

Abstract
Transcription, the process whereby RNA copies are made from sections of the DNA genome, is directed by promoter regions. These define the transcription start site, and also the set of cellular conditions under which the promoter is active. At least in more complex species, it appears to be common for genes to have several different transcription start sites, which may be active under different conditions. Eukaryotic promoters are complex and fairly diffuse structures, which have proven hard to detect in silico. We show that a novel hybrid machine-learning method is able to build useful models of promoters for >50% of human transcription start sites. We estimate specificity to be >70%, and demonstrate good positional accuracy. Based on the structure of our learned models, we conclude that a signal resembling the well known TATA box, together with flanking regions of C-G enrichment, are the most important sequence-based signals marking sites of transcriptional initiation at a large class of typical promoters.
The vast majority of protein coding eukaryotic genes are transcribed using the polII RNA polymerase. A polII promoter consists of a group of transcription factor binding sites clustered around (but primarily upstream of) one or more transcription start sites (Werner 2000). While there has been some success in identifying consensus binding sequences for specific transcription factors, there is still uncertainty about the detailed organization and function of promoters. Therefore, an in silico promoter model should be a powerful tool for genome annotation, and might also give clues about the nature of promoters.
A number of computational methods have been proposed for detecting transcription start sites. The simplest approach is to use a DNA weight matrix to detect the TATA box motif (Bucher 1990), thought to be the core of most eukaryotic promoters. More sophisticated methods use hidden Markov models (Audic and Claverie 1997) or neural networks (Knudsen 1999). Many of these methods were reviewed and evaluated by Fickett and Hatzigeorgiou (1997). All methods were shown to suffer from poor sensitivity, many false positives, and poor positional accuracy. A recent development in promoter recognition is the PromoterInspector program (Scherf et al. 2000). This was trained using a brute-force algorithm to discover a set of sequence motifs overrepresented in promoter regions. It has a much lower false-positive rate than any of the programs reviewed above. However, it only attempts to detect ‘promoter regions’ (defined as regions of the genome containing promoter-like motifs), rather than locating transcription start sites.
We have developed a new program, Eponine, which aims to predict the exact location of transcription start sites (TSS). Eponine models consist of a collection of positioned constraints, each represented by a DNA weight matrix (Bucher 1990). A weight matrix is a simple generative model for a short, ungapped sequence motif. It consists of a series of ‘columns,’ each of which contains a probability distribution over the four symbols of the DNA alphabet. The consensus sequence (the most likely sequence to be generated by the model) is found by simply taking the most likely symbol from each column. Similarly, motifs matching this consensus sequence will receive the highest score when the weight matrix is used to scan genomic sequence.
Weight matrices are good sensors for simple, compact motifs, but are not, on their own, able to model more complex structures where there is some flexibility in the distance between parts of a signal. We built complex models by combining each weight matrix with an associated discrete probability distribution describing its position relative to the TSS. Thus, a score for one of these ‘positioned matrices’ can be calculated as:
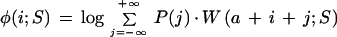 |
where P(j) is a discrete probability distribution; W(x;S) is the weight matrix score, aligning the first column to position x on sequence S; a is the center position of the distribution, relative to the TSS; and i is the position of the true TSS during training, and is varied along the length of the sequence when scanning a sequence with the trained model.
These can be combined to give complex models by taking the weighted sum of a number of these positioned matrix scores. This is equivalent to the well known generalized linear model (GLM) form (McCullagh and Nelder 1983). Such models can be trained using established procedures such as the relevance vector machine (Tipping 2001). The Eponine trainer combines an efficient implementation of the RVM algorithm with a Monte Carlo sampling process for selecting an optimal set of positioned matrices (see Methods).
Linear combination of positioned matrices provides a flexible alternative to approaches which use hidden Markov models to build complex structures from simple sequence motifs (Grundy et al. 1997). The Eponine trainer is able to learn both a set of motifs and a structural model in a single training process, and can potentially build models with overlapping motifs.
RESULTS
Model training was carried out in two stages, each using the RVM-hybrid method. Initial models were trained from the eukaryotic promoter database (EPD)-derived data set. There was some variability between models produced by separate training runs, indicating that insufficient training data were available to support a single, consistent model. Second generation models were trained from the mouse cDNA-derived data set. Not all of these represent full-length mRNAs, so the initial model was used to select a set of 599 traces which were likely to contain true promoters. Training on these selected mouse sequences gave simpler and much more consistent models, as shown in Figure 1. These models consist of four elements: (1) a diffuse preference for CpG enrichment downstream of the start site. This corresponds with the observation that promoters are associated with a CpG island; (2) a TATAAA motif, with a tightly focused distribution centered at position −30 relative to the transcription start site. This corresponds to the widely reported TATA box and (3 and 4) two GC-rich matrices closely flanking the TATA box. These key features can all be recognized in the initial models trained from the EPD dataset, but they are weaker and are combined with various other rules not consistent between training runs.
Figure 1.

Schematic of Eponine core promotor model, showing the constraint distributions and weight-matrix consensus sequences.
To test the positional specificity of the predictor, we ran the final model over the set of EPD entries (Fig. 2a). Predictions were clustered in the interval [−10:20] relative to the annotated start site. This suggests that the model can detect transcription start sites with good positional accuracy, especially since it is very hard to map start sites with perfect accuracy [the stated criterion for accepting entries in EPD is mapping of the TSS within ±5 bp (Perier et al. 2000)]. A smaller peak is observed around −40, which we cannot currently explain, and must assume to be composed of false positives.
Figure 2.


Density of predictions from Eponine relative to the annotated TSSs of (a) EPD entries and (b) chromosome 22 mRNAs. In the latter case, directionality of predictions was ignored (in common with the rest of the chromosome-scale evaluation in this paper).
To investigate the performance of the models for detecting promoters in large pieces of genomic DNA, we ran the same model on the 33.4 Mb assembled sequence of human chromosome 22 (Dunham et al. 1999). Release 2.3 of the chromosome 22 annotation includes 618 distinct genes (excluding pseudogenes). Of this set, 284 have an annotated TSS based on experimentally determined mRNAs, a high proportion of which are expected to be full-length transcripts. At the time of this writing, we believe this to be the largest piece of sequence with such a high proportion of experimentally annotated transcripts, making it ideal for this kind of evaluation. It should be noted, however, that chromosome 22 is a particularly GC-rich chromosome, with a high gene density (Dunham et al. 1999), so results for this chromosome may not translate exactly into results over the whole genome. Similarly, there may be some bias in the subset of genes for which a transcript is annotated. Scanning both strands of this sequence, the Eponine model, with a threshold of 0.999, made 2086 predictions. For 152 of the annotated TSSs, at least one prediction fell within 2kb, giving a sensitivity of 54% (Fig. 2b). Moreover, for 96 of these at least one prediction fell within 100 bases. Lowering the threshold further gave very little increase in the sensitivity (results not shown).
When considering the specificity of Eponine, 2086 predictions over a 33.4 Mb region containing 618 genes might seem to imply a high false-positive rate. However, the predictions were not uniformly distributed, and clustering with a gap tolerance of 1 kb reduced them to 368 clusters (average of 5.6 predictions per cluster). Some of this clustering comes from predictions being made on both strands very close together. Although all of the training examples were presented in the forward orientation only, 47% of forward-strand predictions on chromosome 22 were accompanied by reverse-strand prediction within 100 bases (in the bulk of cases, the reverse-strand prediction was around 60 bases upstream of the forward-strand prediction). If one prediction from each pair is assumed to be a false positive, the number of predictions per cluster would drop to 4.4. Interestingly, other methods of promoter prediction have also resulted in poor strand specificity: in particular, PromoterInspector is reported to be entirely strand-independent. In the present work, the strandedness of predictions was ignored for all methods. The remainder of the clustering could perhaps be explained if Eponine is predicting alternative TSSs used in transcription; however, this needs to be confirmed experimentally.
Measuring the precise specificity of the method is difficult, because we have no information about the position of the TSS for the 334 known genes where there is no full-length mRNA. To estimate specificity, we built a ‘pseudochromosome,’ that is, a long piece of sequence containing the mRNA-annotated genes and the sequence upstream of them, but omitting all known genes without mRNA annotation and pseudogenes (Fig. 3). The result was a 16.4 Mb sequence containing 215 clusters (a total of 1284 individual predictions), giving a specificity of 73.5%. Table 1 shows a comparison of the performance of promoter-finding methods on the pseudochromosome. Three of these methods (Eponine, PromoterInspector, and CpG islands) appear to offer comparable levels of sensitivity. We compared the sets of chromosome 22 promoters detected by the three methods (Fig. 4), and observed that the sets overlap substantially. From a unified set of 199 promoters detected by at least one of the methods, 132 (66%) are detected by all three. Eighty-five promoters (30%) are not detected by any of the methods examined here. We have made efforts to train a separate model on just this subset of promoters; however, initial models are not very consistent, suggesting that this data set is currently too small.
Figure 3.

Construction of the pseudochromosome, selecting only those regions where a full mRNA (transcript) is annotated. In the case where an mRNA-annotated gene is followed by a coding-sequence-only gene in the same orientation, the sequence is cut at the midpoint between the two genes.
Table 1.
Comparison of Various Promoter- and TSS-Detection Methods on the Chromosome 22–Derived Pseudo Chromosome
| Method | Predictions | True positives | False positives | Sensitivity (%) | Selectivity (%) |
|---|---|---|---|---|---|
| Eponine | 215 | 152 | 57 | 53.5 | 73.5 |
| PromoterInspector | 278 | 157 | 100 | 55.3 | 64.0 |
| CpG | 306 | 187 | 116 | 65.8 | 62.1 |
| TATA-2.6 | 540 | 37 | 500 | 13.0 | 7.4 |
| TATA-6.5 | 39869 | 283 | 37581 | 99.6 | 5.7 |
Sensitivity is defined here as the proportion of annotated mRNA starts that are detected by a given method (within 2 kb). Selectivity is the proportion of predictions that are confirmed by the presence of an annotated mRNA start.
PromoterInspector predictions for chromosome 22 (Scherf et al. 2001) were obtained from their web site. These applied to an older assembly of the chromosome and so were mapped onto the latest assembly using SSAHA (Ning et al., 2001). 99.4% of predictions were successfully mapped in this way. CpG islands were extracted from the chromosome 22 annotation repository. Note that this set of CpG island predictions was available to annotators working on this chromosome, so there is some possibility of bias in favor of this method. TATA-box motifs were detected using the log-odds weight matrix published by Bucher (Bucher 1990). The cutoff threshold of −6.5, recommended by Fickett (Fickett and Hatzigeorgiou 1997), gave 84,886 predictions: many more than any other method. We also used the far more stringent threshold of −2.6. This gave 1196 predictions, more in line with the other methods tested.
For Eponine and TATA box predictions, strand information was ignored (i.e., a prediction on the wrong strand will still be considered correct).
PromoterInspector predictions and CpG islands do not provide any information about direction of transcription.
Figure 4.

Intersection of ‘correct’ predictions of promoters by Eponine, PromoterInspector and CpG islands of chromosome 22 mRNAs.
Given the composition of the Eponine TSS model, it might be suggested that better results could be obtained by combining the results of TATA-box and CpG island searching programs. However, since more than 99% of CpG islands in our test set had a TATA box nearby, a straightforward intersection of the two methods will give results very similar to those for CpG islands alone. It may be possible to combine CpG island and TATA box predictors to give better selectivity, but to do so would require careful weighting of the scores of the two methods.
DISCUSSION
We have shown that our hybrid machine-learning method is able to build classification models both with high predictive accuracy and which are suggestive about the sequence requirements for TSSs. Examination of the learned models appears to confirm the classical view that the TATA-box motif is important in eukaryotic transcription initiation. However, we show that the TATA box alone has little or no predictive power for detecting TSSs in genomic DNA. The model suggests that it is the combination of a TATA box with CG-rich ‘flanking’ signals and an overall enrichment in CpG dinucleotides which gives the best indication that a TSS may be present. We hope that the annotation provided by Eponine will be useful for further genome analysis, experimental design, and future promoter research—particularly research into the prevalence of alternative TSSs. We anticipate that our learning method can be used to investigate other features of genomic DNA sequence and may provide new understanding of the sequence requirements that underlie them.
METHODS
RVM-Hybrid Sampling
Classification models were built using our own implementation of the relevance vector machine (RVM) (Tipping 2001). This is a Bayesian method of machine learning which can train probabilistic classification models in generalized linear modal (GLM) form. The RVM is a sparse training algorithm—it takes a set of suggested basis functions and selects the sets which are most helpful in classifying the provided training data, using a ‘pruning’ prior which discards basis functions which do not have enough support from the training data. However, in order to analyze promoters it is necessary to explore an extremely large model space of possible weight matrices and position distributions. To facilitate this, we expanded the RVM implementation to allow sampling from this large rule space. The working set is initialized with weight matrices of lengths 4 to 8, selected at random, and with random, gaussian position distributions. As rules in the initial working set are discarded by the pruning algorithm, new examples are added. These may be produced by the same logic used to initialize the working set, or represent small changes to existing rules. In our implementation, the allowed sampling moves are as follows: (1) adjust the center position of a distribution; (2) adjust the width parameter of a position distribution; (3) adjust the weights in a DNA weight matrix; (4) construct a new DNA probability distribution at random, then add it as a column at one end (randomly chosen) of a weight matrix; and (5) remove a column from one end of a weight matrix.
This gives a hybrid machine-learning approach, combining the RVM with elements of a Monte Carlo sampling approach. Using this hybrid method, a model can be efficiently built from a large space of potential candidate rules.
EPD Training Set
As an initial set of positive examples, we extracted all mammalian promoters from the EPD database (Perier et al. 2000). We then discarded those with less than 500 bases of upstream sequence available, and those located on human chromosome 22. This left 313 sequences, of which 50 were kept aside for test purposes, the remainder forming the training set.
Mouse cDNA-Derived Training Set
In order to build a larger training set, we used the FANTOM collection of full-length enriched cDNAs (Kawai et al. 2001). Because upstream sequence was required, we searched the first 100 bp of each cDNA sequence against the Mouse Genome Project trace repository (http://trace.ensembl.org/) using the SSAHA rapid searching program (Ning et al., 2001). After selecting only those hits with at least 42 bases (three SSAHA words) of exact homology and at least 150 bases of sequence upstream of the mapped mRNA 5′ end, we obtained trace sequences corresponding to 9958 mRNAs.
Negative Examples
To build a classification model, two sets of training data are required. To provide negative training examples, we selected random fragments from human chromosome 20. For all training runs, we supplied an equal number of positive and negative examples.
WEB SITE REFERENCES
http://www.ensembl.org/; predictions for the human genome sequence are now available from Ensembl.
http://www.sanger.ac.uk/Users/td2/eponine/; Eponine information, software downloads, and sequence submission form.
http://servlet.sanger.ac.uk:8080/das/; Sanger Institute distributed annotation system (DAS) server, includes Eponine TSS predictions.
http://trace.ensembl.org/; Mouse Genome Project trace repository.
Acknowledgments
Gene annotations (release 2.3) were produced by the Chromosome 22 Gene Annotation Group at the Sanger Centre and were obtained from the World Wide Web at http://www.sanger.ac.uk/HGP/Chr22 (I. Dunham et al., unpubl.). Mouse whole-genome shotgun data were produced by the Mouse Genome Sequencing Consortium, and obtained via the Ensembl trace repository. T.D. thanks the Wellcome Trust for support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL td2@sanger.ac.uk; FAX 44-1223-494919.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.216102.
REFERENCES
- Audic S, Claverie JM. Detection of eukaryotic promoters using Markov transition matrices. Comput Chem. 1997;21:223–227. doi: 10.1016/s0097-8485(96)00040-x. [DOI] [PubMed] [Google Scholar]
- Bucher P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J Mol Biol. 1990;212:563–578. doi: 10.1016/0022-2836(90)90223-9. [DOI] [PubMed] [Google Scholar]
- Dunham I, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, Ainscough R, Almeida JP, Babbage A, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Fickett JW, Hatzigeorgiou AG. Eukaryotic promoter recognition. Genome Res. 1997;7:861–878. doi: 10.1101/gr.7.9.861. [DOI] [PubMed] [Google Scholar]
- Grundy WN, Bailey TL, Elkan CP, Baker ME. Meta-MEME: Motif-based hidden Markov models of protein families. Comput Appl Biosci. 1997;13:397–406. doi: 10.1093/bioinformatics/13.4.397. [DOI] [PubMed] [Google Scholar]
- Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, Ishii Y, Arakawa T, Hara A, Fukunishi Y, Konno H, et al. Functional annotation of a full-length mouse cDNA collection. Nature. 2001;409:685–690. doi: 10.1038/35055500. [DOI] [PubMed] [Google Scholar]
- Knudsen S. Promoter2.0: For the recognition of PolII promoter sequences. Bioinformatics. 1999;15:356–361. doi: 10.1093/bioinformatics/15.5.356. [DOI] [PubMed] [Google Scholar]
- Laurin NN, Wang SP, Mitchell GA. The hormone-sensitive lipase gene is transcribed from at least five alternative first exons in mouse adipose tissue. Mamm Genome. 2000;11:972–978. doi: 10.1007/s003350010185. [DOI] [PubMed] [Google Scholar]
- McCullagh P, Nelder JA. Generalized linear models. London: Chapman and Hall; 1983. [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier RC, Praz V, Junier T, Bonnard C, Bucher P. The eukaryotic promoter database (EPD) Nucleic Acids Res. 2000;28:302–303. doi: 10.1093/nar/28.1.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherf M, Klingenhoff A, Werner T. Highly specific localization of promoter regions in large genomic sequences by PromoterInspector: A novel context analysis approach. J Mol Biol. 2000;297:599–606. doi: 10.1006/jmbi.2000.3589. [DOI] [PubMed] [Google Scholar]
- Scherf M, Klingenhoff A, Frech K, Quandt K, Schneider R, Grote K, Frisch M, Gailus-Durner V, Seidel A, Brack-Werner R, et al. First pass annotation of promoters on human chromosome 22. Genome Res. 2001;11:333–340. doi: 10.1101/gr.154601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping ME. Spase Bayesian learning and the relevance vector machine. J Machine Learning Res. 2001;1:211–244. [Google Scholar]
- Werner T. Identification and functional modelling of DNA sequence elements of transcription. Brief Bioinform. 2000;1:372–380. doi: 10.1093/bib/1.4.372. [DOI] [PubMed] [Google Scholar]