

Abstract
Our previous finding and the given mechanism of charge and electron transfer in polypeptides are here integrated in a bifunctional model involving electronic charge transfer coupled to special internal rotations. Present molecular dynamics simulations that describe these motions in the chain result in the mean first passage times for the hopping process of an individual step. This “rest and fire” mechanism is formulated in detail—i.e., individual amino acids are weakly coupled and must first undergo alignment to reach the special strong coupling. This bifunctional model contains the essential features demanded by our prior experiments. The molecular dynamics results yield a mean first passage time distribution peaked at about 140 fs, in close agreement with our direct femtosecond measurements. In logic gate language this is a strongly conducting ON state resulting from small firing energies, the system otherwise being a quiescent OFF state. The observed time scale of about 200 fs provides confirmation of our simulations of transport, a model of extreme transduction efficiency. It explains the high efficiency of charge transport observed in polypeptides. We contend that the moderate speed of weak coupling is required in our model by the bifunctionality of peptides. This bifunctional mechanism agrees with our data and contains valuable features for a general model of long-range conductivity, final reactivity, and binding at a long distance.
Charge conductivity in biomolecules has become a general topic of substantial current interest (1, 2). This phenomenon is associated with the fascinating issue to what extent these systems can be classified as molecular wires and thus with the question of the proper mechanism for signal transduction in biomolecules such as proteins. The study of such systems in general is also of interest for the engineering of molecular devices based on the understanding of the transduction of charge by such molecules, thus leading to molecular logic gates—a field of immense current interest.
In previous work we have observed some unusual features for electron transfer and hole transfer in polypeptides. In this work the charge was placed on one end of the polypeptide at the C terminus, after which the charge could migrate to the N-terminal portion of the peptide or not. This process was observed to depend in a very sensitive way on the amino acids present in the chain. After experimenting with some 20 synthetic peptides we arrived at a mechanism demonstrating that each amino acid in zero order contributes semi-independently to the electronic surface of the total polypeptide. This surface could be approximated to first order by the values of the ionization potentials (IPs) of the separate amino acids. The IP of the supermolecule was not directly relevant here. Depending on the amino acids, we could then uniquely predict charge mobility or lack thereof. In fact, we could even insert a high-IP amino acid into the chain and stop charge transport at this site. For these reasons we proposed a hopping mechanism for such a charge transfer process in refs. 3–8.
To understand the pure molecular effect of these charge transfer processes we set out to study pure isolated polypeptides, without attached donors or acceptors, which at worst can compromise the chemistry and at best lead to further complications and additional delays in femtosecond timing studies. In particular, we used only the natural amino acids in peptides in which one of these amino acids contained a natural chromophore. This chromophore was used to introduce the charge into the system by photoexcitation. The charge transfer was thus studied under isolated conditions without a medium—i.e., in the gas phase. The efficiency of these charge transfer processes was extremely high, if allowed, and near zero, if disallowed. It is also interesting to note that a quite similar hopping model has been invoked recently to explain charge transport in DNA (9–18). Our experimental observation showed in 1996 that a hopping mechanism rather that a band model is mandated by the data as was proposed in ref. 5. This proposal has now been cast into a general framework (8).
Recently we undertook direct femtosecond measurements, which confirmed the time scale expected for our model (4). The electronic energy landscape between amino acids typically leads to jumps of some 0.2–0.5 eV between neighboring amino acids. On the other hand, the coupling strength required for such a jump would correspond approximately to 2–5 fs. Our recent direct timing measurements for a whole series of model systems, however, led to single jump times in the 200- to 300-fs range, or a coupling of some 70 meV. Hence we require a model that reconciles these apparently discordant features. We must have intermediate coupling permitting some residence of the charge on each amino acid, but at the same time we must have strong coupling to pass the charge to the next amino acid. An answer to this puzzle has been suggested recently (7). The detailed mechanism for the mobility of a single charge in polypeptide chains was given by Baranov and Schlag (7). The carbamide group of each amino acid is stiff and very loosely hinged to the next amino acid. At this hinge carbon the angles φ and ψ define the orientations of these two amino acids with respect to each other (Fig. 1a). Over a very large range of angles this hinge motion is a nearly free rotation with virtually no potential energy restrictions except at their respective limits. This range is given in the Ramachandran plot (19). Ab initio calculations show that even a pair of two identical amino acids will have a general asymmetry of about 0.6 eV because of the natural asymmetry of the C side and the N side of each amino acid. The interesting discovery of Baranov and Schlag was, however, that any such energy difference is not constant but rather strongly dependent on the angle between the adjacent carbamide groups. The N and O orbitals appear to switch. For the ionized species in a small range of ψ and φ, when the carbonyl groups of the neighboring amino acids are only about 2.87 Å apart, the electronic energy difference reaches a minimum. For the symmetric approach of carbonyls from the other side and the other ion a similar state exists. These two states at this point are isoenergetic with little or no energy barrier between them. At this angle they are also strongly correlated and form one hybridized state. We refer to this as the firing state for charge hopping because this strongly facilitates the charge movement to the next amino acid site in the chain. Hence our model suggests that we indeed have strong coupling but only in a small range of ψ and φ of the Ramachandran angles leading to charge hopping between neighbors (20, 21). The rest of the time the amino acid can freely rotate, within the bounds of the Ramachandran plot. The result is a bifunctional model with both weak and strong coupling that now reconciles the apparent discordance mentioned above.
Figure 1.
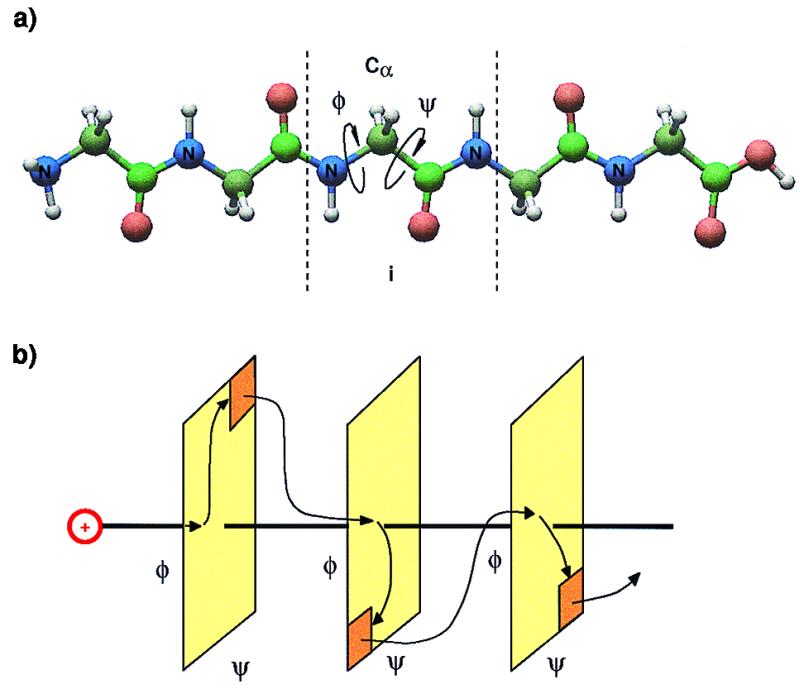
Charge transport in a polypeptide. (a) Polypeptide chain. Charge is first created on the donor and then hops through the amino acid chain until reaching the acceptor. (b) On each amino acid, the motion of the rotors is mapped into a Ramachandran plot. Here a simple two-dimensional (2D) area in phase space represents the hinge, i.e. the junction of two amino acids. The exit or gate part (orange) is the charge ratchet position. After the motion of rotors reaches the gate part, charge jumps to the next amino acid. The iteration of the previous procedure makes the charge hops to the final site.
In this paper we propose a combined mechanical and electronic mechanism for this migration involving a charge hopping process in this intermediate coupling regime and now test this by performing detailed molecular dynamics (MD) simulations. According to the Ramachandran plot, the torsional angles ψ and φ near the Cα atom can readily move inside broad limits with great ease. The firing region in accordance with the above is now defined as a certain subregion of this Ramachandran plot in which the O–O distance between neighboring amino acids approaches a definite critical value. This is taken as the narrow range of angles leading to strong correlation (21), hence transfer of charge from one amino acid to the next—i.e., firing. Only at this point is the electronic surface between neighboring amino groups isoenergetic. The transition rate between two nearby amino acids is then given by the first passage time for these two carbonyl groups to rotate into this isoenergetic firing position. The charge is now easily transferred between degenerate states. This behavior is mapped into a motion on the 2D Ramachandran plot or a phase space defined by ψ and φ. One can, therefore, describe this problem as an entropy-driven escape process in phase space. We now perform MD simulations¶ with this firing process inside a 2D phase space as a waiting process after which firing occurs. The MD calculations here investigate the internal rotational motions required to get into the next firing configuration. We are now able to calculate the charge transport rate of each individual step in a polypeptide chain.
We first consider a polypeptide chain of N amino acids. Each amino acid has a Cα atom—i.e., a hinge between the amino acids. At each hinge there are two torsional angles, φi−1,i and ψi,i+1. Each pair of (φi−1,i, ψi,i+1) constitutes a 2D phase space that is specific to a given pair of amino acids as shown in Fig. 1b. The charge is initially injected into a carbamide group, and its energy is transferred to the two rotational degrees of freedom of the hinge. At the same time this charge is waiting in carbamidei until the carbamidei+1 rotates to a certain angle and distance. Then the charge is transferred with zero barrier height. The electronic transition rate, hopping between two nearby critical structures, is now on the time scale of electronic correlation—i.e., it is rotated into one hybridized state. In this mechanism the charge is initially excited to an electronic excited state and the excess energy is then carried by the charge. When it moves to an adjacent carbamide group the charge dumps part of its energy into the rotational degrees of freedom of the next carbamide group because of energy conservation. The rotational period on the hinge is about 150 fs. In our MD calculations the charge jumps to the next amino group and seeks the next transition. The MD simulation shows that the motion inside the phase space is stochastic rather than coherent. This process will iterate until the charge reaches the terminal. Here chemical reaction can occur for the polypeptide chain. There is a substantial distance between the place of charge introduction and chemical reaction, with no apparent changes in between. We are here confronted with a most interesting feature of chemical reactions at a distance—an aspect of general significance in chemistry.
For any given amino acid there is a production of the ion at some normal inactive configuration of the amino acid. The ion then proceeds to an electronically low energy state, which happens when the carbonyl groups of adjacent amino acids are approximately 2.9–3.0 Å apart. At this point the configuration reaches a hybridized state and the charge migrates to the next neighbor. The amino acid now is neutralized and returns to its normal configuration, at which point the process starts over at the next site. Hence the system goes from a resting state to a firing state. Note that no single model calculation of a matrix element between neighboring amino acids can be successful here.
To model the charge hopping rate, we must consider the motion of the torsion angles moving to a firing configuration. The hinge rotation of φ and ψ angles is here derived from the electronic energy. The MD simulation is performed with the Gly3 peptide, although longer chains give similar results. In fact, 20-member sections cut from natural biomolecules have recently been shown to give quite similar results (unpublished results). The energy is introduced into the hinge rotations as the result of initial excess electronic energy, typically 0.2–0.5 eV, within 20 fs. This energy is introduced locally into the 2D phase space, the rest of the system being at 300 K. Within such a short time, the molecule will not vibrate, but it can rotate on the Cα carbon. Starting from this moment, we measure the time needed for the relative distance between oxygen atoms of adjacent amino acids to come into a firing position. We consider the electron as dumping its energy to the next set of rotational degrees of freedom after firing. A practical solution of this complex MD simulation task is to consider a substitute system that inserts some 170 meV by heating the molecule to 2000 K (or 4000 K), watching the rotational motions for times up to 1 ps as a safe time before intramolecular vibrational redistribution (IVR) can set in. This puts RT into the 2 internal rotations. We then ask the question of the time required to reach the first critical state or “collision” leading to firing.
The important and most interesting result from our MD simulations is that firing occurred in approximately 140 fs (Fig. 2). When the temperature is increased from 2000 K (≈0.170 eV) to 4000 K (≈0.340 eV), the two rotors increase their rotational frequencies by the expected factor of 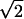 ). When this computer experiment is repeated for a number of samplings, a distribution of passage times results (Fig. 3a). On the right-hand side of the first passage time distribution curve at 2000 K (see Fig. 3a) one observes a plateau. This is just due to firings produced from thermal motions of the heat bath at this high temperature. Similar results occur at 4000 K (Fig. 3c). The plateau is an artifact of these simulations because the heat bath of all vibrations is in reality at 300 K; therefore the plateau from data at 300 K (Fig. 4) should be used. In other words, when we use the concept of 2000 K temperature we mean this to be the energy added locally to only the two nearly free rotations of the amino acid carrying the charge. Vibrational degrees of freedom set in at a much later time than rotational degrees of freedom. The other important implication of the first passage time distribution is the yield for charge transfer. Note that the first passage time curve terminates completely before the end of the 1-ps time. The possibility of inefficiencies should generate a much flatter first passage time curve with a long tail. Surprisingly, our first passage time curve was found to have no such tail but rather a Gaussian distribution with a fast decay. This suggests that all of the stochastic motions in the Ramachandran plot with this energy will eventually reach the exit gate with high efficiency and the yield should be near unity (Fig. 5). It should be noted that on this time scale there are no intramolecular vibrational redistribution (IVR) channels yet open.
). When this computer experiment is repeated for a number of samplings, a distribution of passage times results (Fig. 3a). On the right-hand side of the first passage time distribution curve at 2000 K (see Fig. 3a) one observes a plateau. This is just due to firings produced from thermal motions of the heat bath at this high temperature. Similar results occur at 4000 K (Fig. 3c). The plateau is an artifact of these simulations because the heat bath of all vibrations is in reality at 300 K; therefore the plateau from data at 300 K (Fig. 4) should be used. In other words, when we use the concept of 2000 K temperature we mean this to be the energy added locally to only the two nearly free rotations of the amino acid carrying the charge. Vibrational degrees of freedom set in at a much later time than rotational degrees of freedom. The other important implication of the first passage time distribution is the yield for charge transfer. Note that the first passage time curve terminates completely before the end of the 1-ps time. The possibility of inefficiencies should generate a much flatter first passage time curve with a long tail. Surprisingly, our first passage time curve was found to have no such tail but rather a Gaussian distribution with a fast decay. This suggests that all of the stochastic motions in the Ramachandran plot with this energy will eventually reach the exit gate with high efficiency and the yield should be near unity (Fig. 5). It should be noted that on this time scale there are no intramolecular vibrational redistribution (IVR) channels yet open.
Figure 2.

Energy dissipation process. This is one representative trajectory where the distance between O atoms is plotted. It shows a minimum at 130 fs, the position where firing occurs.
Figure 3.

(a) First passage time distribution. This is the distribution of 3-Å collisions between two carbonyl groups near the second Cα atom of Gly3 polypeptide. The first collision is due to rotary motion into the two hinge angles. The flat part shows the collision due to thermal motion. (b) This curve is fitted with Gaussian [0.02⋅exp−(x − 0.2657)2/(2⋅0.072)/(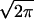 ⋅ 0.07)] and sigmoid [0.004⋅(2.0/(1.0 + exp(−(x − 0.41)/0.0085)))] functions. Note that the x-axis is already scaled by 560 fs. (c) First passage time curve for Gly3 at 4000 K. The maximum of the distribution shifts by the expected
⋅ 0.07)] and sigmoid [0.004⋅(2.0/(1.0 + exp(−(x − 0.41)/0.0085)))] functions. Note that the x-axis is already scaled by 560 fs. (c) First passage time curve for Gly3 at 4000 K. The maximum of the distribution shifts by the expected 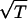 .
.
Figure 4.

First passage time curve for Gly3 at 300 K. This curve shows the thermal distribution for a heat bath at room temperature. The successful configurations are about 0.5%, i.e., a quiescent OFF state.
Figure 5.

Trajectories of the stochastic motion inside the Ramachandran plot. All of the trajectories end in a defined subspace of the total phase space. This region occupies 10% of the MD Ramachandran plot and is close to our ab initio computation.
In Fig. 6a we plot a typical motion of the two angles φ and ψ in time to investigate a possible time structure. But the Fourier transform of this function (Fig. 6b) shows only one strong peak at ω = 0, confirming that we may describe the motions as a stochastic process even in this short time regime.
Figure 6.

(a) φ + ψ vs. time. Here we portray a typical run for the sum of the torsional angles vs. time. Within such short time, with many-body effects, stochastic behavior is displayed. (b) Power spectrum of the time correlation function of the sum of the torsional angles. This figure shows a delta function at ω = 0.
We observe in these MD computer experiments that the charge transport process can be mapped into a stochastic motion on a Ramachandran plot with an exit gate. A theoretical estimation of the mean first passage time can be developed alternatively by solving a statistical theory of a two-rotor motion. The rotor motion inside the Ramachandran plot is similar to a 2D quantum mechanical particle in a box. Our model has the Hamiltonian H = (Pφ2/2Iφ) + (Pψ2/2Iψ), where Pi are the conjugate momenta and Ii are the moments of inertia, i = φ and ψ. The reaction flux passing through the exit region is counted as a successful escaping and its mean first passage time =
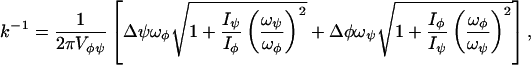 |
where Vφψ = φψ − ΔφΔψ. Here we assume the exit as a cubic with boundary length Δφ, where φ is the box size inside the Ramachandran plot. ωφ and ωψ are the rotational frequencies of the rotors (we also assume both of the rotors are the same). Further theoretical work will be published elsewhere.
To make sure that our picture of random motion in phase space is universal we increased the number of Gly residues to 10 (Fig. 7). The difference between Gly10 and Gly3 is small and demonstrates the interesting result that the larger attached structure does not impede the motion. This observation shows that the electronic energy is transferred into rotational degrees of freedom and is localized in the same (φi−1,i, ψi,i+1) pair with similar times for a more complex polypeptide chain.
Figure 7.

First passage time distribution of a Gly10 polypeptide. This curve was simulated at 2000 K and shows essentially the same distribution as the Gly3 results.
We should also consider such MD calculations at 300 K. These first passage times are shown in Fig. 4. We observe here that even for runs of some 2,000 points only 30 firings are seen. This means that the chain at 300 K is essentially quiescent, firing is not present at 300 K. Only 0.5% of the starting configurations produce the O–O firing collision within the required short time, and even most of them are just background fluctuations. The charge transport has essentially not occurred. This then is a clear indication of no transport—but this would not be expected in the absence of a signal input. In the language of logic switching circuits this is the characteristic of a logic OFF state (23–26). On the contrary, at 2000 K almost 100% of the simulated configurations show the short-time O–O collision, and the charge transport process is totally turned on. Note that the energy for this turn-on corresponds to only about 170 meV. This moves the charge transduction from an OFF state to an ON state. In other words, if the peptide is triggered by a typical redox site such as Cu+–Cu2+, this 153 meV (≈1720 K) is sufficient to initialize the charge transport process of the peptide chain and produce an ON state. At room temperature, in the absence of the redox trigger, the peptide chain cannot be triggered thermally. This is, of course, as it should be. Again, here is the interesting feature of a logic gate: the peptide is in OFF state at 300 K (≈0.027 eV) and in ON state when provided with an input of 170 meV of local energy. Such a small range between ON and OFF states is a remarkable result of switching logic.
In conclusion, our model for charge transport implies two quite different processes, rotation and firing. This is a bifunctional model. Basically the charge placed on a “wiggling” amino acid is initially weakly coupled to the next amino acid on the chain. This motion is taken to be a nearly free rotation of the two rotation angles ψ and φ on the hinge carbon between adjacent amino acids as defined by the Ramachandran plot. As such these are near zero phonons not coupled to the many molecular vibrations. At a certain configuration reached in approximately 140 fs there is orbital degeneracy to the next amino acid, leading to strong coupling. Hence it is a “rest and fire” bifunctional mechanism. Direct femtosecond measurements on a number of systems by us (4) directly confirm this resting time of 200–300 fs. This is the weak coupling part of the mechanism. The firing time on this time scale is instantaneous when the critical configuration of the two carbamide groups is reached. Here is the strong coupling part of the mechanism. The energy of the rotors was taken from the 0.2–0.5 eV provided by the change in electronic energies between neighboring sites corresponding to some 2000 K locally added to just these two rotors. This energy in the MD results appears to lead to completely efficient firing with a distribution peaked at 140 fs. In this paper we have shown that MD calculations of peptide chains based on the above model produce a mean first passage time distribution with results directly corroborating our experimental femtosecond results and producing a coupling strength consistent with our model. The distance between the oxygen atoms of neighboring carbamide groups was taken as an approximation to the critical firing configuration of Baranov and Schlag (7). When this distance approached 3 Å in the simulation this “collision” was taken as the firing configuration. A series of several thousand trajectories thus provided a distribution of first passage times to firing. This distribution has a Gaussian shape with a mean first passage time of 140 fs and a spread of 80 fs. This number is robust, even for longer attached chains. MD calculations show excellent agreement with experimental direct timing result. This proposed bifunctional molecular coupling scheme is an excellent agreement with our model.
Note that a theoretical calculation of any single matrix element between amino acids sites would not produce these results. Instead the matrix element switches drastically depending on the wiggling state of the peptide.
Interestingly, one might speculate about possible medium effects. First, this model predicts that electronic signal transduction is impeded if this mechanical wiggling is impeded—i.e., a sluggish external medium would stop electron and charge transfer. Conversely, shielding the peptide with a mechanically flexible medium would be expected to be useful. Water as an environment would be more complex, and as yet little is known about water at this short time. The dielectric relaxation would be expected just to shift the baseline. Medium relaxation effects, in turn, are expected only beyond approximately 2 ps (27) and hence would not affect short-time behavior. There is, however, a 20- to 50-fs ballistic motion that has been suggested (28), but this is thought to be an inertial motion of each solvent molecule acting independently, probably again having rotational librational character quite similar to the peptide itself. This motion will be expected only to lengthen the 130-fs result found here to approximately 200 fs, well within the range of the conclusion reached here. More interesting might be changes expected in transduction because of environmental effects such as hydrophobic interactions or long-range transmission of charge. This could produce tight bindings at distant sites (22).
At room temperature (300 K) MD calculations produce firing in the manner of a logic gate in only 0.5% of all trajectories. This result demonstrates that the room temperature molecule in the absence of an external trigger is in an essential logic OFF state. When supplied with a small trigger, such as from a typical redox couple (Cu+–Cu2+) with only 0.153 V this commences highly efficient firing, and nearly every charge is transmitted down the chain. Thus the molecule is now converted to an ON state. Note that such a small potential difference can switch this molecular system from an OFF to an ON state. This is a small trigger even by semiconductor standards and readily provides prescriptions for interesting logic gates.
In summary, the detailed MD calculations carried out here on several natural polypeptides fully support both our femtosecond timing measurements and our model in which signal transduction is presented as a bifunctional mechanism of alternating weak and strong coupling. This combines a very special mechanical motion of the peptide chain with an electronic surface jumping process. Our “rest and fire” behavior as a general mechanism for electron and charge transport in biomolecules such as polypeptides is hereby confirmed. This signal transduction is activated between an OFF state and an ON state by only a small trigger potential. There are good reasons why this model may also be generally applicable to many other biomolecular systems. Such a mechanism could also form the basis of new molecular logic gates.
Acknowledgments
This work was supported by the Volkswagen Foundation and the Taiwan/Germany program at the NSC/Deutscher Akademischer Austausdienst. Partial Support of the SF 377 is gratefully acknowledged.
Abbreviations
- 2D
two-dimensional
- MD
molecular dynamics
Footnotes
Dynamics calculations were performed with discover (Biosym/MSI of San Diego) using the consistent valence forcefield (CVFF).
References
- 1.Jortner J, Bixon M, editors. Electron Transfer: From Isolated Molecules to Biomolecules, Advances in Chemical Physics. New York: Wiley; 1999. [Google Scholar]
- 2.Jortner J, Bixon M, editors. Electron Transfer: From Isolated Molecules to Biomolecules, Advances in Chemical Physics. New York: Wiley; 1999. [Google Scholar]
- 3.Weinkauf R, Aicher P, Wesley G, Grotemeyer J, Schlag E W. J Phys Chem. 1994;98:8381–8391. [Google Scholar]
- 4.Weinkauf R, Schanen P, Yang D, Soukara S, Schlag E W. J Phys Chem. 1995;99:11255–11265. [Google Scholar]
- 5.Weinkauf R, Schanen P, Metsala A, Schlag E W, Burgle M, Kessler H. Phys Chem. 1996;100:18567–18585. [Google Scholar]
- 6.Schlag E W, Lin S H, Weinkauf R, Rentzepis P M. Proc Natl Acad Sci USA. 1998;95:1358–1362. doi: 10.1073/pnas.95.4.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baranov L Ya, Baranov L Ya, Schlag E W. Z Naturforsch. 1999;54a:387–396. [Google Scholar]
- 8.Remacle F, Levine RD, Schlag E W, Weinkauf R. J Phys Chem. 1999;103:10143–10158. [Google Scholar]
- 9.Rajski S R, Kumar S, Roberts R J, Barton J K. J Am Chem Soc. 1999;121:5615–5616. [Google Scholar]
- 10.Meggers E, Michel-Beyerle M E, Giese B. J Am Chem Soc. 1998;120:12950–12955. [Google Scholar]
- 11.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle M E. Proc Natl Acad Sci USA. 1998;95:12759–12765. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wan C, Fiebig T, Kelley S O, Treaday C R, Baron J K, Zewail A H. Proc Natl Acad Sci USA. 1999;96:6014–6019. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis F D, Letsinger R L. J Biol Inorg Chem. 1998;3:215–221. [Google Scholar]
- 14.Okada A, Chernyak V, Mukamel S. J Phys Chem A. 1998;102:1241–1251. [Google Scholar]
- 15.Marcus R A. Adv Chem Phys. 1999;106:1–6. [Google Scholar]
- 16.Marcus R A. J Electroanal Chem. 1997;438:251–259. [Google Scholar]
- 17.Turro N J, Barton J K. J Biol Inorg Chem. 1998;3:201–209. [Google Scholar]
- 18.Basilevsky M V, Chudinov G E, Rostov I V, Liu Y P, Newton M D. THEOCHEM J Mol Struct. 1996;371:191–203. [Google Scholar]
- 19.Ramachandran G N, Sasisekharan V. Adv Protein Chem. 1968;23:283–438. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 20.Serrano-Andres L, Fulscher M P. J Am Chem Soc. 1998;120:10912–10920. [Google Scholar]
- 21.Cederbaum L S, Zobeley J. Chem Phys Lett. 1999;307:205–210. [Google Scholar]
- 22.Davies A M, Teague S J. Angew Chem Int Ed. 1999;38:736–749. doi: 10.1002/(SICI)1521-3773(19990315)38:6<736::AID-ANIE736>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 23.Fox M A. Acc Chem Res. 1999;32:201–207. [Google Scholar]
- 24.Piotrowiak P. Chem Soc Rev. 1999;28:143–150. [Google Scholar]
- 25.Davis W B, Svec W A, Ratner M A, Wasielewski M R. Nature (London) 1998;396:60–63. [Google Scholar]
- 26.Collier C P, Wong E W, Belohradsky M, Raymo F M, Stoddart J F, Kuekes P J, Williams R S, Heath J R. Science. 1999;285:391–394. doi: 10.1126/science.285.5426.391. [DOI] [PubMed] [Google Scholar]
- 27.Yoshihara K. In: Electron Transfer: From Isolated Molecules to Biomolecules, Part Two, Advances in Chemical Physics. Jortner J, Bixon M, editors. Vol. 107. New York: Wiley; 1999. pp. 371–402. [Google Scholar]
- 28.Jimenez R, Fleming G R, Kumar P V, Maroncelli M. Nature (London) 1994;369:471–473. [Google Scholar]