

Summary
The transcription factor NF-κB (nuclear factor kappa enhancer binding protein) controls many processes including immunity, inflammation and apoptosis. Studies of the NF-κB pathway have revealed new functions for ubiquitination in addition to its well-known role in targeting protein degradation. Ubiquitin is involved in at least three steps in this pathway: degradation of the NF-κB inhibitor IκB, processing of NF-κB precursors, and activation of the IκB kinase (IKK) through a degradation-independent mechanism.
Introduction
In this post-genomic era filled with cutting-edge technologies, it is refreshing to look back at the history of ubiquitin and NF-κB as both were discovered through an ingenious combination of simple “bucket” biochemistry and a clear mind1–5. Since these classic experiments, both ubiquitin and NF-κB have taken center stage in biology, as each controls a vast array of cellular and organismal processes ranging from the birth and death of a cell to the neuronal behavior of an organism. Again, it was through biochemistry that the essential role of ubiquitination in NF-κB activation was revealed6–8. Ubiquitin targets not only IκB for degradation, but also the NF-κB precursors, p105 and p100, for processing into mature forms by the proteasome. More surprisingly, a critical role for ubiquitination in activating protein kinases in the NF-κB pathway through a degradation-independent mechanism has also been discovered9–11. Thus, the convergence of ubiquitin and NF-κB research has not only helped elucidate the biochemical mechanism underlying NF-κB activation, but also broadened our understanding of ubiquitin as a versatile signaling tag. As several extensive reviews of ubiquitination in the NF-κB pathway are available12,13, here I will summarize the current view of the traditional role of ubiquitin in the NF-κB pathway, and then focus on recent advances in understanding the non-traditional role of ubiquitin in this pathway.
The traditional role of ubiquitin in the NF-κB pathway
NF-κB is a dimeric transcription factor consisting of Rel family members, which include Rel-A (also known as p65), c-Rel, Rel-B, p50 and p5214. p50 and p52 are derived from the larger precursors p105 and p100, respectively, through proteolytic processing by the proteasome. All NF-κB proteins contain a highly conserved Rel-homology domain (RHD) that is responsible for DNA binding, dimerization, nuclear translocation and interaction with the IκB proteins. The IκB proteins, including IκBα, β and ɛ, bind to NF-κB via ankyrin repeats and block its nuclear import and thereby, its transcriptional activity. The C-termini of p105 and p100 also contain the IκB-like ankyrin repeats that must be degraded in order to generate the mature Rel subunits.
The NF-κB activation pathways are broadly classified as the canonical and non-canonical pathways, depending on whether activation involves IκB degradation or p100 processing (Figure 1)15. In the canonical pathway, which is the predominant NF-κB signaling pathway, stimulating cells with an agonist such as tumor necrosis factor α (TNFα) or interleukin-1β (IL-1β) activates the IKK complex that is composed of two catalytic subunits IKKα and IKKβ and a regulatory subunit NEMO (also known as IKKγ). Genetic experiments have demonstrated that IKKβ, but not IKKα, phosphorylates IκB proteins at two N-terminal serine residues. This signal-induced phosphorylation targets IκB for polyubiquitination and subsequent degradation by the proteasome7, thus releasing NF-κB. The non-canonical pathway of NF-κB activation operates mainly in B cells in response to stimulation of a subset of the TNF receptor superfamily, including receptors for BAFF, lymphotoxin-β (LTβ) and CD40 ligand. Stimulation of these receptors activates the protein kinase NIK, which in turn activates IKKα. IKKα then phosphorylates p100 at two C-terminal serine residues leading to the selective degradation of its IκB -like domain by the proteasome16,17. The mature p52 subunit and its binding partner Rel-B translocate into the nucleus to regulate gene expression.
Figure 1. The NF-κB signaling pathways.
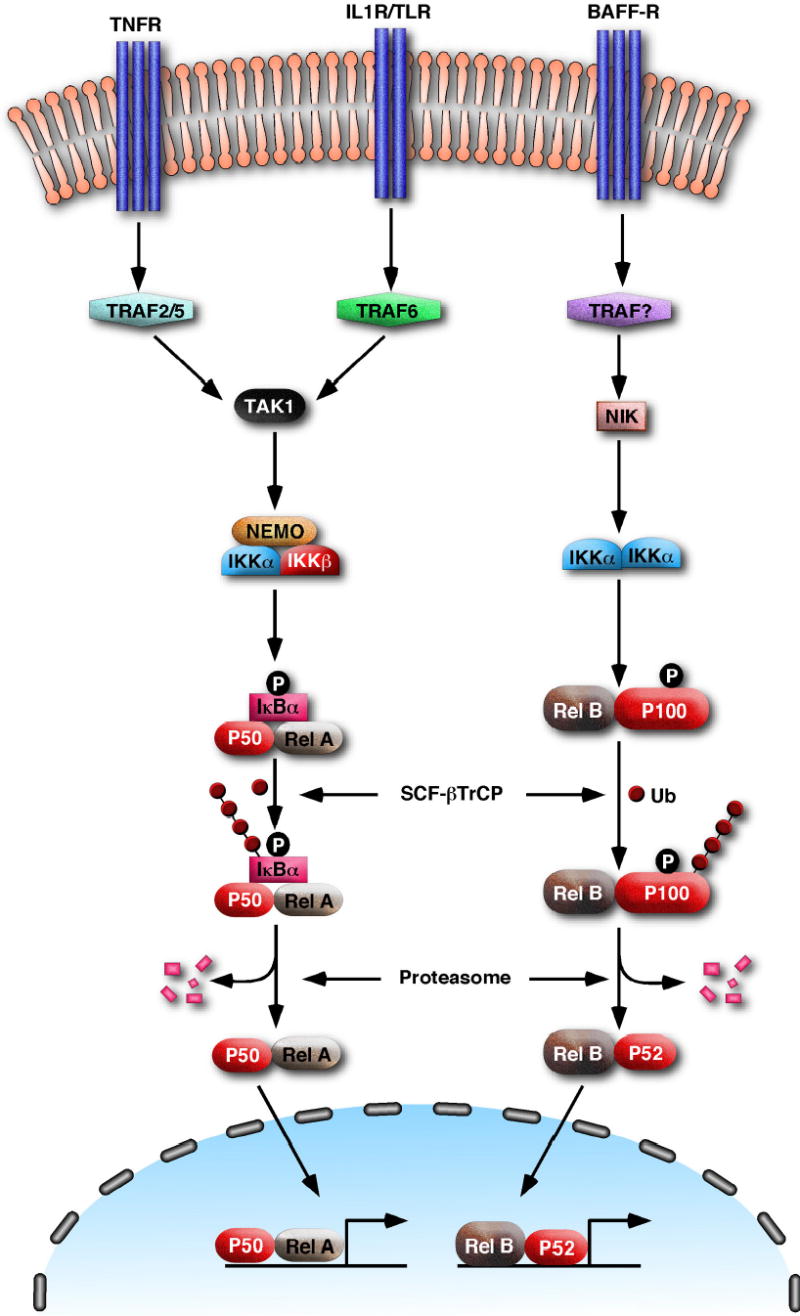
In the canonical pathway (left), stimulation of TNF receptors (TNFR), IL-1 receptors (IL-1R) or Toll-like receptors (TLR) with their cognate ligands activates TRAF proteins and subsequently the kinase TAK1, which phosphorylates and activates IKKβ. IKKβ then phosphorylates IκB resulting in its ubiquitination by the SCF-βTrCP ubiquitin ligase complex and subsequent degradation by the proteasome. The NF-κB dimer consisting of p50 and Rel-A can then enter the nucleus to regulate the expression of targets genes involved in inflammation, immunity and cell survival. In the non-canonical pathway, a subset of receptors belonging to the TNFR superfamily, such as the B cell receptor for BAFF (BAFF-R), activates the kinase NIK through an unknown mechanism. NIK then phosphorylates IKKα, which in turn phosphorylates the NF-κB precursor p100. p100 is subsequently polyubiquitinated and then processed to the mature subunit p52 by the proteasome. p52 and its binding partner Rel-B translocate to the nucleus to turn on genes that are important for the maturation of B cells.
The ubiquitin-proteasome pathway plays a crucial role in both the canonical and non-canonical pathways of NF-κB activation. Ubiquitination is a reversible covalent modification catalyzed by three enzymatic steps18. In the first step, ubiquitin is activated by a ubiquitin-activating enzyme (E1) in an ATP-dependent reaction. In the second step, the activated ubiquitin is transferred to a ubiquitin-conjugating enzyme (E2 or Ubc), forming an E2-Ub thioester. Finally, in the presence of a ubiquitin-protein ligase (E3), ubiquitin is attached to a target protein through an isopeptide bond between the carboxyl terminus of ubiquitin and the ɛ-amino group of a lysine residue in the target protein. Ubiquitin contains seven lysines, which can be attached to another ubiquitin in a highly processive reaction to form a polyubiquitin chain. Typically, a polyubiquitin chain that targets a protein for degradation by the proteasome is linked through lysine-48 (K48) of ubiquitin19. However, ubiquitin chains linked through other lysines of ubiquitin have also been found in cells20. In particular, K63-linked polyubiquitin chains have recently been found to regulate DNA repair and protein kinase activation through a degradation-independent mechanism (see below).
Ubiquitination of IκB is carried out by an E2 of the Ubc4/5 family7,9,21 and the SCF-βTrCP E3 ligase (Skp1-Cul1-F-box ligase containing the F-box protein βTrCP)22–26. Two βTrCP proteins, βTrCP1 and βTrCP2, have been found in mammalian cells. Genetic deletion of βTrCP1 in mice leads to only modest retardation of IκBα degradation, suggesting that βTrCP1 and βTrCP2 may have redundant functions27. Indeed, silencing of both βTrCP1 and βTrCP2 expression by RNAi block IκBα degradation. βTrCP1 and βTrCP2 bind specifically to the phosphorylated form of IκB through their C-terminal WD40 repeats, and to the rest of the SCF complex through the F-box. The SCF complex contains the RING domain protein Roc1/Rbx1, which binds to Ubc4/5, allowing this E2 to ubiquitinate IκB at two conserved N-terminal lysine residues. The polyubiquitinated IκB remains associated with NF-κB, but is selectively degraded by the 26S proteasome, while NF-κB itself is spared7.
The ubiquitin-proteasome pathway is also responsible for the processing of p105 and p100 to p50 and p52, respectively. p105 can be processed either co-translationally or post-translationally, both requiring the proteasome6,28. Co-translational processing of p105 appears to be a constitutive process that does not require phosphorylation or ubiquitination and is likely the major source of p50, which is constitutively present in unstimulated cells. Some agents such as phorbol ester (PMA) and lipopolysaccharides (LPS) can stimulate post-translational processing of p105 by activating IKK, which phosphorylates p105 at a C-terminal domain29. On the other hand, the processing of p100 is tightly regulated by the non-canonical pathway of NF-κB activation and depends on both phosphorylation and ubiquitination. As discussed above, stimulation of certain receptors of the TNFR family on B cells activates IKKα, which phosphorylates p100 at two C-terminal serine residues (Figure 1). This phosphorylation relieves the inhibition of p100 processing by a C-terminal death domain, and recruits the SCF-βTrCP ligase to polyubiquitinate p100 at a specific lysine 16,17,30,31. Subsequently, the C-terminal IκB-like domain is selectively degraded by the proteasome, generating the mature p52 subunit.
How does the proteasome partially degrade a protein? More specifically, what defines where degradation starts and stops? A plausible model for regulated ubiquitin/proteasome-dependent processing has recently been proposed 32. According to this model (Figure 2), the ubiquitin tag on a substrate recruits the proteasome to an internal unstructured sequence such as the glycine rich region (GRR) of p105 and p10033. It is postulated that this region forms a hairpin-like loop that inserts into the proteolytic chamber of the 20S proteasome. Proteolysis begins at the loop and then proceeds in both the N- and C-terminal directions of the target protein. In most cases, the N- and C-terminal domains can be completely unfolded by the 19S subcomplex of the proteasome and threaded into the proteolytic chamber so that the target is completely degraded. In the cases of substrates such as p105 and p100, the RHD domain is a tightly folded structure that may be difficult to unfold. Thus, after initiating proteolysis from the putative GRR loop of p105 or p100, the proteasome chews along the C-terminal polypeptide until it reaches the end, but comes to a halt at the N-terminus when it encounters the tightly folded RHD structure.
Figure 2. A model for the processing of p100 by the proteasome.
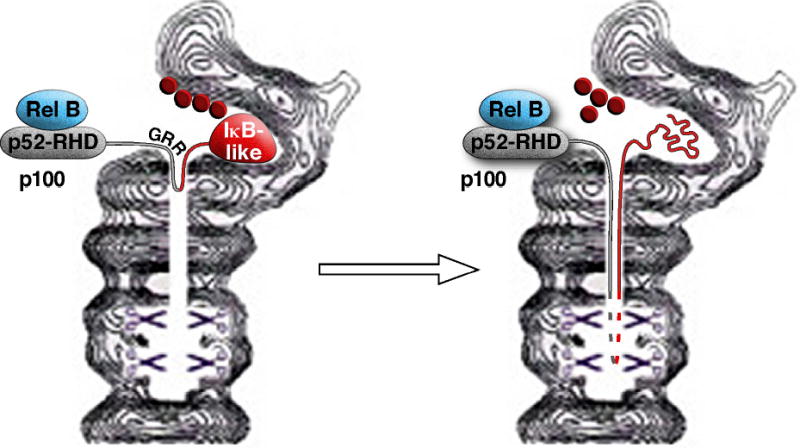
Polyubiquitinated p100 is recruited to the proteasome, allowing the flexible glycine rich region (GRR) to insert into the proteasome as a loop. Degradation of the polypeptide proceeds in both the N- and C-terminal directions. The degradation of the C-terminus proceeds to the end, whereas the degradation along the N-terminus stops when the tightly folded Rel-homology domain (RHD) cannot be unfolded to allow insertion into the proteasome. The resulting p52 subunit and its dimeric partner Rel-B is then released from the proteasome.
The non-traditional role of ubiquitin in activating protein kinases
Serendipity played a major role in the discovery of a ubiquitination-dependent IκB kinase complex9. In 1995, a cell-free system was established to demonstrate the signal-induced ubiquitination of IκBα7. This system was later used to identify the enzymes involved in IκB ubiquitination by separating HeLa cytosolic extracts into two fractions, dubbed fraction I and II1,9. Surprisingly, each fraction alone no longer supported IκBα phosphorylation but when both were combined, or when fraction I was replaced with ubiquitin and Ubc4 or Ubc5, IκBα phosphorylation was restored. Further purification of fraction II led to the identification of a 700 kDa kinase complex whose activity can be regulated by polyubiquitination in a degradation-independent manner9.
A major issue concerning the early work on IKK activation by ubiquitin was the lack of a connection between ubiquitination and signaling pathways known to activate IKK. This issue was largely resolved by the discovery that TRAF (TNF receptor associated factor) proteins are ubiquitin E3 ligases10. TRAF proteins play a pivotal role in signaling pathways for NF-κB activation from many cell surface receptors including the TNF receptor superfamily (TNFR), IL-1 receptors (IL-1R) and Toll-like receptors (TLRs)34. Seven members of this family have been identified in the human genome. With the exception of TRAF1, all TRAF proteins contain an N-terminal RING domain followed by several zinc finger domains. Among all TRAF proteins, TRAF2 and 6 have been most intensively studied. TRAF2 is recruited to the TNF receptor complex through binding to the adaptor protein TRADD35. Both TRADD and TRAF2 also interact with the receptor interacting protein RIP, a protein kinase that signals in the TNF pathway through a kinase-independent mechanism. TRAF6 is essential for IKK and JNK activation in the IL-1 and TLR pathways36,37. In these pathways, stimulation of the receptors leads to the recruitment of MyD88, an adaptor protein that further recruits the protein kinase IRAK4 and IRAK1. IRAK1 then binds to TRAF6, which subsequently activates downstream signaling cascades including those of IKK and JNK.
IKK activation by the TRAF6 and TRAF2 ubiquitin ligases
In a series of biochemical experiments aiming at elucidating the mechanism of IKK activation by TRAF6, two TRAF6-regulated IKK activators (TRIKAs) were identified10,11. TRIKA1 is an E2 enzyme complex consisting of Ubc13 and a Ubc-like protein Uev1A10,38. This E2 functions with TRAF6, a RING domain E3, to synthesize a K63-linked polyubiquitin chain on target proteins including NEMO and TRAF6 itself. TRIKA2 is a trimeric complex consisting of the protein kinase TAK1 and two adaptor proteins TAB1 and TAB211,39. TAK1 phosphorylates IKKβ at two serine residues in the activation loop, thereby activating IKK. TAB2 and its related protein TAB3 contain a highly conserved novel zinc finger (NZF) domain that binds preferentially to K63-linked polyubiquitin chains40. Mutation of the NZF domain abolishes the ability of TAB2 and TAB3 to activate TAK1 and IKK, whereas the replacement of the NZF domain with a heterologous ubiquitin-binding domain restores the function of TAB2 and TAB3, suggesting that ubiquitin-binding by the NZF domain is necessary for activating TAK1 and IKK. These studies have led to the following model of IKK activation by TRAF6-mediated polyubiquitination (Figure 3). Upon ligand binding to IL-1R or TLRs, TRAF6 is recruited to the receptor complexes and forms oligomers. TRAF6 oligomerization activates its ligase activity, leading to K63-linked polyubiquitination of targets including TRAF6 itself. Ubiquitinated TRAF6 recruits TAB2 and activates the TAB2-associated TAK1 kinase, which then phosphorylates and activates IKK. Ubiquitin-activated TAK1 also phosphorylates and activates MKK kinases such as MKK6, which in turn activates the JNK and p38 kinase pathway11. Thus, TRAF6-mediated polyubiquitination initiates protein kinase cascades through a degradation-independent mechanism. Recent structural studies have shown that the K48- and K63-linked ubiquitin chains adopt very different configurations, potentially providing the molecular basis for the distinct signaling functions of these polyubiquitin chains41.
Figure 3. A model for IKK activation by TRAF6 ubiquitination.
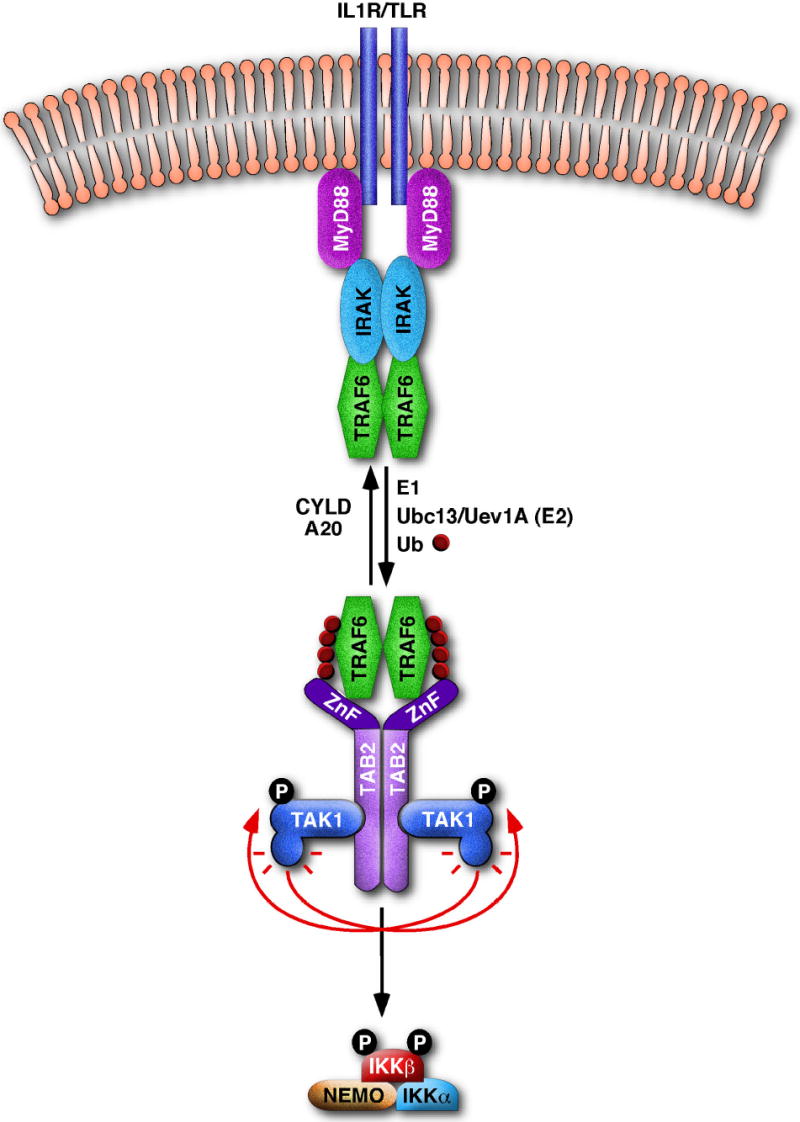
Stimulation of IL-1R or TLR leads to the recruitment of MyD88, IRAK and TRAF6 to the receptor complex. This may facilitate TRAF6 oligomerization and activate its ubiquitin ligase activity, leading to K63-linked polyubiquitination of targets including Nemo (not shown) and TRAF6 itself. This polyubiquitination reaction requires E1 and Ubc13/Uev1A, and can be reversed by deubiquitination enzymes such as CYLD and A20. Ubiquitinated TRAF6 is recruited to the TAK1/TAB2 complex through binding of K63-linked polyubiquitin chains to the NZF domain of TAB2 as well as by the direct interaction between TRAF6 and TAB2. This binding may facilitate the dimerization or oligomerization of the TAK1/TAB2 complex, promoting its autophosphorylation and TAK1 activation. TAK1 then phosphorylates IKKβ, resulting in its activation.
Like TRAF6, TRAF2 is also a RING domain ubiquitin ligase (Figure 4)40,42–44. A dominant negative mutant of Ubc13 has been shown to block NF-κB activation by TRAF2 or TNFα10. Overexpression of TRAF2 leads to its polyubiquitination by Ubc13/Uev1A, and TRAF2 ubiquitination appears to be important for the activation of JNK42. A recent study showed that TRAF2 ubiquitination is essential for JNK, but not p38 or NF-κB, activation by TNFα44. This is likely due to the redundant role of TRAF2 and TRAF5 in NF-κB activation by TNFα, as TRAF2-deficient cells are defective in JNK but not NF-κB activation45, whereas TRAF2/TRAF5 double knockout cells are defective in both JNK and NF-κB activation46. TRAF2 and/or TRAF5 may also ubiquitinate RIP, which is essential for NF-κB activation40,47. It has been shown that TNFα induces RIP polyubiquitination, and that ubiquitinated RIP associates with the TNF receptor as well as TAB240,48,49. Thus, it is possible that polyubiquitination of RIP recruits the TAK1/TAB2 complex, which subsequently activates IKK. However, definitive proof that RIP polyubiquitination is required for TNFα signaling is still lacking.
Figure 4. A central role for ubiquitin in multiple signaling pathways.
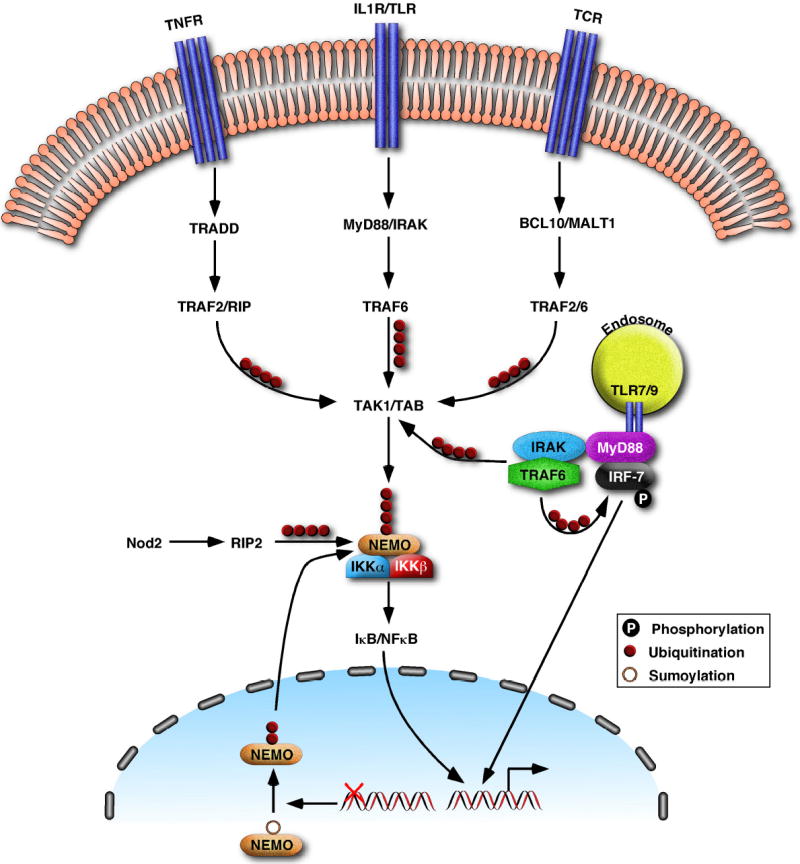
Polyubiquitination by TRAF2 and TRAF6 not only mediates signaling by TNFR, IL1R and TLR (see Fig. 3) in the innate immunity pathways, but also plays an important role in adaptive immune responses by activating IKK in response to stimulation of TCR. In T cells, BCL10 and MALT1 activate the ligase activity of TRAF6 (and possibly TRAF2), to catalyze the K63-linked polyubiquitination of NEMO and TRAF6 itself. These ubiquitination events mediate the activation of TAK1 and IKK. NOD2 and RIP2, which trigger innate immune responses against intracellular bacteria, also facilitate the K63-linked polyubiquitination of NEMO. DNA damage leads to the sumoylation and subsequent ubiquitination of NEMO in the nucleus. The ubiquitinated NEMO then exits the nucleus and associates with the other subunits of the cytosolic IKK complex, resulting in IKK activation. Some receptors of the TLR family, including TLR7, 8 and 9, are localized in the endosomal membrane. Stimulation of TLR7/8 and TLR9 by viral RNA and CpG DNA, respectively, leads to the activation of both NF-κB and IRF7, which coordinately regulate interferon production. IRF7 binds to MyD88, which is associated with the endosomal TLRs as well as IRAK and TRAF6. TRAF6 polyubiquitination is required for the activation of IRF7, presumably by promoting the phosphorylation and subsequent nuclear translocation of IRF7.
Roles of the TAK1 kinase complex in IKK and JNK activation
Since TAK1 knockout mice are early embryonic lethal, it has not been possible to determine whether this kinase is essential for NF-κB activation in mice50. However, multiple lines of evidence support a crucial role for the TAK1 complex in IKK and JNK activation. First, TAK1 phosphorylates IKKβ and MKK6 at key serine residues within the activation loop, leading to their activation 11. Second, silencing of TAK1 expression by RNAi or inhibition with a specific kinase inhibitor in mammalian cells drastically reduced IKK activation and completely blocked JNK activation by IL-1β and TNFα40,51,52. Third, mutations of TAK1 in Drosophila melanogaster, or silencing of TAK1 in a D. melanogaster cell line, blocked IKK and JNK in response to bacterial challenge53–55. Thus, independent genetic and biochemical studies have indicated that TAK1 is important for IKK and JNK activation in both invertebrates and vertebrates. With regard to the role of TAB2 in the NF-κB pathway, a recent study showed that a TAB2-deficient cell line has normal NF-κB activation in response to IL-1β56. However, this is likely due to the redundant functions of TAB2 and its closely related homologue TAB3, as RNAi against TAB2 and TAB3 in mammalian cells abrogated IKK activation by IL-1β and TNFα. Moreover, mutations in the D. melanogaster homologue of TAB2 led to severe defects in antibacterial responses (D. Ferrandon, personal communication). Interestingly, most of these mutations have been mapped to the ubiquitin-binding domain of dTAB2, suggesting that ubiquitin-mediated activation of TAK1 and IKK is an evolutionarily conserved mechanism. The TAK1 kinase complex also contains TAB1; however, the role of TAB1 in the NF-κB pathway is not clear, as reconstitution experiments showed that TAB1 is dispensable for IKK activation in vitro11. In addition, there is no evidence that RNAi or genetic ablation of TAB1 impairs IKK activation57. Furthermore, there is no apparent homologue of TAB1 in the Drosophila genome. Thus, TAB1 may not be involved in NF-κB activation, but in the regulation of other pathways such as the Wnt or TGFβ pathway58,59. In fact, TAK1 was first isolated as a TGFβ-activated kinase, but its mechanism of activation in this pathway is poorly understood60.
While TAK1 and TAB2 clearly play a major role in innate immune responses in D. melanogaster, the phenotypes of dTAK1 and dTAB2 mutants are not as severe as those of dIKK mutants 53. Similarly, although RNAi against TAK1 or TAB2 and TAB3 severely impaired IKK activation, there is still residual IKK activity (approximately 20–30%). While some of this residual activity may be because depletion by RNAi is incomplete, it is also likely that there is another minor pathway contributing to IKK activation. One possible activator of IKK is p62, a UBA domain containing protein that binds to TRAF6 and RIP61–63. Mice lacking p62 are partially defective in prolonged NF-κB activation by RANK, a TNFR that activates IKK through TRAF6 in osteoclasts64. Interestingly, p62 forms a complex with an atypical PKC (ζPKC), representing potentially another example of a ubiquitin receptor-containing kinase complex similar to the TAB2/TAK1 complex. Another potential activator of IKK is MEKK3, as murine cells lacking MEKK3 are partially defective in NF-κB activation in response to certain stimuli65,66. It is also possible that the residual IKK activation does not require an upstream kinase, but is induced by the oligomerization of IKK itself. In this case, TRAF6 or TRAF2 and RIP may somehow interact with IKK/NEMO to induce its oligomerization, causing IKK to autophosphorylate itself, resulting in its activation.
Ubiquitin signaling in adaptive immunity
Recent studies have revealed that ubiquitin signaling plays an important role in adaptive immunity, particularly T cell activation (Figure 4). Stimulation of T cell receptors (TCR) activates a tyrosine kinase cascade that subsequently activates the serine/threonine kinase PKCθ. PKCθ then induces the formation of a protein complex composed of CARMA1, BCL10 and MALT1, which are recruited to the lipid rafts where TCR resides67,68. Two studies have shown that BCL10 and MALT1 induce the K63-linked polyubiquitination of NEMO by Ubc13/Uev1A, leading to IKK activation69,70. However, there is a discrepancy concerning the E3 ubiquitin ligase for NEMO between these studies. In one study, MALT1 directly ubiquitinates NEMO at K399, despite the fact that MALT1 does not contain any known E3 domain69. In the other study, MALT1 was found to recruit TRAF6 via its TRAF6-binding sites, and TRAF6, in turn ubiquitinates NEMO70. The latter study showed that MALT1 also induces auto-ubiquitination of TRAF6, which then recruits the TAK1/TAB2 complex to activate IKK. This entire process from MALT1-mediated polyubiquitination to IκB phosphorylation was reconstituted in vitro using purified recombinant proteins70. TRAF6 and TRAF2 are redundant in the TCR pathway, as RNAi of both TRAF2 and TRAF6 effectively blocked IKK activation and IL-2 production by TCR stimulation, whereas RNAi of either TRAF alone was less effective70. However, it remains to be seen whether TRAF2 and TRAF6 are required for T cell activation in vivo.
Emerging evidence for the regulatory role of ubiquitin in diverse signaling pathways
NF-κB is regulated by a large variety of stimuli, including microbial pathogens and DNA damaging agents, which have recently been shown to activate IKK through a ubiquitin-dependent but degradation-independent mechanism. An elegant example was provided from the study of DNA damage-induced IKK activation71. DNA damaging agents, especially those that generate double strand breaks, such as topoisomerase inhibitors and gamma irradiation, can activate NF-κB in certain cancer cells. This DNA damage response, which requires the cytoplasmic IKK complex, may be responsible for the resistance of some cancer cells to chemotherapy. A mechanism by which the nuclear DNA damage signal is transduced to the cytoplasmic IKK complex has recently been uncovered (Figure 4)71. Upon DNA damage, a small fraction of nuclear NEMO is modified by the ubiquitin-like protein SUMO, which further retains NEMO in the nucleus. DNA damage also activates the kinase ATM, which phosphorylates NEMO. This phosphorylation leads to the replacement of SUMO by ubiquitin at two lysine residues (K277/K309) within NEMO. Ubiquitinated NEMO subsequently enters the cytoplasm, where it associates with the rest of the IKK complex, resulting in IKK activation.
Another recent study has provided evidence that K63 polyubiquitination may be involved in IKK activation by NOD1 and NOD2, two cytosolic receptors for intracellular bacteria72. Mutations in NOD2 have been closely linked to Crohn’s disease, an inflammatory bowel disease. NOD1 and NOD2 activate IKK through RIP2, a RIP1-like kinase. Through a process that is still not well understood, NOD2 and RIP2 induce the K63-linked polyubiquitination of NEMO at K285, thereby activating IKK (Figure 4)73. Interestingly, the NOD2 mutations found in Crohn’s disease patients impair NOD2’s ability to activate NF-κB and to induce NEMO polyubiquitination in cell culture experiments73. However, mouse knock-in experiments show that the NOD2 mutations associated with the disease lead to NF-κB activation rather than inhibition74. Thus, the signaling mechanisms of NOD proteins and the potential role of ubiquitination in NOD signaling require further investigation.
The regulatory role of ubiquitin may not be limited to the NF-κB pathway. Indeed, TRAF6 ubiquitination has recently been found to be important for the activation of the transcription factor IRF775. IRF7 regulates the production of type-I interferons in response to stimulation of certain Toll-like receptors (TLR7, 8 & 9) by viral RNA or bacterial DNA. IRF7 is activated through its phosphorylation by an unknown kinase, which is in turn activated by the MyD88-IRAK4-TRAF6 complex (Figure 4). Recent studies have shown that MyD88 and TRAF6 bind to IRF7, inducing its phosphorylation and nuclear translocation. Interestingly, Ubc13 and TRAF6 ubiquitination are required for IRF7 activation75, suggesting that K63 polyubiquitination of as yet unidentified targets may be involved in the activation of an IRF7 kinase.
Regulation of IKK activation by deubiquitination
Like phosphorylation, ubiquitination is a reversible covalent modification. Two deubiquitinating enzymes (DUBs) have recently been shown to play an important role in suppressing NF-κB activation at a step upstream of IKK. One of these DUBs is the cylindromatosis tumor suppressor protein CYLD, which inhibits IKK activation by cleaving K63-linked polyubiquitin chains on several proteins including TRAF2, TRAF6 and NEMO76–78. Mutations within the DUB domains of CYLD, which are found in patients with cylindromas, lead to the enhanced activation of NF-κB, thereby contributing to the pathogenesis of the tumors. A recent study found that CYLD also inhibits JNK activation79, consistent with the idea that CYLD interferes with TRAF ubiquitination or TAK1 activation, which functions upstream of both IKK and JNK. However, this study found that CYLD inhibits IKK activation by IL-1β, LPS and CD40, but not TNFα, whereas previous studies showed that CYLD inhibits IKK activation by TNFα76–78. The reason for this discrepancy is not clear.
Another DUB that acts in this pathway is A20, an NF-κB induced protein that inhibit NF-κB in a negative feedback loop80–82. A20 deficient mice develop severe inflammatory diseases in multiple organs partly due to the enhanced and prolonged activation of NF-κB by proinflammatory stimuli including TNFα and LPS83. A20 contains a novel OTU type deubiquitination enzyme domain at the N-terminus, and several zinc finger domains in the remainder of the protein. The OTU domain disassembles K63-linked polyubiquitin chains from RIP (in the TNFα pathway)81 and TRAF6 (in the LPS pathway)80, thereby suppressing IKK activation. Moreover, A20 also functions as a ubiquitin ligase through its zinc finger domains to assemble K48-linked polyubiquitin chains on RIP after the K63-linked chains are removed by the OTU domain81. K48-linked polyubiquitination targets RIP for degradation by the proteasome, further diminishing IKK activation. Thus, A20 provides a dramatic example that a switch in polyubiquitin chains can profoundly influence the signaling and degradation of targets. A20 also inhibits the activation of the transcription factor IRF3 by RNA viruses84. In this case, the role of ubiquitin is less clear, as overexpression of an A20 mutant lacking the OTU domain still inhibits IRF3. It appears that A20 binds directly to and inhibits TBK1, an IKK-like kinase that phosphorylates IRF3.
Conclusions and Perspectives
The past decade has witnessed tremendous progress in elucidating the intertwining between ubiquitination and phosphorylation in the NF-κB pathway. It is now well established that phosphorylation is a prerequisite for ubiquitination of IκB and the NF-κB precursors p100 and p105. The identification of the ubiquitination machinery, especially the ubiquitin ligase complex containing βTrCP, which binds specifically to phosphorylated substrates, provides the molecular basis for the signal-dependent degradation of IκB and processing of p100 and p105. Recent studies have also identified several ubiquitin pathway components that function upstream of IKK, including the TRAF proteins as E3s, Ubc13/Uev1A as E2s, TAB2 and TAB3 as ubiquitin receptors, and CYLD and A20 as DUBs (Table 1). These results lend strong support to the thesis that ubiquitination can also regulate phosphorylation by activating IKK through a proteasome-independent mechanism.
Table 1. Protein and enzymes involved in ubiquitin-mediated activation of IKK.
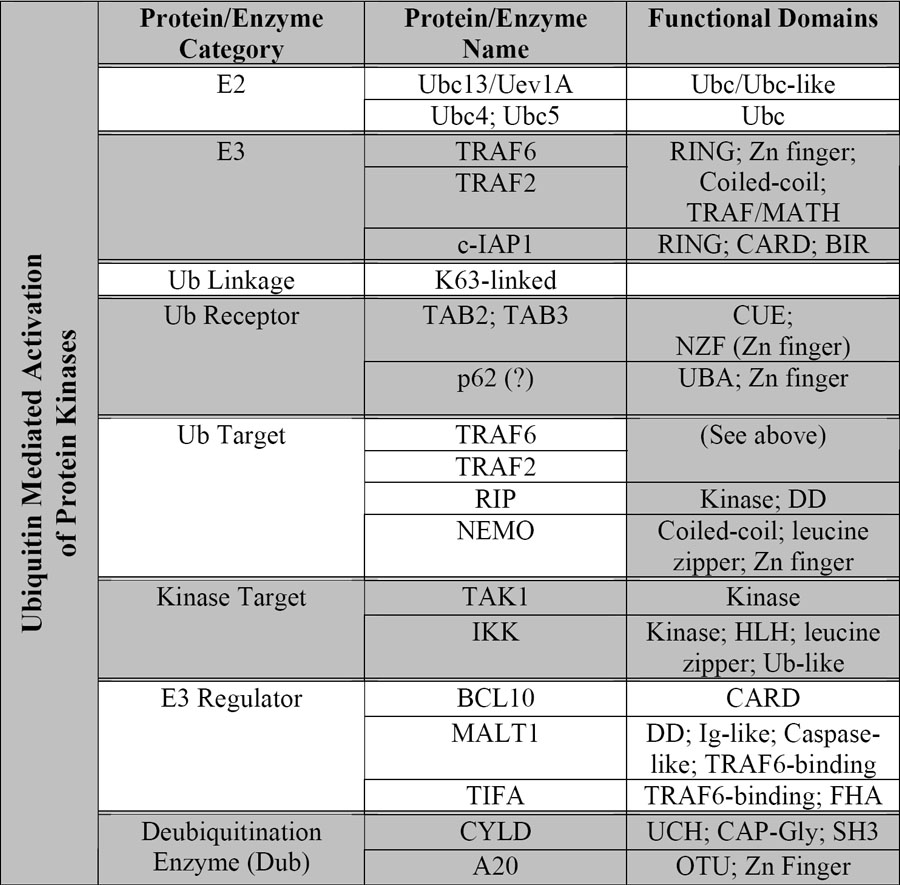
Protein name abbreviations: c-IAP1: cellular inhibitor of apoptosis 1; RIP: receptor interacting protein; NEMO: NF-κB essential modulator; MALT1: mucosal associated lymphoid tissue 1; TIFA: TRAF-interacting protein with a forkhead-associated domain. CYLD: cylindromatosis suppressor; Protein domain abbreviations: Ubc: ubiquitin conjugating enzyme; RING: really interesting new gene; CUE: cue1 homologous; NZF: novel zinc finger; UBA: Ub association; DD: death domain; HLH: helix-loop-helix; CARD: caspase activation and recruitment domain; Ig-like: immunoglobulin like; FHA: forkhead-associated; UCH: ubiquitin C-terminal hydrolase; CAP-Gly: cytoskeleton-associated protein; SH3: Src homology-3; OTU: ovarian tumor type cysteine protease.
While the traditional role of ubiquitin in degrading and processing NF-κB inhibitors is widely accepted, there are several important issues that remain to be addressed. For example, what is the mechanism underlying the highly processive polymerization of ubiquitin to form polyubiquitin chains? How are different E2 selected to a common SCF ubiquitin ligase scaffold to ubiquitinate distinct substrates? Is the assembly of a specific SCF complex regulated by the availability of substrates? How is ubiquitinated IκB, but not NF-κB present within the same complex, selectively degraded by the proteasome? How do p100 and p105 escape complete degradation by the proteasome? Are there additional proteins that escort the ubiquitinated IκB or p105/p100 to the proteasome?
Much also remains to be learnt about the non-traditional role of ubiquitin in the NF-κB pathway. Specifically, how does binding of a ubiquitinated protein (e.g, TRAF6 or RIP) to a ubiquitin receptor (e.g, TAB2 or TAB3) activate the receptor-associated kinase (e.g. TAK1)? What is the function of Nemo ubiquitination? Does NIK activation in the non-canonical pathway of NF-κB activation involve TRAF proteins and/or ubiquitin? Are there other NF-κB activation pathways that utilize ubiquitin as a signaling molecule for IKK activation? Are there other E2s (e.g., Ubc4/5) and E3s that may regulate IKK? In more general terms, is ubiquitin signaling involved in activating other enzymes including other kinases and ATPases? In this regard, the 19S proteasome contains multiple ATPases and ubiquitin binding proteins. Does the binding of polyubiquitinated protein substrates to the 19S proteasome activate its associated ATPase activity? If so, ubiquitin regulation of enzymes may be a unified mechanism underlying both the traditional and non-traditional functions of this protein tag.
Acknowledgments
Research in the Chen laboratory is supported by grants from NIH (R01-GM63692), the Welch Foundation (I1389) and American Cancer Society (RSG0219501TBE). Z.J.C is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in Pathogenesis of Infectious Diseases.
References
- 1.Hershko A, Ciechanover A, Rose IA. Resolution of the ATP-dependent proteolytic system from reticulocytes: a component that interacts with ATP. Proc Natl Acad Sci U S A. 1979;76:3107–10. doi: 10.1073/pnas.76.7.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciechanover A, Heller H, Elias S, Haas AL, Hershko A. ATP-dependent conjugation of reticulocyte proteins with the polypeptide required for protein degradation. Proc Natl Acad Sci U S A. 1980;77:1365–8. doi: 10.1073/pnas.77.3.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46:705–16. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 4.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell. 1986;47:921–8. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle PA, Baltimore D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-kappa B transcription factor. Cell. 1988;53:211–7. doi: 10.1016/0092-8674(88)90382-0. [DOI] [PubMed] [Google Scholar]
- 6.Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell. 1994;78:773–85. doi: 10.1016/s0092-8674(94)90482-0. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–97. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 8.Chen, Z. & Maniatis, T. Role of the Ubiquitin-Proteasome Pathway in NF-kB Activation, 303–322 (Plenum Press, New York, 1998).
- 9.Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–62. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 10.Deng L, et al. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–61. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 11.Wang C, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–51. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 12.Deng, L. & Chen, Z. Role of ubiquitin in NF-kB signaling, 139–160 (Kluwer, 2003).
- 13.Ben-Neriah Y. Regulatory functions of ubiquitination in the immune system. Nat Immunol. 2002;3:20–6. doi: 10.1038/ni0102-20. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 15.Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–5. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- 16.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–9. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 17.Senftleben U, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–9. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 18.Pickart CM. Back to the future with ubiquitin. Cell. 2004;116:181–90. doi: 10.1016/s0092-8674(03)01074-2. [DOI] [PubMed] [Google Scholar]
- 19.Chau V, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 20.Peng J, et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–6. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 21.Alkalay I, et al. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1995;92:10599–603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margottin F, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1:565–74. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 23.Winston JT, et al. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999;13:270–83. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yaron A, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396:590–4. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 25.Spencer E, Jiang J, Chen ZJ. Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev. 1999;13:284–94. doi: 10.1101/gad.13.3.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998;391:493–6. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- 27.Guardavaccaro D, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4:799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 28.Lin L, DeMartino GN, Greene WC. Cotranslational biogenesis of NF-kappaB p50 by the 26S proteasome. Cell. 1998;92:819–28. doi: 10.1016/s0092-8674(00)81409-9. [DOI] [PubMed] [Google Scholar]
- 29.Ciechanover A, et al. Mechanisms of ubiquitin-mediated, limited processing of the NF-kappaB1 precursor protein p105. Biochimie. 2001;83:341–9. doi: 10.1016/s0300-9084(01)01239-1. [DOI] [PubMed] [Google Scholar]
- 30.Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene. 2004;23:2540–7. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- 31.Fong A, Sun SC. Genetic evidence for the essential role of beta-transducin repeat-containing protein in the inducible processing of NF-kappa B2/p100. J Biol Chem. 2002;277:22111–4. doi: 10.1074/jbc.C200151200. [DOI] [PubMed] [Google Scholar]
- 32.Rape M, Jentsch S. Productive RUPture: activation of transcription factors by proteasomal processing. Biochim Biophys Acta. 2004;1695:209–13. doi: 10.1016/j.bbamcr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 33.Lin L, Ghosh S. A glycine-rich region in NF-kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol Cell Biol. 1996;16:2248–54. doi: 10.1128/mcb.16.5.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung JY, Park YC, Ye H, Wu H. All TRAFs are not created equal: common and distinct molecular mechanisms of TRAF-mediated signal transduction. J Cell Sci. 2002;115:679–88. doi: 10.1242/jcs.115.4.679. [DOI] [PubMed] [Google Scholar]
- 35.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–5. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 36.Lomaga MA, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–24. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naito A, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–62. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- 38.Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell. 1999;96:645–53. doi: 10.1016/s0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- 39.Ninomiya-Tsuji J, et al. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–6. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 40.Kanayama A, et al. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–48. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 41.Varadan R, et al. Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J Biol Chem. 2004;279:7055–63. doi: 10.1074/jbc.M309184200. [DOI] [PubMed] [Google Scholar]
- 42.Shi CS, Kehrl JH. Tumor necrosis factor (TNF)-induced germinal center kinase-related (GCKR) and stress-activated protein kinase (SAPK) activation depends upon the E2/E3 complex Ubc13-Uev1A/TNF receptor-associated factor 2 (TRAF2) J Biol Chem. 2003;278:15429–34. doi: 10.1074/jbc.M211796200. [DOI] [PubMed] [Google Scholar]
- 43.Xia ZP, Chen ZJ. TRAF2: a double-edged sword? . Sci STKE. 2005:pe7–2005. doi: 10.1126/stke.2722005pe7. [DOI] [PubMed] [Google Scholar]
- 44.Habelhah H, et al. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-kappaB. Embo J. 2004;23:322–32. doi: 10.1038/sj.emboj.7600044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeh WC, et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–25. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 46.Tada K, et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001;276:36530–4. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 47.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279:33185–91. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 48.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12:301–11. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 49.Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity. 2003;18:655–64. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 50.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 51.Ninomiya-Tsuji J, et al. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem. 2003;278:18485–90. doi: 10.1074/jbc.M207453200. [DOI] [PubMed] [Google Scholar]
- 52.Takaesu G, et al. TAK1 is critical for IkappaB kinase-mediated activation of the NF-kappaB pathway. J Mol Biol. 2003;326:105–15. doi: 10.1016/s0022-2836(02)01404-3. [DOI] [PubMed] [Google Scholar]
- 53.Vidal S, et al. Mutations in the Drosophila dTAK1 gene reveal a conserved function for MAPKKKs in the control of rel/NF-kappaB-dependent innate immune responses. Genes Dev. 2001;15:1900–12. doi: 10.1101/gad.203301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silverman N, et al. Immune activation of NF-kappaB and JNK requires Drosophila TAK1. J Biol Chem. 2003;278:48928–34. doi: 10.1074/jbc.M304802200. [DOI] [PubMed] [Google Scholar]
- 55.Chen W, White MA, Cobb MH. Stimulus-specific requirements for MAP3 kinases in activating the JNK pathway. J Biol Chem. 2002;277:49105–10. doi: 10.1074/jbc.M204934200. [DOI] [PubMed] [Google Scholar]
- 56.Sanjo H, et al. TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol Cell Biol. 2003;23:1231–8. doi: 10.1128/MCB.23.4.1231-1238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komatsu Y, et al. Targeted disruption of the Tab1 gene causes embryonic lethality and defects in cardiovascular and lung morphogenesis. Mech Dev. 2002;119:239–49. doi: 10.1016/s0925-4773(02)00391-x. [DOI] [PubMed] [Google Scholar]
- 58.Shibuya H, et al. TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science. 1996;272:1179–82. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- 59.Shibuya H, et al. Role of TAK1 and TAB1 in BMP signaling in early Xenopus development. Embo J. 1998;17:1019–28. doi: 10.1093/emboj/17.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamaguchi K, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–11. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 61.Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271:20235–7. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- 62.Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. Embo J. 1999;18:3044–53. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. Embo J. 2000;19:1576–86. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duran A, et al. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell. 2004;6:303–9. doi: 10.1016/s1534-5807(03)00403-9. [DOI] [PubMed] [Google Scholar]
- 65.Huang Q, et al. Differential regulation of interleukin 1 receptor and Toll-like receptor signaling by MEKK3. Nat Immunol. 2004;5:98–103. doi: 10.1038/ni1014. [DOI] [PubMed] [Google Scholar]
- 66.Yang J, et al. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001;2:620–4. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- 67.Thome M, Tschopp J. TCR-induced NF-kappaB activation: a crucial role for Carma1, Bcl10 and MALT1. Trends Immunol. 2003;24:419–24. doi: 10.1016/s1471-4906(03)00177-7. [DOI] [PubMed] [Google Scholar]
- 68.van Oers NS, Chen ZJ. Cell biology. Kinasing and clipping down the NF-kappa B trail. Science. 2005;308:65–6. doi: 10.1126/science.1110902. [DOI] [PubMed] [Google Scholar]
- 69.Zhou H, et al. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature. 2004;427:167–71. doi: 10.1038/nature02273. [DOI] [PubMed] [Google Scholar]
- 70.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 71.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell. 2003;115:565–76. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 72.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–82. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 73.Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004;14:2217–27. doi: 10.1016/j.cub.2004.12.032. [DOI] [PubMed] [Google Scholar]
- 74.Maeda S, et al. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–8. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 75.Kawai T, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–8. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 76.Brummelkamp TR, Nijman SM, Dirac AM, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 77.Kovalenko A, et al. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature. 2003;424:801–5. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 78.Trompouki E, et al. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature. 2003;424:793–6. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 79.Reiley W, Zhang M, Sun SC. Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem. 2004;279:55161–7. doi: 10.1074/jbc.M411049200. [DOI] [PubMed] [Google Scholar]
- 80.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 81.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 82.Evans PC, et al. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378:727–34. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–4. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-kappaB and ISRE and IFN-beta promoter. FEBS Lett. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]