

Abstract
Monolayer mixtures of dihydrocholesterol and phospholipids at the air–water interface are used to model membranes containing cholesterol and phospholipids. Specific, stoichiometric interactions between cholesterol and some but not all phospholipids have been proposed to lead to the formation of condensed complexes. It is reported here that an externally applied electric field of the appropriate sign can destabilize these complexes, resulting in their dissociation. This is demonstrated through the application of an electric field gradient that leads to phase separations in otherwise homogeneous monolayers. This is observed only when the monolayer composition is close to the stoichiometry of the complex. The electric field effect is analyzed with the same mean field thermodynamic model as that used previously to account for pairs of upper miscibility critical points in these mixtures. The concentrations of dihydrocholesterol, phospholipid, and complex vary strongly and sometimes discontinuously in the monolayer membrane in the field gradient. The model is an approximation to a two-dimensional liquid in which molecules freely exchange between free and complexed form so that the chemical potentials are constant throughout the membrane. The calculations are illustrated for a complex of about 15 molecules, composed of 5 cholesterol molecules and 10 phospholipid molecules.
Keywords: monolayers, membranes, chemical activity, phase separations, sphingomyelin
An understanding of the chemical activity of cholesterol in cell membranes has been a challenging problem for many years (1). Studies of cholesterol containing lipid monolayers at the air–water interface have the potential to shed light on this problem because the lateral interactions between molecules in monolayers, lipid bilayers, and biological membranes are likely to be similar at comparable molecular densities. The monolayer systems have the special advantages of simplicity, and the ease with which intermolecular interactions can be changed by changes of molecular density (2).
In recent work it was shown that a mean field thermodynamic model can be used to account for reported multiple upper miscibility critical points in monolayers composed of phospholipids and cholesterol, or dihydrocholesterol (DChol) (3, 4). [DChol is often used instead of cholesterol to minimize air oxidation. The phase behavior of the two steroids in mixtures with phospholipids is very similar (5).] The phospholipids employed include phosphatidylcholines, phosphatidylserines, and sphingomyelin. The model employs thermodynamic parameters describing liquid–liquid immiscibility and complex formation. The term “condensed complex” is used to describe a cooperative interaction between cholesterol and phospholipids. This cooperativity of complex formation follows directly from the parameters used to model the observed phase diagrams. The cooperativity is also implied by molecular area vs. membrane composition measurements. The model has been extended to include the concurrent formation of complexes with more than one stoichiometry, but as yet there is no experimental evidence for multiple stoichiometries in monolayers (6).
The idea behind using an electric field gradient for this study is simple. It is possible to produce external electric fields in lipid monolayers that are of the order-of-magnitude of 105 volts/cm near an electrode. (This is of the same order as transbilayer electric fields when the transbilayer potential difference is of order 100 millivolts.) Dipole densities in lipid monolayers are known from a variety of studies, and are of the order of one debye per 100 Å2 (7–9). For a molecule of area 100 Å2, the energy of interaction between this field and the molecule is of order 3 × 10−23 joules. This is small compared to kT at room temperature (3 × 10−21 joules). Thus, one does not expect a significant redistribution of molecules in the presence of a field gradient unless the monolayer is close to a phase transition or immiscibility phase boundary (10–12). However, a “large” complex might reasonably have a large dipole moment, in which case the applied electric field gradient might lead to an observable non-uniform distribution of molecules in the field gradient. This effect is expected to increase in proportion to the change in dipole density and the number of molecules in the complex. In the present work, we report that, in a number of Dchol-phospholipid mixtures, this effect is even larger than anticipated because of a field-induced phase separation related to the field destabilization of the complexes.
Experimental
Egg sphingomyelin (Egg-SM), dimyristoyl phosphatidylcholine (DMPC), dimyristoyl phosphatidylserine (DMPS), and dipalmitoyl phosphatidylcholine (DPPC) were obtained from Avanti Polar Lipids; cholesterol (Chol) and DChol were obtained from Sigma. The fluorescent dye, Texas red dihexadecanoyl-glycero-phosphoethanolamine (TR-DHPE), was obtained from Molecular Probes. TR-DHPE partitions preferentially among the phases, thereby providing contrast. The lipid mixtures used in this electric field study were composed of DChol and various phospholipids. Many experiments, especially in the case of Egg-SM, were carried out with Chol as well as DChol to be sure that the sterols are substantially equivalent. Except for sphingomyelin, all of the lipid mixtures have been studied earlier (3, 4). Their phase diagrams display cusps indicative of complex formation. Illustrative phase diagrams together with average area per molecule measurements are given in Fig. 1. The arrows indicate putative stoichiometric compositions.
Figure 1.

Phase diagrams showing liquid–liquid miscibility critical points and average molecular area measurements. (a, b, and c) Phase diagrams for mixtures of DChol (mole fraction x0) and phospholipids (mole fraction 1 − x0). (a) The phospholipid is a 2∶1 molar mixture of DMPS and DMPC. (b) The phospholipid is DMPS. (c) The phospholipid is Egg-SM. In a, b, and c, the plotted data points represent the transition pressures that mark the disappearance of two-phase coexistence during monolayer compression and thus define a phase boundary between a two-phase coexistence region at low pressures and a one-phase region at higher pressures. Stripe superstructure phases, which represent a proximity to a critical point (32), were observed at the transitions marked by filled circles and not at those marked by the open circles. The two-phase coexistence region corresponding to low DChol mole fraction is referred to as α, and the two-phase coexistence region corresponding to high DChol mole fraction is referred to as β. (d) Average molecular area for the lipid mixture of a at a pressure of 3 mN/m. (e) Average molecular area for the lipid mixture of b at a pressure of 25 mN/m. (f) Average molecular area for the lipid mixture of c at a pressure of 22 mN/m. Typical errors for the above measurements are 3–8%. The arrows indicate putative stoichiometric compositions.
All substances were used without further purification. Lipid mixtures with 0.5 mol% of TR-DHPE were spread from a 1 mg/ml chloroform solution on the air–water interface of a 9- × 2.5-cm Teflon trough that had a movable barrier to modulate the surface pressure. All experiments were carried out at room temperature on a subphase of 2 mM KCl in distilled water. The electric field was applied by using a setup described in detail in earlier work (7, 11) and is schematically shown in Fig. 2. Capillary tubes (borosilicate glass, diameter of 1.5 mm) were pulled on a Sutter Instruments (Novato, CA) micropipette puller to narrow the tip of the tube to ≈15–20 μm. A 1-mm diameter platinum wire with a 5-μm tungsten wire fused to the end of it was threaded through the glass tube. The glass tube was then bent over a gas burner to the desired shape of a hook. The Teflon trough had a groove milled into it, which housed the electrode. The tip of the glass tube with the electrode inside it poked through the subphase.
Figure 2.

Schematic for the experimental setup. An inhomogeneous electric field is generated at the air–water interface by applying a potential V0 to an insulated tungsten wire. The radius of the insulated electrode is a, about 6 μm. The electric field strength perpendicular to the surface of the interface is denoted ɛ(r).
For the geometry depicted in Fig. 2, it has been shown that the electrode produces a field in the monolayer plane given by
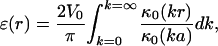 |
1 |
where ɛ(r) = ɛ is the component of the electric field perpendicular to the monolayer at the air–water interface, r is the radial distance from the center of the electrode, a is the radius of the electrode, V0 is the applied potential, and κ0 is the zeroth-order modified Bessel function of the second kind (7, 11). Epifluorescence microscopy and methods described previously (13, 14) were used to observe coexisting liquid phases in the presence and absence of a field.
Results
Fig. 3 shows micrographs of monolayers containing DChol and a phospholipid mixture composed of a 2∶1 ratio of DMPS and DMPC. The phase diagram and molecular area measurements for this system are shown in Fig. 1. As seen in Fig. 3a, at a monolayer surface pressure of 21 mN/m, well above the two critical pressures (3.5 and 12 mN/m), a uniform gray phase is observed in the absence of an electric field. When the electrode is made positive at 150 volts at this same high pressure, two liquid domains are formed in a narrow band around the electrode (Fig. 3b). This effect is not seen when the electrode is negative. For an applied voltage of 150 V, it takes 10–20 minutes for the domains to form around the electrode. The domains disappear completely within 10–15 minutes after the field is turned off. The domain formation around the electrode is also not seen except in a narrow composition range for this system, 30–35 mol% DChol. This phenomenon was also observed in other mixtures, such as Egg-SM/DChol (for 22–28% DChol), DMPS/DChol (for 25–40 mol% DChol), and 1∶1 DMPC∶DPPC/DChol (for 30–45 mol% DChol). The effect was the strongest in the 2∶1 DMPS:DMPC/DChol and Egg-SM/DChol mixtures and was much weaker in the other two lipid mixtures. All of these systems exhibit phase diagrams with cusps in the composition range displaying the electric field effect. Two other mixtures (DMPC/DChol and 3∶1 DMPC∶DPPC/DChol) were also studied and showed no effect under the electric field.
Figure 3.

Epifluorescence micrographs of a lipid monolayer consisting of DMPS, DMPC, and DChol in the absence and presence of an inhomogeneous electric field. The monolayer contains 21.6 mol% DMPC, 43.2 mol% DMPS, 35 mol% DChol, and 0.2 mol% TR-DHPE. In a, the surface pressure is 21 mN/m, well above the critical pressures for the system (see Fig. 1a), and there is no electric field. The monolayer appears uniformly gray around the electrode. In b, a positive potential of 150 volts has been applied, and domain formation can be seen in a narrow band around the electrode.
The Model
The experiments were planned on the basis of the following extension of a previously described mean field thermodynamic model (4, 15, 16) to include the effects of an electric field gradient. As before, we consider a mixture of two components, C (cholesterol or DChol) and P (phospholipid), that react to form a complex CqPp,
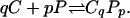 |
2 |
The values of p and q are taken to be small integers that are relatively prime (see later). The chemical potential of each component is pressure-dependent (11, 17) and is given by
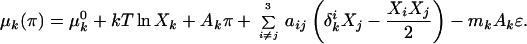 |
3 |
Here, μk0 is the chemical potential of k at the reference pressure of zero, Xk is the mole fraction of component k, Ak is the molecular area of k, aij is the interaction energy between i and j normalized with respect to kTr (where Tr is a reference temperature), and δki is the Kronecker delta function defined as δki = {1 for i = k; 0 for i ≠ k}. The matrix {aij} is symmetric. The indices k = 1, 2, 3 refer to cholesterol, phospholipid, and complex, respectively.
When these chemical potentials are incorporated in the expression for the free energy,
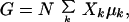 |
4 |
one obtains the following free energy as described in earlier work, except for the addition of the electric field term.
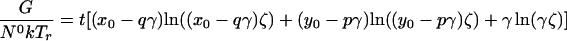 |
5 |
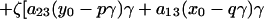 |
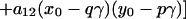 |
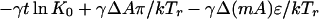 |
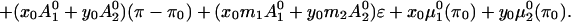 |
Here, N0 is the total number of molecules before reaction, π is the surface pressure, π0 is some reference surface pressure, t = T/Tr, x0, and y0 are the initial mole fractions of C and P, respectively, γ = N3/N0 describes the extent of reaction, and ζ = [1 + (1 − p − q)γ]−1. K0 is the equilibrium constant of the reaction at zero pressure (π = 0) and zero field strength, and ΔA = A30 − qA10 − pA20 is the area change of the reaction. The interaction parameters aij are pressure-dependent and can be written as aij = 2 + a′ij(π − πij), where πij is the critical pressure of a hypothetical nonreacting mixture of i and j. In principle, the interaction terms are likewise field-dependent, but we assume this dependence is negligible. Also, mk is the dipole moment density of component k, and ɛ is the radius-dependent electric field strength defined in Eq. 1. The change in molecular dipole moment due to the reaction is
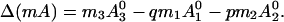 |
6 |
The direction of a dipole mk is defined as plus to minus. The relative directions of the field ɛ and mk are then defined to be consistent with the energy in Eqs. 3 and 5.
The following procedure is used to calculate the effect of the field gradient on the monolayers. At large distances from the electrode, the free energy in Eq. 5 can be expressed as a function of three variables, G = G(x0, γ, π(∞)), where π(∞) is the monolayer pressure far from the electrode. At the experimental pressure, this free energy is minimized with respect to the degree of reaction γ for each composition x0. The compositions X1(∞), X2(∞), X3(∞) are then found for each x0. The compositions closer to the electrode, X1(r), X2(r), X3(r), are determined by iteration using the constancy of the chemical potentials in Eq. 3. That is, Eq. 3 is linearized in the composition variables, and the field strength is changed in small increments. For reasons given later, the radial dependence of the pressure is neglected, π(r) ≅ π(∞).
Previous data (4) have suggested that the reaction is more cooperative than that described by Eq. 2, when p and q are restricted to be small integers that are relatively prime. Accordingly, the reaction is written as
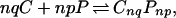 |
7 |
where n is an oligomerization parameter. The change in the molecular dipole moment due to this reaction is then increased:
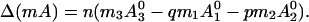 |
8 |
The equilibrium constant K for this reaction is
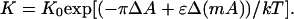 |
9 |
One expects to observe an electric field effect if ɛΔ(mA) in Eq. 9 is significant compared to kT. It should be noted with respect to this expression, for the equilibrium constant, the pressure varies with distance from the electrode:
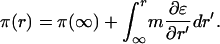 |
10 |
This effect destabilizes the complex when the electrode is positive, but this destabilization is small because the maximum change in pressure is no more than 0.1 mN/m for the parameters used.
In general, calculated concentrations of the various components vary continuously with distance from the electrode. However the experiments show, as illustrated in Fig. 3b, that the electric field gives rise to phase separation with two distinct phase boundaries. Fig. 4 is given to illustrate how this can come about. The solid line in this figure gives a phase diagram calculated by using n = 5, q = 1, p = 2, and the other parameters, as given in the legend. It will be seen that the calculated phase diagram is similar to the experimental diagrams in Fig. 1. (Until both experimental and theoretical uncertainties are explored, there is no point in attempting to obtain better agreement.) The gray line gives the phase diagram obtained when the equilibrium constant for complex formation is set equal to zero. The assumed critical pressure for this no-complex phase diagram is 15 mN/m. This critical pressure appears in the free energy (Eq. 5) used in the calculation of the phase diagram with complex, but has little effect on the shape of the diagram. In other words, from the observed phase diagrams with complex and at zero field, there is no way to deduce this no-complex critical pressure. The observed field-induced phase separations are interpreted as caused by the destabilization of the complex when the field direction is such as to oppose complex formation. In the absence of complex, there is a phase separation related to the no-complex phase diagram. Calculated compositions are given in Fig. 5 using the same parameters as used for Fig. 4. The two distinct phase boundaries are evident from jumps in composition. An important experimental result is that the field-induced phase separation is only observed for monolayers with compositions close to the stoichiometric composition: in this case, q = 1, p = 2. This is also found in the calculations, as shown in Fig. 5.
Figure 4.

Calculated phase diagrams as a function of the initial mole fraction of cholesterol (Chol), x0, for a binary reactive mixture of Chol and phospholipids for different equilibrium constants K0. This calculation considers multimeric complexes generated by the reaction nqC + npP ⇌ CnqPnp. The parameters used are for room temperature (t = 1), p = 2, q = 1, a′ = −0.33 m/mN, b′ = −0.5 m/mN, c′ = −0.4 m/mN, π12 = 15 mN/m, π13 = 6 mN/m, π23 = 3.3 mN/m, A10 = 40 Å2/molecule, A20 = 70 Å2/molecule, and A30 = n⋅140 Å2/molecule. The calculations are carried out for n = 5 by using Eq. 5 and procedures described earlier (4). The black line depicts the phase diagram when K0 = 2, which corresponds to a phase diagram in the absence of any field (r = ∞). The gray line depicts the phase diagram when K0 = 0, when the complex is destabilized, leaving behind a no-complex binary system.
Figure 5.

Calculated concentration profiles for cholesterol, phospholipid, and complex as a function of distance from the electrode for different initial compositions of Chol (x0). The calculations are carried out by using the parameters of Fig. 4 for n = 5, π = 9 mN/m, m1 = 2.9 debye/100 Å2, m2 = 1 debye/100 Å2, m3 = 2.7 debye/100 Å2, K0 = 2, and V0 = 150 volts. The electric field as a function of r is calculated by using Eq. 1. In a, x0 = 0.33, corresponding to the cusp stoichiometry of Fig. 4, whereas in b, x0 = 0.28. The sharp jumps in composition occur at phase boundaries.
All of the calculations are carried out for monolayers assumed to act as binary mixtures whereas most of the experiments for technical reasons have been carried out by using ternary or even more complex mixtures. Preliminary experimental as well as theoretical work in this laboratory has shown that this pseudobinary mixture approximation is sometimes good, sometimes not, depending on the phospholipids involved.
Discussion
The thermodynamic model for the formation of condensed complexes in lipid monolayers containing Dchol and phospholipids was based on phase diagrams and molecular area data such as those shown in Fig. 1 (4). We reasoned that cooperative complex formation would likely result in a substantial change in molecular dipole moment, and this change could be detected through an electric field effect. It was anticipated that the electric field would change the complex concentration in the vicinity of the electrode, and this would give rise to a continuous change in fluorescence intensity in this region. What was not anticipated was an even larger effect: namely, a field-induced phase separation in the vicinity of the electrode. Indeed, the experiments were carried out at pressures well above the known critical pressures to avoid field-induced phase separations of the sort previously described (10, 11). What could not be anticipated was the immiscibility of DChol and phospholipids in the absence of complex. That is, when the complex is largely destabilized by an electric field, the remaining DChol–phospholipid mixture can give rise to two immiscible liquids if the pressure of the system is below the no-complex critical pressure of this mixture. There is no a priori means of knowing the value of this critical pressure because the corresponding immiscibility critical pressure has no large effect on the phase diagrams when a complex is present. Calculated zero-field phase diagrams with and without complex are illustrated in Fig. 4.
The electric field effect is not symmetric. The phase separation is only seen when the electrode is positive. This separation gives rise to the three regions in Fig. 3b. These three regions are identified as the complex phase, the DChol-rich phase, and the phospholipid-rich phase. They are analogous but not identical to the three distinct phases observed when experiments are carried out in the absence of an applied field and below the critical pressures. In previous work, we have found that the fluorescent probe always partitions most strongly into the phospholipid-rich phase compared to the complex phase, and preferably into the complex phase as compared to the most DChol-rich phase (3, 4). Accordingly the outermost region (away from the electrode) is the phase with the condensed complex, next (moving in) is the dark, most DChol-rich phase, and finally (next to the electrode) are the punctuate droplets of the phospholipid-rich phase. The relative positions of the two phases closest to the electrode is in accord with previous measurements on co-existing domains of DChol-rich and phospholipid-rich domains in which it is found that the DChol-rich domain is repelled most strongly by a positive electrode (3, 4). Accordingly, the dipole density of this DChol-rich phase is larger and directed downward into the plane of the monolayer relative to that of the phospholipid-rich phase. Given that the outer complex phase is repelled by the positive electrode, it follows that complex formation results in an increase of dipole moment.
One may imagine the observed distribution of phases in Fig. 3b as arising in the following manner. In the absence of a field, one has a homogeneous phase, as in Fig. 3a. When the field is turned on, the molecular complexes near the electrode are destabilized relative to the reactants. Under these circumstances, close to the electrode, one then has a mixture of DChol and phospholipid. If, as illustrated in Fig. 4, the monolayer pressure is such that the mixture is below the no-complex phase boundary for phase separation, there will be a phase separation into domains of finite size, and these then would move electrophoretically in the field gradient (7). Experimentally, motion is seen but not easily resolved, perhaps because the phase separation and electrophoresis proceed simultaneously. After the field is removed, and during an approximately 15-minute period, both the DChol-rich region and the phospholipid-rich regions detach from the electrode. The phospholipid-rich domains become circular with well defined boundaries. They drift away and eventually become part of the complex phase. The dark DChol-rich liquid also moves away from the electrode, but the domain shapes are not so well defined.
An important result is that, for all four mixtures of phospholipids with DChol, these field-induced changes are only observed over limited ranges of DChol concentration. The range is 30–35% in the case of Fig. 1b. These results clearly reflect the stoichiometry of the complex. This sensitivity of the field effect to membrane composition is seen in the simulations in Fig. 5.
The destabilization of the complexes by the electric field can be discussed in terms of Eq. 4. In order for the complex to be destabilized by the positive electrode, it is necessary that Δ(mA) be positive. As seen in Fig. 1d–f, the change in molecular area on complex formation is clearly negative (A30 − qA10 − pA20 < 0). It follows that there must be a more than compensating increase in positive dipole density on complex formation. For a given change in dipole density, the size of the change in dipole moment is proportional to the number of molecules in the complex. It has been suggested that the terminal methyl groups of the fatty acid chains and cholesterol make significant contributions to the dipole densities in lipid monolayers (18), accounting for both the magnitude and sign of the observed density differences (5). The present work describes complexes in which this dipole density is increased even further on complex formation. This is of course consistent with a more organized structure, where on average terminal methyl groups have their three-fold axes more nearly perpendicular to the monolayer.
The calculations shown in Figs. 4 and 5 show how plausible values of the various parameters can be used to account for the results. The only restriction on the parameters we have chosen is that they be consistent with the experimental phase diagrams, molecular area measurements, prior work on dipole densities, and the model. In general, the data are qualitatively well represented by a cooperativity parameter n = 5.
As noted earlier, there is a long history of proposals for “complex” formation as well as for lattice-like structures in monolayers and bilayers (1, 19–31). We make no distinction between a complex and a molecular lattice with short-range order. Our proposal for the formation of condensed complexes differs from earlier work by others in that it is based on thermodynamic parameters that lead to pairs of upper miscibility critical points in lipid monolayers. At lower pressures, these parameters describe effective “repulsions” between cholesterol and phospholipid, between cholesterol and complex, and between phospholipid and complex. The repulsions as well as oligomerization enhance the cooperativity of complex formation. As is evident from this work as well as earlier work (4), complex formation is strongly cooperative even in the homogeneous phase well above the critical pressures. The term “condensed complex” is used to reflect both the cooperativity and area contraction associated with complex formation. It is anticipated that it will be possible to apply this model to bilayers and biological membranes by using parameters obtained from monolayer phase diagrams as a guide.
Acknowledgments
It is a pleasure to acknowledge helpful discussions with S. Keller, J. Klingler, T. Anderson, and J. Groves. This work was supported by the National Science Foundation and the National Institutes of Health Biotechnology Program.
Abbreviations
- DChol
dihydrocholesterol
- Chol
cholesterol
- DMPS
dimyristoyl phosphatidylserine
- DMPC
dimyristoyl phosphatidylcholine
- DPPC
dipalmitoyl phosphatidylcholine
- Egg-SM
egg sphingomyelin
- TR-DHPE
Texas red dihexadecanoyl-glycero-phosphoethanolamine
References
- 1.Feingold L. Cholesterol in Membrane Models. Ann Arbor, MI: CRC Press; 1993. [Google Scholar]
- 2.McConnell H M. Annu Rev Phys Chem. 1991;42:171–195. [Google Scholar]
- 3.Radhakrishnan A, McConnell H M. J Am Chem Soc. 1999;121:486–487. [Google Scholar]
- 4.Radhakrishnan A, McConnell H M. Biophys J. 1999;77:1507–1517. doi: 10.1016/S0006-3495(99)76998-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benvegnu D J, McConnell H M. J Phys Chem. 1993;97:6686–6691. [Google Scholar]
- 6.Anderson, T. G. & McConnell, H. M. (2000) Colloids Surf. A, in press.
- 7.Klingler J F, McConnell H M. J Phys Chem. 1993;97:2962–2966. [Google Scholar]
- 8.McConnell H M, Rice P A, Benvegnu D J. J Phys Chem. 1990;94:8965–8968. [Google Scholar]
- 9.Heckl W M, Miller A, Möhwald H. Thin Solid Films. 1988;159:125–132. [Google Scholar]
- 10.Lee K Y C, Klingler J F, McConnell H M. Science. 1994;263:655–658. doi: 10.1126/science.8303272. [DOI] [PubMed] [Google Scholar]
- 11.Lee K Y C, McConnell H M. Biophys J. 1995;68:1740–1751. doi: 10.1016/S0006-3495(95)80351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wirtz D, Fuller G G. Phys Rev Lett. 1993;71:2236–2239. doi: 10.1103/PhysRevLett.71.2236. [DOI] [PubMed] [Google Scholar]
- 13.Subramaniam S, McConnell H M. J Phys Chem. 1987;91:1715–1718. [Google Scholar]
- 14.Hirshfeld C L, Seul M. J Phys France. 1990;51:1537–1552. [Google Scholar]
- 15.Talanquer V. J Chem Phys. 1992;96:5408–5421. [Google Scholar]
- 16.Corrales L R, Wheeler J C. J Chem Phys. 1989;91:7097–7112. [Google Scholar]
- 17.Groves, J. T., Boxer, S. G. & McConnell, H. M. (2000) J. Phys. Chem., in press.
- 18.Vogel V, Möbius D. Thin Solid Films. 1988;159:73–81. [Google Scholar]
- 19.Albrecht O, Gruler H, Sackmann E. J Colloid Interface Sci. 1981;79:319–338. [Google Scholar]
- 20.Engelman D M, Rothman J E. J Biol Chem. 1972;247:3694–3697. [PubMed] [Google Scholar]
- 21.Finean J B. Experientia. 1953;9:17–19. doi: 10.1007/BF02147697. [DOI] [PubMed] [Google Scholar]
- 22.Gershfeld N L. Biophys J. 1978;22:469–488. doi: 10.1016/S0006-3495(78)85500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller-Landau F, Cadenhead D A. Chem Phys Lipids. 1979;25:315–328. [Google Scholar]
- 24.Parassassi T, Giusti A M, Raimondi M, Gratton E. Biophys J. 1995;68:1895–1902. doi: 10.1016/S0006-3495(95)80367-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Presti F T, Pace R J, Chan S I. Biochemistry. 1982;21:3831–3835. doi: 10.1021/bi00259a017. [DOI] [PubMed] [Google Scholar]
- 26.Tang D, van Der Meer W, Cheng S-Y S. Biophys J. 1995;68:1944–1951. doi: 10.1016/S0006-3495(95)80371-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandenheuvel F A. J Am Oil Chem Soc. 1963;40:455–471. [Google Scholar]
- 28.Virtanen J A, Ruonala M, Vauhkonen M, Somerharju P. Biochemistry. 1995;34:11568–11581. doi: 10.1021/bi00036a033. [DOI] [PubMed] [Google Scholar]
- 29.Wang M M, Sugar I P, Chong P L. Biochemistry. 1998;37:11797–11805. doi: 10.1021/bi980290k. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen M, Miao L, Ipsen J H, Zuckermann M J, Mouritsen O G. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Top. 1999;59:5790–5803. doi: 10.1103/physreve.59.5790. [DOI] [PubMed] [Google Scholar]
- 31.Somerharju P, Virtanen J A, Cheng K H. Biochim Biophys Acta. 1999;1440:32–48. doi: 10.1016/s1388-1981(99)00106-7. [DOI] [PubMed] [Google Scholar]
- 32.Keller S L, McConnell H M. Phys Rev Lett. 1999;82:1602–1605. [Google Scholar]