanism can overcome the inhibitor resistance of human mesotrypsin. The rational for this hypothesis was the observation that mesotrypsin hydrolyzes the reactive-site peptide bond of canonical trypsin inhibitors in a substrate-like manner [5]. Cleavage of the reactive site peptide-bond is an essential part of the serpin inhibitory mechanism (see Fig 1), suggesting that mesotrypsin might be subject to inhibition by serpins. The results indicate that mesotrypsin is completely resistant to wild-type α1AT and this resistance solely depends on the mesotrypsin-specific Arg198 residue. The complete resistance is surprising, since canonical trypsin inhibitors exhibit a reduced but still significant affinity toward mesotrypsin and thus they behave as weakly binding, competitive inhibitors [4, 5]. Clearly, the combination of the suboptimal P1 Met residue in wild-type α1AT and the steric obstruction of the S2′ site by Arg198 in mesotrypsin prevents formation of the initial Michaelis complex. In contrast to wild-type α1AT, the Pittsburgh variant inhibited mesotrypsin via the classic serpin mechanism with a rapid association rate and high kinetic stability. Thus, serpins inhibit mesotrypsin if substrate-like hydrolysis of the reactive-site peptide bond can occur. In this respect, the Arg358-Ser359 reactive-site peptide bond satisfies the restricted S1′ specificity of mesotrypsin, and explains the efficient inhibition by α1AT Pittsburgh.
While this manuscript was in preparation two studies were published that support our conclusions. First, mesotrypsin was shown to cleave selectively the Arg79-Thr80 and Arg97-Thr98 peptide bonds in the lipid bound form of human myelin basic proteins [29], which is in perfect agreement with the S1’ subsite specificity determined here. Second, using the 4-methylumbelliferyl 4-guanidinobenzoate substrate analogue, thermodynamic analysis demonstrated significant structural rearrangements during the acylation step in mesotrypsin, which were absent in the R198G (chymo# R193G) mutant [30]. These results are consistent with our proposal that the restricted S1’ subsite specificity of mesotrypsin is the result of conformational changes during substrate binding, which are dependent on Arg198.
EXPERIMENTAL PROCEDURES
Materials
N-CBZ-Gly-Pro-Arg-p-nitroanilide and 4-methylumbelliferyl 4-guanidinobenzoate HCl (MUGB) were purchased from Sigma (St. Louis, Missouri). Ni-NTA agarose, mouse tetra-His antibody and SG13009 competent cells were from Qiagen (Valencia, California). Anti-mouse IgG HRP conjugate was from Promega (Madison, Wisconsin). Human α1AT purified from plasma was purchased from Calbiochem (San Diego, California) and Sigma. Recombinant human pro-enteropeptidase was from R & D Systems (Minneapolis, Minnesota). Pro-enteropeptidase (0.07 mg/mL stock solution) was activated with 50 nM human cationic trypsin in 0.1 M Tris-HCl (pH 8.0), 10 mM CaCl2 and 2 mg/ml bovine serum albumin (final concentrations) for 30 min at room temperature. Staphylococcus aureus V8 Protease (Endoproteinase GluC) was from New England Biolabs (Ipswich, Massachusetts).
Nomenclature
The genetic abbreviations PRSS1 (protease, serine, 1), PRSS2 and PRSS3 are used to denote human cationic trypsinogen, anionic trypsinogen, and mesotrypsinogen, respectively. Note that mesotrypsin is also referred to as trypsin 4 or trypsin IV in the literature. Amino acid residues in the trypsinogen sequences are numbered according to their position in the native pre-proenzyme, starting with Met1. Where indicated by the chymo# abbreviation, the conventional chymotrypsin numbering is used. Amino-acid numbering of α1AT starts with the first amino-acid of the mature native form (Glu1), according to the convention in the literature.
Expression and purification of human trypsinogens
Construction of expression plasmids for human cationic trypsinogen (PRSS1), anionic trypsinogen (PRSS2) and mesotrypsinogen (PRSS3) and engineering of the R198G mesotrypsin mutant were described previously [5, 31–33]. Mutation R122A was introduced into the PRSS1 and PRSS2 genes by site-directed mutagenesis using the overlap-extension PCR method. Recombinant trypsinogens were expressed in E. coli Rosetta (DE3) as inclusion bodies and following in vitro refolding zymogens were purified on an ecotin affinity column, as reported previously [5, 31–33]. Trypsinogens (2 μM concentration) were activated with human recombinant enteropeptidase (10 ng/mL final concentration), in 0.1 M Tris-HCl (pH 8.0), 10 mM CaCl2, and 2 mg/ml bovine serum albumin (BSA) for 1 h at 37 oC. Trypsin concentration was then determined with active site titration using 4-methylumbelliferyl 4-guanidinobenzoate HCl [34].
Expression and purification of α1-antitrypsin
The pQE30-vector based expression plasmids for wild-type α1AT and α1AT Pittsburgh were kind gifts from Peter Gettins' laboratory. The recombinant α1AT expressed from these plasmids corresponds to the M2 natural allele, but contains 7 stabilizing mutations (F51L, T59A, T68A, A70G, M374I, S381A, and K387R) that hinder polymerization and the C232S mutation that prevents intermolecular disulfide bond formation [35, 36]. In addition, the native N terminus of EDPQG has been replaced with the MRGSHHHHHHGS sequence, which includes a 6-His tag. Mutations of Thr11 (to Asn, Asp, Gly, Ile, Met, Pro, Phe, Ser, and Tyr) and Asp12 (to Val and Ile) were introduced with PCR mutagenesis. α1AT was expressed in E. coli SG13009. Cultures were grown to an OD600 nm of 0.6–0.8, and induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside for 3 h. Cells were harvested, resuspended in 20 mL 50 mM NaCl, 50 mM Tris-HCl (pH 8.0), 1 mM EDTA and 1 mM PMSF and disrupted by sonication. The cell lysate was clarified by centrifugation and the supernatant was loaded onto a Ni-NTA affinity column (Qiagen), which was pre-equilibrated with the same buffer. The column was washed with a stepwise imidazole gradient (10 mM, 50 mM, and 250 mM imidazole in 50 mM Na-phosphate (pH 7.4) and 250 mM NaCl), and α1AT eluted at 50 mM and 250 mM imidazole concentrations. Fractions (5 mL) were collected, analyzed by SDS-PAGE, and typically 1–2 fractions were pooled and dialyzed against 20 mM Tris-HCl (pH 8.0) at 4 C°. Affinity-purified α1AT was then loaded onto a MonoQ column equilibrated with 20 mM Tris-HCl (pH 8.0), and the column was developed with a 0–0.5 M NaCl gradient. The peak corresponding to α1AT eluted at about 200 mM NaCl concentration. Fractions (1 mL) were analyzed by SDS-PAGE, and pooled fractions (5–10 mL) were dialyzed against 20 mM Tris-HCl (pH 8.0) at 4 C°. The concentration of α1AT was determined from the ultraviolet absorbance at 280 nm, using a theoretical extinction coefficient of 19,940 M−1 cm−1.
Inhibition assays
Rates of complex association
The apparent association rates between trypsins and serpins were determined with discontinuous or continuous assays, as indicated. These assays measure the observed association rate (kobs), which is multiplied with the stoichiometry of inhibition (SI) to obtain the association rate constant (ka). In the discontinuous assays, trypsin (10 nM concentration) and α1AT were incubated under pseudo first-order conditions (inhibitor/enzyme ratio > 10) in 0.1 M Tris-HCl (pH 8.0), 10 mM CaCl2, 2 mg/mL BSA, at room temperature. Inhibitors were used at 100 nM concentration to measure the reaction of anionic trypsin with wild-type α1AT or mesotrypsin with α1AT Pittsburgh. Rates of inactivation of cationic trypsin and R198G-mesotrypsin by wild-type α1AT were determined using 1 μM inhibitor concentrations. Aliquots were removed at regular intervals and residual trypsin activity was determined. The observed pseudo-first order rate constant, kobs, was determined from the slope of linear fits of semi-logarithimc plots of ln(vt/v0) versus time of inhibition, where v0 is the maximal, uninhibited enzyme activity and vt is the residual trypsin activity; using the equation (−kobs) × (time) = ln (vt/v0). The second-order rate constant was then calculated by dividing the pseudo first-order rate constant with the inhibitor concentration.
The rate of association between cationic and anionic trypsins and α1AT Pittsburgh was too rapid to measure with the discontinuous assay, therefore a continuous assay and progress curve analysis was used to determine kobs. Briefly, 10 or 20 nM trypsin (final concentration) was incubated in the absence of inhibitor or in the presence of 10 or 20 nM α1AT Pittsburgh in 70 or 140 μM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate, in 0.1 M Tris-HCl (pH 8.0), 10 mM CaCl2, and 2 mg/ml BSA. The increase in absorbance at 405 nm was monitored continuously for 10 min. The four progress curves (2 trypsin concentrations with 2 substrate concentrations) generated were used in a single fitting session to determine the kobs using the KINSIM-FITSIM [37, 38] or KinTekSim (KinTek Corporation) programs, and the following two equations.
| (1) |
| (2) |
E, enzyme, S, substrate, P, product and I, inhibitor. First, the progress curves obtained in the absence of the inhibitor were used to fit the k1, k−1 and k2 parameters. Next, these rate constants were fixed and the kobs values were calculated from the progress curves obtained in the presence of inhibitors.
Rates of complex dissociation
To determine the first-order dissociation rate constants, trypsin-serpin complexes were prepared by incubating 0.5 μM trypsin with 1.5 μM α1AT at room temperature in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, and 2 mg/ml BSA until complete inhibition was observed (~1–5 min). Complexes were then diluted 125–1000-fold into 800 μM N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate in the same buffer to achieve complex concentrations of 0.25, 0.5, 1, 2, and 4 nM. Liberation of p-nitroaniline, as a measure of trypsin activity, was monitored continuously at 405 nm for 60 min. The parabolic curves were then fitted with the second-order polynomial function y = a+ bx + cx2; where y is absorbance in mOD units and x is time in seconds, as described in [39] and discussed in [40]. In this equation, "a" represents the initial "background" absorbance and the "bx" term describes the increase in absorbance owing to free trypsin present at the beginning of the measurement. The absorbance increase due to trypsin released from complexes is described by the term "cx2", where c = ½ × (initial complex concentration) × (dissociation rate constant) × (turnover number). The fitted "c" coefficients were plotted as a function of initial complex concentration, and the slopes of linear fits were used to calculate kdiss. The linearity of the plots also confirmed that no complex re-association took place during the assay and true dissociation constants were determined. The turnover number was determined from calibration curves prepared under the same experimental conditions as used for the dissociation assays. Initial rates of substrate hydrolysis were plotted against the molar enzyme concentrations, and the slope of the linear fit gave the turnover number.
Stoichiometry of inhibition (SI)
A fixed concentration of trypsin (20 nM) was incubated with increasing concentrations of α1AT (4–2000 nM) in 0.1 M Tris-HCl (pH 8.0), 1 mM CaCl2, and 2 mg/mL BSA at room temperature until the reaction reached completion. For wild-type α1AT, 20 min reaction time was used, with the exception of cationic trypsin, which was allowed to associate for 3 hours. For α1AT Pittsburgh the incubation time was 10 min. Residual trypsin activity was determined with 0.1 mM (final concentration) N-CBZ-Gly-Pro-Arg-p-nitroanilide substrate. Residual trypsin activity was plotted against inhibitor concentration and the equivalence point was determined from the y-intercept of the extrapolation of the linear portion of the titration curve. The stoichiometry of inhibition was then calculated as the inhibitor/enzyme combining ratio at the equivalence point.
Trypsin activity assays
Trypsin activity was measured at room temperature (25 C°) in 0.1 M Tris-HCl (pH 8.0) buffer containing 1 or 10 mM CaCl2, as indicated, using the chromogenic trypsin substrate N-CBZ-Gly-Pro-Arg-p-nitroanilide at 0.1 – 0.14 mM final concentration. The liberation of the yellow p-nitroaniline was followed at 405 nm in a SpectraMax Plus384 microplate reader (Molecular Devices). Where indicated, 2 mg/mL BSA was included in the trypsin activity assays.
Inactivation of α1-antitrypsin with Staphylococcus aureus V8 protease
To abolish the inhibitory activity of α1AT, the reactive center loop was digested with the Glu-specific V8 protease. This treatment results in a single specific cleavage of the Glu354-Ala355 peptide-bond and complete loss of α1AT activity [13, 14]. α1AT (10 μM concentration) was digested with 5 nM (final concentration) of V8-protease (New England Biolabs) in 0.1 M Tris-HCl (pH 8.0) overnight at room temperature.
N-terminal processing of α1-antitrypsin by trypsin
Wild-type and mutant α1AT proteins (5 μM concentration) were incubated with the indicated concentrations of trypsins in 0.1 M Tris-HCl (pH 8.0) buffer containing 1 mM CaCl2 at 37 oC. At given times (typically at 0, 1, 5, 10, 20, 30, 45, 60, 90, and 120 min) aliquots were removed from the digestion mixtures and precipitated with 10 % trichloroacetic acid. The precipitate was recovered by centrifugation, dissolved in Laemmli sample buffer containing 100 mM dithiothreitol and heat-denatured at 95 oC for 5 min. Electrophoretic separation was performed on 13 % SDS-PAGE mini gels in standard Tris-glycine buffer and gels were stained with Brilliant Blue R. Gels were dried and subjected to densitometry as described in [33].
Acknowledgments
This work was supported by NIH grant DK058088 to M. S.-T. The authors thank Vera Sahin-Tóth for technical assistance in site-directed mutagenesis and DNA work. Special thanks to Peter Gettins and József Dobó for the antitrypsin expression plasmids.
References
- 1.Rinderknecht H, Renner IG, Carmack C, Friedman R, Koyama P. A new protease in human pancreatic juice. Clin Res. 1978;26:112A. [Google Scholar]
- 2.Rinderknecht H, Renner IG, Abramson SB, Carmack C. Mesotrypsin: a new inhibitor-resistant protease from a zymogen in human pancreatic tissue and fluid. Gastroenterology. 1984;86:681–692. [PubMed] [Google Scholar]
- 3.Nyaruhucha CNM, Kito M, Fukuoka SI. Identification and expression of the cDNA-encoding human mesotrypsin(ogen), an isoform of trypsin with inhibitor resistance. J Biol Chem. 1997;272:10573–10578. doi: 10.1074/jbc.272.16.10573. [DOI] [PubMed] [Google Scholar]
- 4.Katona G, Berglund GI, Hajdu J, Gráf L, Szilágyi L. Crystal structure reveals basis for the inhibitor resistance of human brain trypsin. J Mol Biol. 2002;315:1209–1218. doi: 10.1006/jmbi.2001.5305. [DOI] [PubMed] [Google Scholar]
- 5.Szmola R, Kukor Z, Sahin-Tóth M. Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J Biol Chem. 2003;278:48580–48589. doi: 10.1074/jbc.M310301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt AE, Ogawa T, Gailani D, Bajaj SP. Structural role of Gly193 in serine proteases: investigations of a G555E (Gly193 in chymotrypsin) mutant of blood coagulation factor XI. J Biol Chem. 2004;279:29485–29492. doi: 10.1074/jbc.M402971200. [DOI] [PubMed] [Google Scholar]
- 7.Bobofchak KM, Pineda AO, Mathews FS, Di Cera E. Energetic and structural consequences of perturbing Gly-193 in the oxyanion hole of serine proteases. J Biol Chem. 2005;280:25644–25650. doi: 10.1074/jbc.M503499200. [DOI] [PubMed] [Google Scholar]
- 8.Szilágyi L, Kénesi E, Katona G, Kaslik G, Juhász G, Gráf L. Comparative in vitro studies on native and recombinant human cationic trypsins. Cathepsin B is a possible pathological activator of trypsinogen in pancreatitis. J Biol Chem. 2001;276:24574–24580. doi: 10.1074/jbc.M011374200. [DOI] [PubMed] [Google Scholar]
- 9.Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a novel agonist of protease-activated receptors 2 and 4. J Biol Chem. 2004;279:13532–13539. doi: 10.1074/jbc.M312090200. [DOI] [PubMed] [Google Scholar]
- 10.Grishina Z, Ostrowska E, Halangk W, Sahin-Tóth M, Reiser G. Activity of recombinant trypsin isoforms on human proteinase-activated receptors (PAR): mesotrypsin cannot activate epithelial PAR-1, -2, but weakly activates brain PAR-1. Br J Pharmacol. 2005;146:990–999. doi: 10.1038/sj.bjp.0706410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gettins PG. Serpin structure, mechanism, and function. Chem Rev. 2002;102:4751–4804. doi: 10.1021/cr010170+. [DOI] [PubMed] [Google Scholar]
- 12.Vercaigne-Marko D, Carrere J, Guy-Crotte O, Figarella C, Hayem A. Human cationic and anionic trypsins: differences of interaction with α1-proteinase inhibitor. Biol Chem Hoppe Seyler. 1989;370:1163–1171. doi: 10.1515/bchm3.1989.370.2.1163. [DOI] [PubMed] [Google Scholar]
- 13.Potempa J, Watorek W, Travis J. The inactivation of human plasma α1-proteinase inhibitor by proteinases from Staphylococcus aureus. J Biol Chem. 1986;261:14330–14334. [PubMed] [Google Scholar]
- 14.Nelson D, Potempa J, Travis J. Inactivation of α1-proteinase inhibitor as a broad screen for detecting proteolytic activities in unknown samples. Anal Biochem. 1998;260:230–236. doi: 10.1006/abio.1998.2708. [DOI] [PubMed] [Google Scholar]
- 15.Lewis JH, Iammarino RM, Spero JA, Hasiba U. Antithrombin Pittsburgh: an α1-antitrypsin variant causing hemorrhagic disease. Blood. 1978;51:129–137. [PubMed] [Google Scholar]
- 16.Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. α1-antitrypsin Pittsburgh (358 Met → Arg), a fatal bleeding disorder. N Engl J Med. 1983;309:694–698. doi: 10.1056/NEJM198309223091203. [DOI] [PubMed] [Google Scholar]
- 17.Travis J, Matheson NR, George PM, Carrell RW. Kinetic studies on the interaction of α1-proteinase inhibitor (Pittsburgh) with trypsin-like serine proteinases. Biol Chem Hoppe Seyler. 1986;367:853–859. doi: 10.1515/bchm3.1986.367.2.853. [DOI] [PubMed] [Google Scholar]
- 18.Schellenberger V, Turck CW, Hedstrom L, Rutter WJ. Mapping the S′ subsites of serine proteases using acyl transfer to mixtures of peptide nucleophiles. Biochemistry. 1993;32:4349–4353. doi: 10.1021/bi00067a026. [DOI] [PubMed] [Google Scholar]
- 19.Schellenberger V, Turck CW, Rutter WJ. Role of the S′ subsites in serine protease catalysis. Active-site mapping of rat chymotrypsin, rat trypsin, alpha-lytic protease, and cercarial protease from Schistosoma mansoni. Biochemistry. 1994;33:4251–4257. doi: 10.1021/bi00180a020. [DOI] [PubMed] [Google Scholar]
- 20.Kurth T, Ullmann D, Jakubke HD, Hedstrom L. Converting trypsin to chymotrypsin: structural determinants of S1′ specificity. Biochemistry. 1997;36:10098–10104. doi: 10.1021/bi970937l. [DOI] [PubMed] [Google Scholar]
- 21.Grahn S, Kurth T, Ullmann D, Jakubke HD. S′ subsite mapping of serine proteases based on fluorescence resonance energy transfer. Biochim Biophys Acta. 1999;1431:329–337. doi: 10.1016/s0167-4838(99)00059-x. [DOI] [PubMed] [Google Scholar]
- 22.Stephens AW, Siddiqui A, Hirs CH. Site-directed mutagenesis of the reactive center (serine 394) of antithrombin III. J Biol Chem. 1988;263:15849–15852. [PubMed] [Google Scholar]
- 23.Theunissen HJ, Dijkema R, Grootenhuis PD, Swinkels JC, de Poorter TL, Carati P, Visser A. Dissociation of heparin-dependent thrombin and factor Xa inhibitory activities of antithrombin-III by mutations in the reactive site. J Biol Chem. 1993;268:9035–9040. [PubMed] [Google Scholar]
- 24.Petrassi HM, Williams JA, Li J, Tumanut C, Ek J, Nakai T, Masick B, Backes BJ, Harris JL. A strategy to profile prime and non-prime proteolytic substrate specificity. Bioorg Med Chem Lett. 2005;15:3162–3166. doi: 10.1016/j.bmcl.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rezaie AR, Olson ST. Contribution of lysine 60f to S1′ specificity of thrombin. Biochemistry. 1997;36:1026–1033. doi: 10.1021/bi9620823. [DOI] [PubMed] [Google Scholar]
- 27.Gaboriaud C, Serre L, Guy-Crotte O, Forest E, Fontecilla-Camps JC. Crystal structure of human trypsin 1: unexpected phosphorylation of Tyr151. J Mol Biol. 1996;259:995–1010. doi: 10.1006/jmbi.1996.0376. [DOI] [PubMed] [Google Scholar]
- 28.Kurth T, Grahn S, Thormann M, Ullmann D, Hofmann HJ, Jakubke HD, Hedstrom L. Engineering the S1′ subsite of trypsin: design of a protease which cleaves between dibasic residues. Biochemistry. 1998;37:11434–11440. doi: 10.1021/bi980842z. [DOI] [PubMed] [Google Scholar]
- 29.Medveczky P, Antal J, Patthy A, Kékesi K, Juhász G, Szilágyi L, Gráf L. Myelin basic protein, an autoantigen in multiple sclerosis, is selectively processed by human trypsin 4. FEBS Lett. 2006;580:545–552. doi: 10.1016/j.febslet.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 30.Tóth J, Gombos L, Simon Z, Medveczky P, Szilágyi L, Gráf L, Málnási-Csizmadia A. Thermodynamic analysis reveals structural rearrangement during the acylation step in human trypsin 4 on 4-methylumbelliferyl 4-guanidinobenzoate substrate analogue. J Biol Chem. 281(18):12596–12602. doi: 10.1074/jbc.M512301200. [DOI] [PubMed] [Google Scholar]
- 31.Sahin-Tóth M. Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J Biol Chem. 2000;275:22750–22755. doi: 10.1074/jbc.M002943200. [DOI] [PubMed] [Google Scholar]
- 32.Sahin-Tóth M, Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun. 2000;278:286–289. doi: 10.1006/bbrc.2000.3797. [DOI] [PubMed] [Google Scholar]
- 33.Kukor Z, Tóth M, Sahin-Tóth M. Human anionic trypsinogen. Properties of autocatalytic activation and degradation and implications in pancreatic diseases. Eur J Biochem. 2003;270:2047–2058. doi: 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- 34.Jameson GW, Roberts DV, Adams RW, Kyle WSA, Elmore DT. Determination of the operational molarity of solutions of bovine α-chymotrypsin, trypsin, thrombin and factor Xa by spectrofluorimetric titration. Biochem J. 1973;131:107–117. doi: 10.1042/bj1310107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KN, Park SD, Yu MH. Probing the native strain in α1-antitrypsin. Nat Struct Biol. 1996;3:497–500. doi: 10.1038/nsb0696-497. [DOI] [PubMed] [Google Scholar]
- 36.Lee KN, Im H, Kang SW, Yu MH. Characterization of a human α1-antitrypsin variant that is as stable as ovalbumin. J Biol Chem. 1998;273:2509–2516. doi: 10.1074/jbc.273.5.2509. [DOI] [PubMed] [Google Scholar]
- 37.Barshop BA, Wrenn RF, Frieden C. Analysis of numerical methods for computer simulation of kinetic processes: development of KINSIM--a flexible, portable system. Anal Biochem. 1983;130:134–145. doi: 10.1016/0003-2697(83)90660-7. [DOI] [PubMed] [Google Scholar]
- 38.Zimmerle CT, Frieden C. Analysis of progress curves by simulations generated by numerical integration. Biochem J. 1989;258:381–387. doi: 10.1042/bj2580381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jesty J. The kinetics of formation and dissociation of the bovine thrombin antithrombin III complex. J Biol Chem. 1979;254:10044–10450. [PubMed] [Google Scholar]
- 40.Calugaru SV, Swanson R, Olson ST. The pH dependence of serpin-proteinase complex dissociation reveals a mechanism of complex stabilization involving inactive and active conformational states of the proteinase which are perturbable by calcium. J Biol Chem. 2001;276:32446–32455. doi: 10.1074/jbc.M104731200. [DOI] [PubMed] [Google Scholar]