

Abstract
Secretory vesicles dock at their target in preparation for fusion. Using single-vesicle total internal reflection fluorescence microscopy in chromaffin cells, we show that most approaching vesicles dock only transiently, but that some are captured by at least two different tethering modes, weak and strong. Both vesicle delivery and tethering depend on Munc18-1, a known docking factor. By decreasing the amount of cortical actin by Latrunculin A application, morphological docking can be restored artificially in docking-deficient munc18-1 null cells, but neither strong tethering nor fusion, demonstrating that morphological docking is not sufficient for secretion. Deletion of the t-SNARE and Munc18-1 binding partner syntaxin, but not the v-SNARE synaptobrevin/VAMP, also reduces strong tethering and fusion. We conclude that docking vesicles either undock immediately or are captured by minimal tethering machinery and converted in a munc18-1/syntaxin-dependent, strongly tethered, fusion-competent state.
Keywords: chromaffin, docking, exocytosis, Munc18-1, SNARE
Introduction
Neurons and neurosecretory cells employ conserved mechanisms for regulated secretion of neurotransmitter and hormones. Probably, the most intensively studied gene families involved in these processes are those that encode SNARE proteins (Sollner and Rothman, 1996). Zipping-up of four SNARE domains of the SNARE proteins is recognized as a central molecular mechanism to drive fusion of synaptic vesicles and dense core vesicles (Jahn et al, 2003). Thus, genetic deletion or enzymatic cleavage of SNARE genes/proteins invariably blocks fusion in many different systems (see for review, Rizo and Sudhof, 2002; Toonen and Verhage, 2003), and evidence is accumulating that SNAREs participate in several sequential processes in the exocytosis pathway, leading up to and including the formation of a fusion pore between the vesicle interior and the outside of the cell (Gil et al, 2002; Sørensen et al, 2003; Han et al, 2004; Borisovska et al, 2005). However, the assembly of SNARE complex is not likely to be the first event to occur when secretory vesicles reach their target. Genetic deletion experiments suggest that SNAREs are not necessary to morphologically dock different classes of secretory vesicles at their respective target membranes (Hunt et al, 1994; Broadie et al, 1995; O'Connor et al, 1997; Schoch et al, 2001; Washbourne et al, 2002; Sørensen et al, 2003; Borisovska et al, 2005). Hence, unknown processes distinct from and upstream of SNARE-complex assembly are expected to be involved in capturing arriving vesicles.
One indication for the identity of such upstream machinery was obtained in studies of large dense core vesicles (LDCVs) in neuroendocrine cells. The Sec1/Munc18-like (SM) protein Munc18-1 binds to the t-SNARE syntaxin (Rizo and Sudhof, 2002), and munc18-1 null mutant mice display a complete block of neurotransmission (Verhage et al, 2000). This, together with the reported affinity of Munc18-1 for actin (Bhaskar et al, 2004), prompted us to test its potential role in vesicle docking. Deletion of munc18-1 expression produced a marked defect in LDCVs docking to the plasma membrane in adrenal chromaffin cells (Voets et al, 2001) and somatotrophs of the anterior pituitary (Korteweg et al, 2005), whereas deletion of the SNARE genes SNAP25 or synaptobrevin/VAMP in chromaffin cells did not (Sørensen et al, 2003; Borisovska et al, 2005).
Unfortunately, a more systematic analysis of the protein cascade that orchestrates the reception of secretory vesicles at their target is hampered by the current poor definition of the docking process itself. Docking is typically assessed on the basis of electron micrographs; however, this method does not allow the study of vesicle dynamics in living cells, and thus precludes the identification of different docked states. More recently, total internal reflection fluorescence microscopy (TIRFM) has been exploited to study the dynamics of individual, fluorescent vesicles at the membrane in living cells (Lang et al, 1997; Steyer et al, 1997; Oheim and Stuhmer, 2000; Steyer and Almers, 2001), and it became clear that a complex pattern of vesicle trafficking exists in the submembrane region of secretory cells (Lang et al, 2000; Johns et al, 2001) and synapses (Zenisek et al, 2000). It remains unresolved how this complex pattern relates to the docked and fusion-competent vesicle pools previously characterized by other approaches and which molecular factors are involved.
In order to address these questions, here we have combined TIRFM analysis of submembrane vesicles with molecular manipulations of known docking and fusion proteins, electrophysiological analysis of releasable vesicle pools and ultrastructural morphometry. Thus, we have a complementary set of assays to monitor events from the first arrival of secretory vesicles at the membrane to their final fusion. Given its clear docking phenotype, we used munc18-1 null mutant chromaffin cells as a starting point for molecular manipulations. Using this set of assays, we found that most approaching vesicles do not dock stably, but that two distinguishable tethering states exist and that only the most long-retaining (strongly tethered) state is a prerequisite for fusion. Furthermore, we show that Munc18-1 and the t-SNARE syntaxin are essential for this strongly tethered state.
Results
Docking and secretion defects in the absence of Munc18-1 are rescued by acute expression of the disrupted gene
To be able to study vesicle docking with total internal reflection fluorescence (TIRF) imaging in living cells, we cultured chromaffin cells from munc18-1 null mutant adrenals in which we previously observed a docking defect (Voets et al, 2001). As expected, the docking and secretion defects were preserved in cultured cells, and both could be rescued by acute expression of the disrupted gene (Figure 1A–G). Rescue of the docking phenotype was already complete 4 h after infection. EGFP alone had no detectable effect on vesicle distribution, and the total number of vesicles was similar among all groups (Supplementary Figure S1). The capacitance and amperometric responses to flash uncaging of caged Ca2+ were also restored to control levels within 4 h after reintroduction of the munc18-1 gene (Figure 1E–G and Supplementary Figure S2). All phases of exocytosis (see Voets, 2000) were restored to a similar extent (Figure 1F) and the fusion time constants for different vesicle pools were indistinguishable from control cells (Figure 1G).
Figure 1.
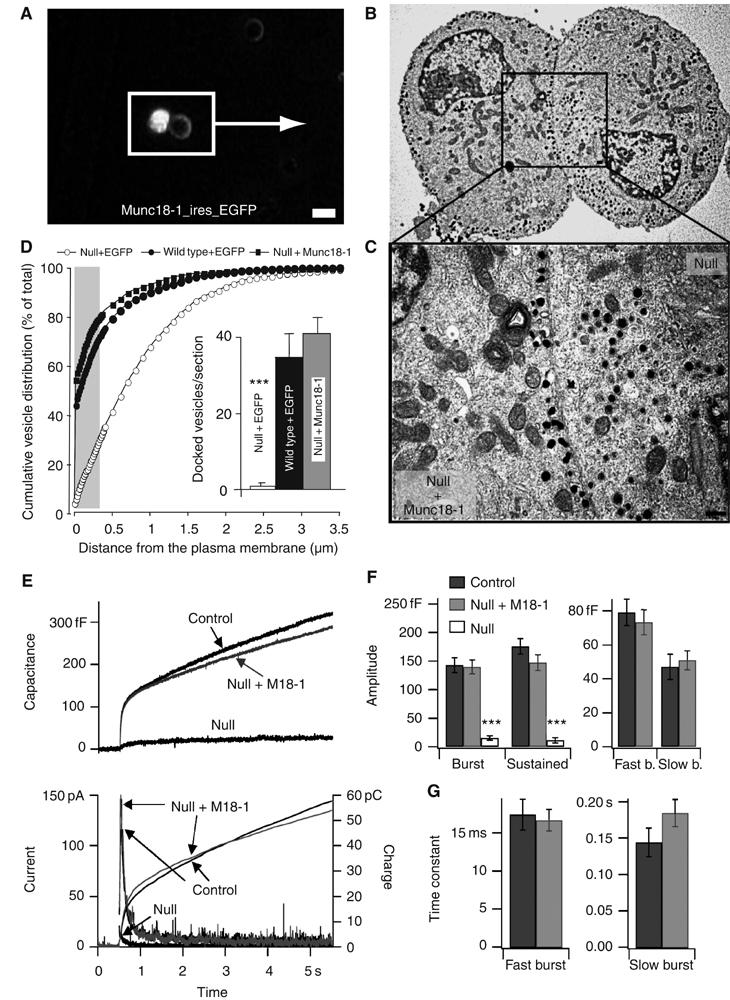
Overexpression of Munc18-1 rescues secretion in munc18-1 null chromaffin cells. (A) Fluorescent image of null mutant cells incubated with SFV munc18-IRES-egfp. The two cells in the white box were retrieved on ultrathin sections in the electron microscope; bar =10 μm. (B) Electron micrographs of the same cells, one of which is infected to induce munc18-1 expression (bright cell in panel A) and shows an increase in docked vesicles. The neighboring uninfected null mutant cell (dark in panel A) shows severely impaired vesicle docking. (C) Magnification of the submembrane region (outlined in panel B); bar =200 nm. (D) Cumulative plots of vesicle distribution in null mutant, wild type cells and null cells after acute overexpression of munc18-1. Inset shows average number ±s.e.m. of morphologically docked vesicles for each experimental condition. The number of vesicles was quantified in the following number of cells (n) and animals (N): null+EGFP: n=19, N=4; wild type+EGFP: n=20, N=4; wild type+Munc18: n=20, N=4 (***P<0.001, Student's t-test with N=4). (E) Secretory responses in munc18-1 heterozygote cells (control, n=36 cells, N=8 animals), in munc18-1 null cells (null, n=21, N=4) and in null cells overexpressing munc18-1 (null+M18-1, n=21, N=5). Secretion was elicited by flash photolysis of caged-Ca2+ (at arrow), which resulted in similar intracellular [Ca2+] steps in all experimental groups (see Supplementary Figure S2). Secretion was assayed simultaneously with membrane capacitance measurements and amperometry, which measure the increase in plasma membrane area and the liberation of catecholamines, respectively, as a result of vesicle fusion. Averaged traces are shown; the individual amperometric spikes are therefore barely recognizable. The amperometric current was integrated over time to obtain the cumulative charge, which mirrors the capacitance increase except for an additional diffusional delay (right ordinate axis); (F) Amplitudes of the different kinetic components of the flash responses. Left: The capacitance increase occurring within the first 0.5 s after the flash and during the following 4.5 s. Right: Following kinetic analysis, the amplitudes of the fast burst component (representing the fusion of the readily releasable vesicle pool) and the slow burst component (representing the fusion of the slowly releasable vesicle pool) are shown. ***P<0.001 (Mann–Whitney). (G) Time constants for fusion of the fast and slow burst components of the flash responses.
Vesicle docking and secretion varies with the amount of Munc18-1 expressed
Given its confirmed role in docking, we asked if the extent of docking and secretion varies as a function of the cellular Munc18-1 level, exploiting the fact that munc18-1 heterozygous null mutant mice have a reduced munc18-1 expression (in adrenals exactly 50% reduction; Voets et al, 2001). Conversely, we used Semliki Forest viral overexpression of munc18-1-IRES-egfp in wild type cells. The extent of munc18-1 overexpression was 10- to 20-fold higher than endogenous expression, based on semiquantitative analysis of immunocytochemical stainings (data not shown). Reduced expression of Munc18-1 in chromaffin cells from heterozygous mice led to a proportional reduction in morphologically docked vesicles (Figure 2A–C). Overexpression of Munc18-1, on the other hand, produced a significant increase in the number of vesicles docked at the target membrane (Figure 2A–C). The total number of vesicles remained unaltered (Supplementary Figure S1). Hence, under- or overexpression resulted in parallel changes in the number of docked vesicles.
Figure 2.
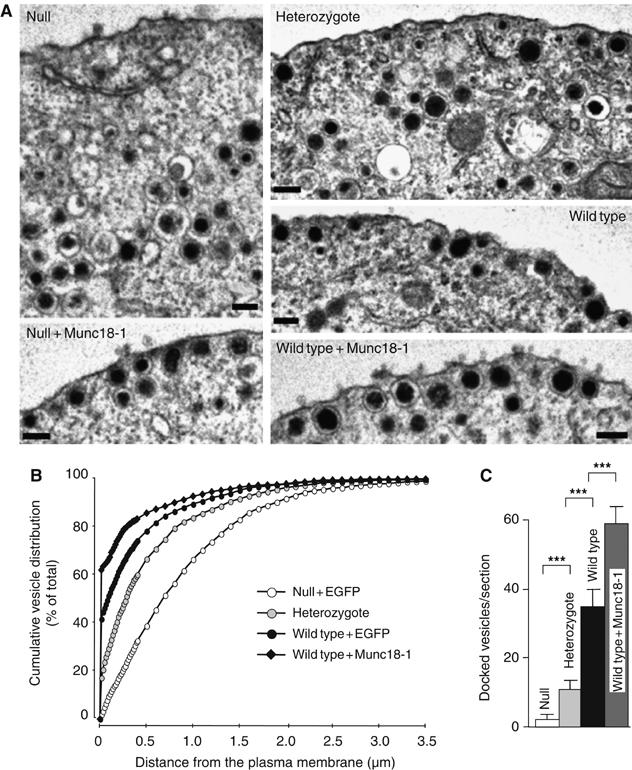
The number of docked vesicles is influenced by Munc18-1 expression level. As the expression level of Munc18-1 rises, the number of morphologically docked vesicles at the plasma membrane increases. (A) Electron micrographs of the submembrane area of munc18-1 null mutants overexpressing egfp or munc18-1, munc18-1 heterozygotes, and wild type cells overexpressing egfp or munc18-1. Bars, 100 nm. (B) Average number ±s.e.m. of morphologically docked vesicles for each experimental condition (***P<0.001, Student's t-test with N=4). (C) Cumulative plots of vesicle distribution for each experimental condition (except null+Munc18-1, which is shown in Figure 1D, and has the same distribution as wild type+EGFP). The number of vesicles was quantified in the following number of cells (n) and animals (N): null+EGFP: n=19, N=4; heterozygotes: n=20, N=4; wild type+EGFP: n=20, N=4; wild type+Munc18: n=20, N=4. Neither over- nor underexpression of Munc18-1 affected the total number of secretory vesicles in chromaffin cells (Supplementary Figure S1).
Although docking was reduced in the heterozygous chromaffin cells, the secretory responses to flash photolysis of caged-Ca2+ were close to control responses in amplitude (Figure 3A–C and Supplementary Figure S3). Kinetic analysis revealed that the sustained component was significantly decreased in the heterozygotes (Figure 3B), but this reduction was rather small. However, munc18-1 overexpression markedly increased secretion in capacitance measurements and amperometry (Figure 3D–F). Increases were observed in both burst and sustained phases of release (Figure 3E), suggesting that an early secretory step was potentiated. Thus, the number of morphologically docked and, to some extent, fusogenic vesicles varies with the amount of Munc18-1 expressed. This identifies Munc18-1 not only as an essential docking factor, but also as a rate-limiting, positive factor in the secretory cascade.
Figure 3.
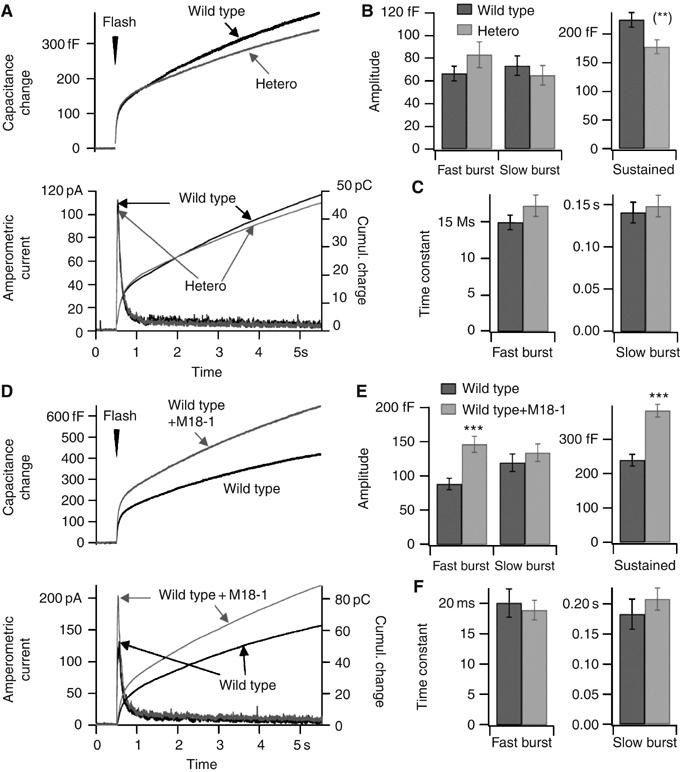
Modulation of secretion by gene dose: exocytosis in munc18-1 heterozygote cells and in wild type cells overexpressing Munc18-1. (A) Secretory responses in munc18-1 heterozygote cells (hetero, n=60, N=5) and wild type cells (wild type, n=58, N=5). For explanation, see the legend to Figure 1E and F and Supplementary Figure S3. (B) Left: Amplitudes of the fast and the slow burst components. Right: Sustained rate of secretion, measured between 0.5 and 5 s after the flash stimulation. (**) Mann–Whitney test: P<0.01. (C) Time constants for fusion of the fast and slow burst components of the flash responses. (D) Secretory responses in non-infected wild type cells (n=50, N=7) and in wild type cells overexpressing Munc18-1 (wild type+Munc18-1, n=52, N=7). (E, F) Amplitudes and time constants of different kinetic components of the flash responses; as in panels C and D. ***P<0.001 (Mann–Whitney).
Three docking states can be distinguished, two of which are Munc18-1 dependent
To investigate docking in living cells, we used TIRF imaging of secretory vesicles labeled with neuropeptide Y fused to Venus (NPY-Venus; Nagai et al, 2002). Within the layer illuminated by the evanescent wave (d1/e=120±10 nm in our setup), many secretory vesicles were detectable in all genotypes (Figure 4A–C), but the total number was two-fold lower in the munc18-1 null mutant (0.13±0.01 versus 0.24±0.03 vesicles/μm2 in the wild type, P<0.005; Figure 4D). This reduction is in accordance with the morphological docking defect observed in the electron microscope (see Figure 1D, the shaded area indicates the 3d1/e TIRF illuminated volume). Acute expression of munc18-IRES-NPY-Venus in null cells completely restored the number of detected vesicles (0.22±0.03 vesicles/μm2; Figure 4C and D).
Figure 4.
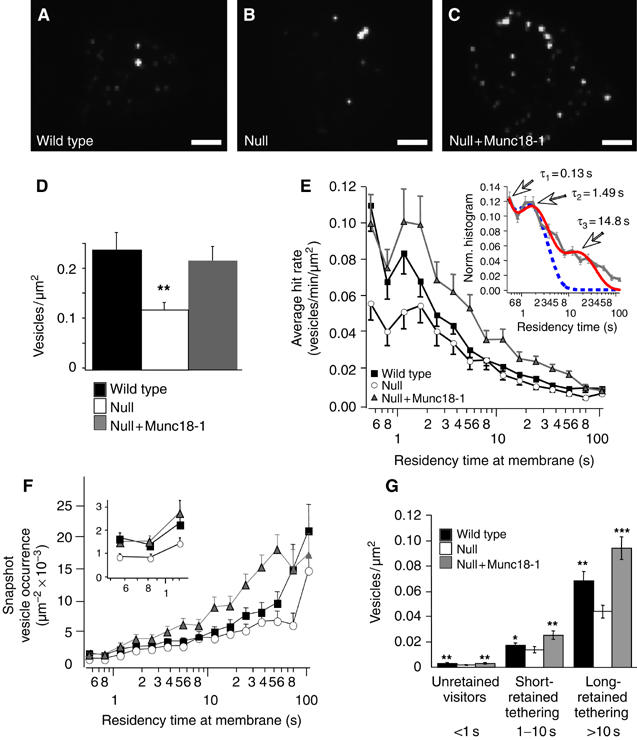
Dynamics of NPY-Venus-labeled vesicles in the submembrane region. Appearance of footprints with NPY-Venus-labeled granules in wild type (A), null (B) and null mutant cells expressing munc18-1-IRES-NPYVenus (C). Scale bar is 5 μm. Density of granules per unit area was noticeably smaller in null mutant cells. (D) Quantification of the density of labeled vesicles at the footprint of chromaffin cells in different genotypes, estimated from the average projection images (A–C). The number of vesicles was significantly smaller for null mutant cells than the other genotypes. (E) Lifetime distribution of vesicles hitting the plasma membrane during 180 s (600 frames) observations at 3.3 Hz. Frequencies are normalized to membrane unit area of the cell's footprint and observation time and logarithmically binned. Inset: Fitting the normalized residency time histogram of munc18-1-overexpressing cells with models of two (dotted blue line) or three (solid red line) exponentially distributed lifetime states suggests at least three different residency states: visitors (lifetime <1 s), short-retained vesicles (1–10 s) and long-retained vesicles (>10 s). In both genotypes, most vesicles only visit the TIRF plane (visitors) but significantly less do so in the null mutant compared to wild type cells. The long-retained state has very low frequency, and only becomes apparent owing to its reduction in the null mutant cells (see also panels F and G). Total number of vesicles is n=2997 (N=35), 2398 (N=30) and 3536 (N=34) for wild type, null and null+Munc18-1 cells, respectively. (F) Average vesicle abundance at any given time during image acquisition shows the significant contribution of long-retained vesicles to a snapshot analysis of docked vesicles and supports the existence of three distinct docking states. The inset zooms in on the short residency times corresponding to the unretained visitors (<1 s). (G) Average vesicle abundance at any given time during image acquisition grouped according to their residency times at the membrane (unretained: <1 s; short-retained and long-retained tethered: 1–10 and >10 s, respectively). The majority of vesicles found on TIRF images belonged to the tethered states (residency times between 1 and 180 s). * and ** indicate statistically significant differences between null and wild type (or null+Munc18-1) at P<0.05 and 0.01 levels (Wilcoxon test). Note that t-test does not detect differences between null and wild type for the short-retained tethering bin (P>0.24).
To analyze the dynamic behavior of individual vesicles, we acquired TIRF images with 0.3 s interval for 3 min, and measured the number of vesicles that appeared in the TIRF field and their residency times at the membrane (see Materials and methods). To show all events in one histogram, the residency times were logarithmically transformed and their distribution was displayed using logarithmically increasing bin sizes (Figure 4E). By logarithmic transformation, exponentially distributed residency or lifetimes result in a skewed distribution with a peak at the time constant of each individual lifetime state (Sigworth and Sine, 1987). Different states can be distinguished as distinct peaks in the distribution. The first state, with a lifetime shorter than 1 s most likely represents vesicles that move in and out of the TIRF plane unretained by tethering forces at the plasma membrane (visitors). The majority of arriving vesicles in all genotypes belonged to this category but the number of visitors in null mutant cells was significantly lower compared to wild type. A second state (between 1 and 10 s in Figure 4E) is characterized by a distinct, longer lifetime and therefore indicates a form of minimal retention of these vesicles at the target: short-retained, or weak tethering. A third, more long-retained state is suggested by the pronounced tail in the residency time distributions (>10 s in Figure 4E). Fitting of the normalized histograms with two or three exponentially distributed lifetime states indeed supports the existence of three states with average lifetimes of 0.13 (τ1), 1.49 (τ2) and 14.8 (τ3) s (Figure 4E, inset). Interestingly, the distribution in munc18-1 null mutant cells suggested that especially the visitors and the long-retained vesicles occurred less frequently than in control cells (cf. Figure 4F).
To correlate the residence time distribution in Figure 4E with our (snapshot) electron microscopical analysis of fixed cells, we calculated the average abundance of every vesicle bin at any given time during the image recording (vesicle snapshot occurrence, see Materials and methods; Figure 4F). Based on the histogram and curve fit in Figure 4E, vesicles were grouped into three classes (visiting vesicles with residence times <1s; short-retained tethered, between 1 and 10s; and long-retained tethered >10 s). In Figure 4G, the average abundance of these three groups at any time is plotted for the three genotypes. Whereas the visiting vesicles dominate the residence time analysis (Figure 4E), the long-retained tethered vesicles dominate in single snapshots (Figure 4F), as applies to electron micrographs. Changes in cellular Munc18-1 level changed the ratio between the three groups. The long-retained tethered state was reduced by 1/3 in the absence of Munc18-1 and rescued by acute munc18-1 expression. Despite the fact that relatively few vesicles are captured in this state, more than 75% of all vesicles seen at the membrane footprint in wild type cells belonged to this class, owing to their long lifetime (Figure 4G). Transient occurrences (unretained visitors, <1 s) were also significantly decreased in munc18-1 null mutant cells (45% compared to wild type). Hence, the number of vesicles arriving per unit of time was reduced (Figure 4E), suggesting a distinct aspect of munc18-1 function in the delivery of vesicles to the target. This phenotype was again rescued by acute expression of the disrupted gene (Figure 4G). Together, these data identify different docking states. Most vesicles move in and out of the TIRF plane apparently without being retained, but some vesicles are retained for shorter or longer times by local tethering forces at the target. Munc18-1 most strongly affects vesicle delivery rate and long-retained tethering. The munc18-1-dependent changes in morphological docking in electron micrographs (Figures 1 and 2) may be explained by the combination of these changes in delivery rate and tethering.
Autocorrelation analysis of vesicle movement reports tethering forces
The decrease in long-retained vesicles in the absence of Munc18-1 may be influenced by the reduced vesicle delivery rate. To find independent evidence for Munc18-1's role in long-retained tethering, we examined the jittering movement of long-retained vesicles at the target membrane. Owing to the exponential decay of the evanescent wave, relative changes in the position of fluorescent vesicles perpendicular to the target (i.e. Z-position) can be quantified with extraordinary precision by analyzing changes in fluorescence intensity (Figure 5A). It was previously shown by autocorrelation analysis of such jittering movement that vesicles near the target show a tendency to reverse the direction of movement within a characteristic time τ (Johns et al, 2001). This change in the direction of axial movement is represented by a negative component in averaged autocorrelation functions (ACFs) of fluctuations in relative Z-velocity (ΔZ−ACF, see Materials and methods) and discriminates this behavior from pure diffusion or uncorrelated noise sources. Tethering or restricting forces are expected to reverse vesicles that start to diffuse away from the target membrane and thus result in a negative component in ΔZ-ACF analysis. We indeed observed a negative component in ΔZ-ACF in the 0.2–1 s range (Figure 5B), confirming the previously reported tendency of vesicles near the target to reverse direction (Johns et al, 2001). Immobile controls (40-nm fluorescent beads and vesicles in fixed cells) did not show this negative component, which excludes system artifacts (Figure 5B). We defined a single parameter to quantify this negative persistence of autocorrelation (NPA) by summing the first two points of the ΔZ-ACF that allowed us to compare different populations of vesicles and cells with different genotypes. Both theoretically and practically (Qian et al, 1991), the ACF approaches zero for larger τ and therefore this specific time window is assigned to the NPA. The NPA for live cells is a factor 100 larger than for fixed cells and fluorescent beads (Figure 5C) and the NPA increases when vesicles exhibit behavior distinct from random movement (Johns et al, 2001, and our computer simulations, not shown).
Figure 5.
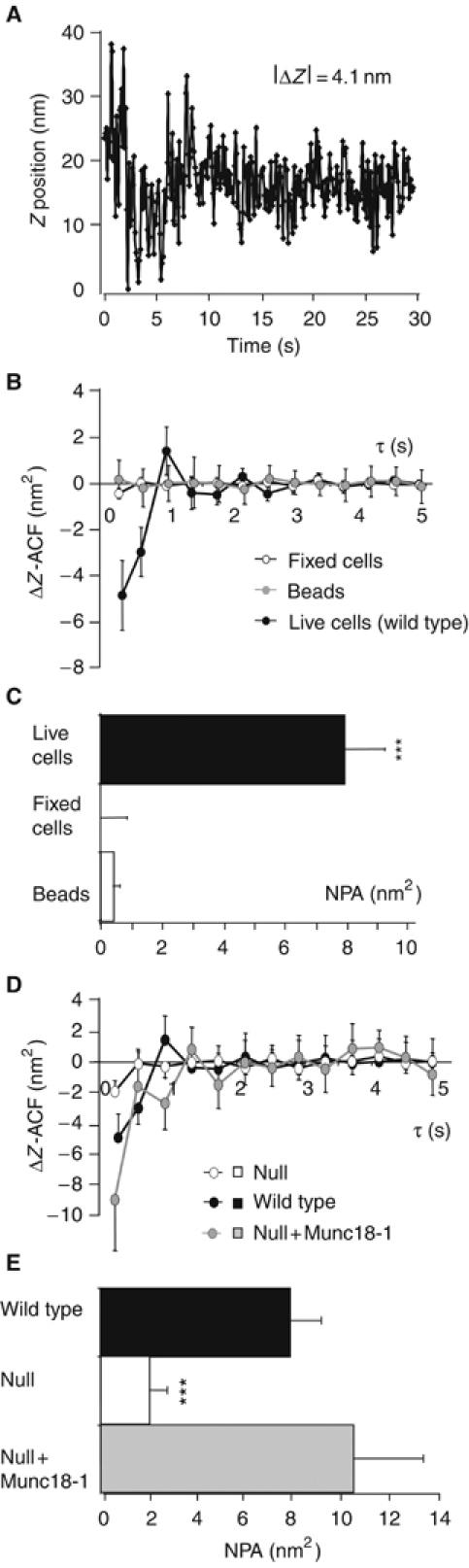
Autocorrelation analysis reports tethering forces that are lost in the absence of Munc18-1. (A) Example of vesicle trajectory perpendicular to the plasma membrane (relative Z-position; see Materials and methods) during 30 s image acquisition at 30 Hz. (B) Velocity ACF (ΔZ-ACF) of vesicle movement in wild type chromaffin cells compared to fixed cells and fluorescent beads. The negative points in the beginning of the ACF reflect changes in the direction of Z-movement. These changes are absent in fixed cells and fluorescent beads. (C) The NPA for fixed cells, beads and live cells transfected with NPY-Venus. The absolute value of negative amplitude rises when the restricted movement of an object can be reliably resolved from the uncorrelated fluorescence noise. The NPA was calculated for 113 vesicles in fixed cells, 127 beads and 244 vesicles in wild type live cells. (D) ΔZ-ACF of wild type and munc18-1 null mutant vesicles reveals an increased freedom in vesicle movement in the absence of Munc18-1, which results in a more than eight-fold reduction of the negative ACF component in these cells. (E) Munc18-1 introduction on the null background rescues the NPA to wild type levels. *** indicates statistically significant differences as compared to wild type or rescued cells at P<0.001 (Wilcoxon test).
Normal tethering forces are lost in the absence of Munc18-1
Subsequently, we performed the autocorrelation analysis in munc18-1 null mutant cells. Strikingly, the negative component in the ΔZ-ACF was strongly reduced (Figure 5D) and the NPA was approximately four-fold lower than in wild type cells (Figure 5E). This may in principle indicate very strong and inflexible binding to the target, as in fixed cells (see Figure 5C), resulting in a shift of the negative correlation to much shorter correlation times, but more likely indicates the absence of strong tethering forces, which is consistent with the lifetime analyses (Figure 4F and G). Rescue of null mutant cells with Semliki Forest particles expressing munc18-1-IRES-NPY-Venus fully restored the NPA (Figure 5E).
Fluorescent vesicles in the null mutant were on average further away from the target than in wild type cells (see Figures 1B–D and 4D). Because vesicle movement is progressively restricted at shorter distances to the target (Oheim and Stuhmer, 2000; Johns et al, 2001), we examined whether differences in average distance could explain differences in NPA. We found that also for vesicle populations at a similar distance to the target (i.e. of similar fluorescence intensity), the NPA was significantly smaller in mutant cells than in wild type cells and rescued null cells (not shown). Hence, the lifetime and autocorrelation analysis both indicate that tethering mechanisms that restrict vesicle movement near their target are absent in the munc18-1 null mutant.
Munc18-1-dependent reduction of the actin cortex may cause increased vesicle delivery
Chromaffin cells display a distinct submembrane actin cytomatrix, and its removal is known to increase morphological docking in chromaffin cells (Vitale et al, 1995). Munc18-1 is present at the plasma membrane in a discrete punctate pattern that partially overlaps with the F-actin cortex (data not shown; Tsuboi and Fukuda, 2006) To examine whether the observed decrease in vesicle delivery to the membrane in the absence of Munc18-1 (Figure 4E–G) is a result of interference of Munc18-1 with the actin cortex, we stained cells (wild type, null, overexpression) for F-actin using fluorescently labeled phalloidin (Figure 6A). As a control for munc18-1 overexpression, also wild type cells expressing egfp only were examined. The actin cortex was thinner and fenestrated in wild type and munc18-1-overexpressing cells compared to munc18-1 null cells. Quantification revealed a marked reduction of intact F-actin in the presence of Munc18-1, which scaled with the expression level (Figure 6A, bar diagram).
Figure 6.
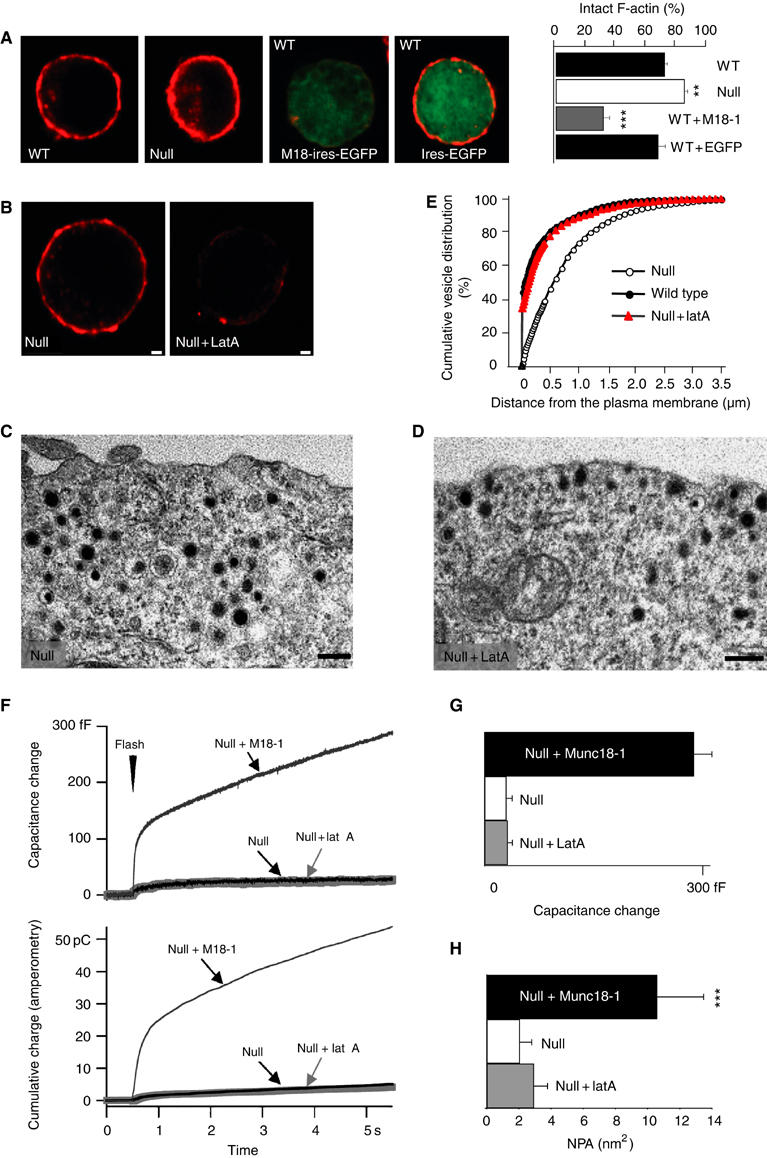
Lat A increases the number of morphologically docked vesicles, but does not rescue secretion and NPA in munc18-1 null mutant cells. (A) Phalloidin (red) staining of wild type (WT), munc18-1 null mutant (null) and WT cells infected with munc18-1-IRES-egfp or IRES-egfp only. The bar graph shows the quantification of the intactness of the phalloidin ring for the different genotypes (see Materials and methods) in the following number of cells (n) and animals (N) for each condition: wild type: n= 118, N=12; wild type+EGFP: n=73, N=4; wild type+Munc18: n=91, N=5; null: n=112, N=14. ** indicates statistically significant difference, P<0.01 and ***P<0.001. Bars, 1 μm. (B) Equatorial optical confocal sections of munc18-1 null mutant cells stained with rhodamine-phalloidin before (null) and after incubation with Lat A (null+Lat A). Bars, 1 μm. (C, D) Electron micrographs of undocked (C) and docked vesicles (D) at the plasma membrane in munc18-1 null cells without or with application of Lat A, respectively. (E) Cumulative plots of vesicle distribution in munc18-1 null cells with or without Lat A and wild type cells. The number of vesicles was quantified in the following number of cells (n) and animals (N) for each condition: null: n=23, N=6; null+Lat A: n=20, N=3; wild type: n=26, N=6. (F) Secretory responses of munc18-1 null cells (null, n=30), and null cells after treatment with Lat A (null+Lat A, n=32). For comparison, we included the secretory response of null cells rescued with Munc18-1 expression from Figure 1E and F. (G) The capacitance increase occurring within the first 0.5 s after the flash and during the following 4.5 s. (H) Lat A treatment fails to restore the NPA in munc18-1 null mutant cells, despite the fact that the number of vesicles near the target membrane was similar in the treated null mutant and the wild type/rescued cells. The NPA was calculated for 137 vesicles in null mutant cells, 155 vesicles in null mutant cells treated with Lat A and 135 vesicles in rescued cells. *** indicates statistically significant differences between rescued versus null and null plus Lat A at P<0.001 (Wilcoxon test).
Actin cytoskeleton interference rescues morphological docking but not tethering
As mentioned above, it is conceivable that the Munc18-1-induced increase in functional docking (long-retained, large NPA) is merely a consequence of increased vesicle delivery to the membrane. Therefore, we tested whether actin removal alone, using the actin depolarizing drug Latrunculin A (Lat A), is sufficient to restore functional docking in munc18-1 null cells. Lat A disrupted the submembrane cytomatrix (Figure 6B) and allowed more vesicles in the submembrane area (Figure 6C and D), resulting in a similar amount of morphologically docked vesicles as in untreated wild type cells (Figure 6E), whereas the total number of vesicles remained unaffected (Supplementary Figure S1). Hence, Lat A rescued the morphological docking phenotype in the null mutant cells. However, vesicle fusion remained severely impaired and no significant enhancement over the marginal secretion in the null mutant was observed in capacitance measurements and amperometry (Figure 6F and G). Moreover, the reduced NPA in the null mutant was also not rescued by Lat A treatment (Figure 6H). Thus, although the morphological docking phenotype in munc18-1 null mutant cells was completely restored and a normal number of vesicles were morphologically docked, Lat A was completely ineffective in restoring stringent tethering mechanisms and the exocytosis of these vesicles in the absence of Munc18-1.
The target SNARE syntaxin, but not vesicle SNAREs, is necessary for normal tethering
Munc18-1 is a hydrophilic protein with no inherent affinity for membranes. It is unlikely that this protein in itself is capable of tethering lipid vesicles at the target. In order to identify the additional factors involved in tethering, we analyzed NPAs in chromaffin cells deficient for several candidate proteins by proteolytic cleavage using viral overexpression of clostridial neurotoxin light chains. Deletion of the vesicle and target SNARE proteins using tetanus toxin (TeNT) and botulinum toxin C (BoNT-C), respectively, completely abolished secretion as observed before in many systems (data not shown; Jahn and Niemann, 1994). In line with previous observations in squid giant terminals (Hunt et al, 1994; O'Connor et al, 1997), deletion of synaptobrevin/VAMP, but not syntaxin, led to an increase in the total number of vesicles (Supplementary Figure S1). As suggested previously, this increase may reflect an increased vesicle biogenesis and/or half-life. With equal numbers of vesicles in each group, we found that deletion of syntaxin, but not synaptobrevin/VAMP, reduced the NPA significantly (Figure 7B). Given the fact that BoNT-C cannot cleave already assembled SNARE-complexes (Hayashi et al, 1994), the reduction of NPA upon syntaxin cleavage may be an underestimation. Hence, target SNAREs are necessary for tethering of docked vesicles, but the vesicle SNARE synaptobrevin/VAMP is only required for subsequent steps leading to the final fusion. Interestingly, cleavage of syntaxin did not significantly change the lifetime distribution compared to wild type cells (Supplementary Figure S4). This suggests that Munc18-1 affects the vesicle hit rate independently of its interaction with syntaxin.
Figure 7.
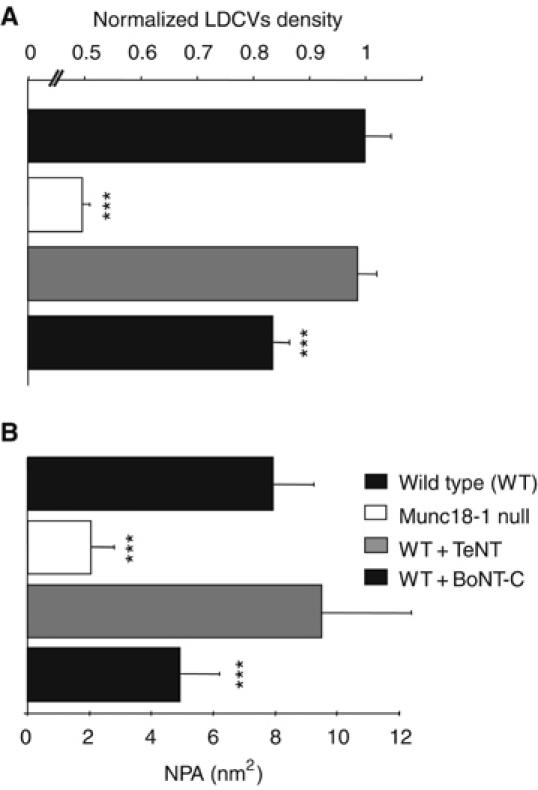
tSNARE but not vSNARE function is necessary for normal tethering forces. (A) Number of vesicles per unit area detected in the TIRF plane after BoNT-C but not TeNT light chain overexpression was decreased to approximately 83% of wild type. The vesicle density was normalized to littermate wild type cells. *** indicates P<0.001 (Student's t-test). (B) NPA calculated for vesicles in wild type (n=244) and munc18-1 null cells (n=137) and in wild type cells transfected with wither tetanus neurotoxin (TeNT, n=144) or botulinum neurotoxin-C (BoNT-C, n=142) light chain. *** indicates significant differences at P<0.005 compared to wild type (Wilcoxon test).
Discussion
In this study, we examined the molecular events that harbor approaching secretory vesicles at their target. We provide evidence for at least three distinct docking states (Figure 8) and show that movement jittering analysis in living cells can be used to fingerprint a tethered state essential for vesicle fusion. Genetic deletions, proteolytic cleavage and pharmacological manipulations implicate Munc18-1 and the t-SNARE syntaxin in specific stages of vesicle docking.
Figure 8.
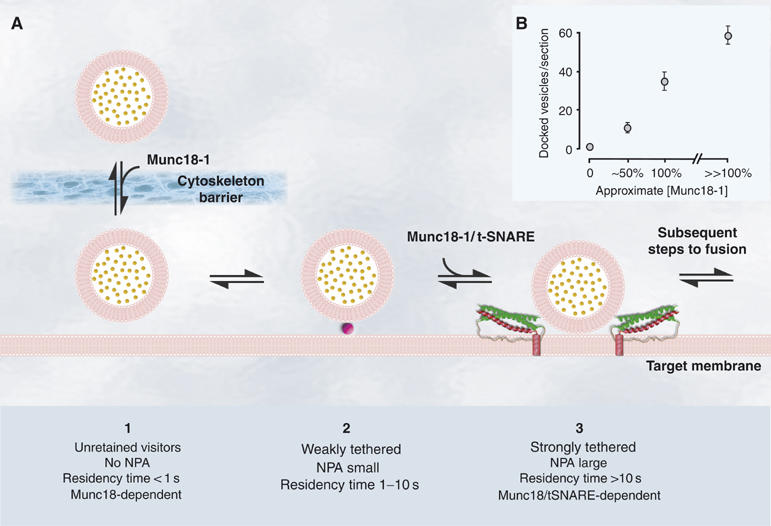
Cartoon of different proposed docking states and their main characteristics and the correlation between Munc18-1 expression levels and docking. (A) Cartoon showing the main characteristics of the three distinct docking states. Purple ball indicates as yet unknown Munc18-1- and syntaxin 1-independent minimal tethering machinery. Red spiral represents t-SNARE syntaxin 1 α-helix and green spirals SNAP-25 α-helices. (B) The number of morphologically docked vesicles was plotted as a function of the Munc18-1 expression level as determined by quantitative analysis of protein expression levels (data not shown) of the null mutant (0%), munc18-1 heterozygote (50%), wild type (100%) and wild type+Munc18-1 (>100%). Note that the horizontal axis is not linear.
Using TIRF imaging, we were able to study single vesicle behavior close to the membrane. We introduced residency timing as a means to discriminate different docking states. This analysis showed that the majority of vesicles appearing in the TIRF plane were visible only very transiently (less than 1 s), suggesting that they were not retained or tethered by the target (unretained visitors; see Figure 8A). Tethering mechanisms only captured a small number of arriving vesicles. Thus, successful capture of arriving vesicles is a relatively rare event. Among the vesicles that were captured at the target, half had characteristic residency times between 1 and 10 s (short-retained, weakly tethered; Figure 8A). A third, long-retained state appeared as a long tail in the residency time diagram, which could not be properly fitted by assuming only two residence states (Figure 4E, inset). The existence of this state was confirmed by two subsequent observations. First, Lat A treatment rescued morphological docking in munc18-1 null cells, but the morphologically docked vesicles could not fuse and had an unaltered, low NPA, indicating the existence of two distinct tethered states. Second, deletion of munc18-1 severely decreased the number of vesicles with residence times longer than 10 s, whereas the number of vesicles staying between 1 and 10 s at the membrane was only marginally affected. For these reasons, we propose to discriminate three classes of vesicles near the target: (a) unretained visitors, (b) short-retained or weakly tethered and (c) long-retained or strongly tethered (see Figure 8). The latter state may actually consist of multiple states that cannot be resolved with the current methodology. Although few arriving vesicles are captured into the strongly tethered state, owing to their long lifetime they make a large contribution to the number of vesicles present at the membrane. The secretion capacity was very low in munc18-1-deficient cells, whereas weak tethering was only marginally affected in these cells. Therefore, we conclude that weak tethering cannot support secretion and that the infrequently occurring, strongly tethered state is a prerequisite in preparation for fusion. Weak tethering may represent unknown, minimal tethers, which are stabilized and/or converted into strong tethers by the actions of Munc18-1.
The combination of munc18-1 null mutation and Lat A treatment to depolymerize actin indicated that a considerable discrepancy can occur between morphological and functional docking. Upon dissociation of the submembrane actin cytoskeleton in the munc18-1 null mutant cells, many vesicles reached the target and electron micrographs were indistinguishable from wild type cells, but the NPA was still very low and secretion remained fully inhibited. These data indicate that morphological docking is not sufficient to support secretion, but that Munc18-1-dependent strong tethering is a prerequisite in preparation for fusion. Moreover, this also indicates that morphological assessment of docking may not reveal important functional docking defects.
In addition to its role in establishing a strongly tethered state, Munc18-1 appears to be involved also in regulating the vesicle delivery rate (Figure 4G). In a linear scheme (Figure 8), this implies that Munc18-1 will affect the size of the total population of vesicles close to the membrane, which contributes to an explanation of the docking phenotype in munc18-1 null cells observed in electron micrographs (Figure 1). This aspect of Munc18-1 function may relate to its proposed association with the cytoskeleton (Bhaskar et al, 2004), which may influence cytoskeletal stability or penetrance and hereby vesicle delivery. Indeed, we found that the cortical actin ring was thicker in munc18-1 null cells, whereas Munc18-1 overexpression thinned and fenestrated this ring. These results demonstrate that there are at least two functions of Munc18-1 in secretion: to regulate overall vesicle delivery, probably by influencing the actin cytoskeleton, and to induce the high-affinity tethered state fingerprinted by a high NPA.
The collective consequence of the two functions of Munc18-1 in chromaffin cells is that the cellular Munc18-1 level dictates morphological docking (summarized in Figure 8B) and the secretion capacity: in the null mutant, the distribution of vesicles did not significantly deviate from a random distribution and secretion was blocked, whereas upon overexpression docking increased concomitantly with an increased secretion capacity. Together, these data identify Munc18-1 as an essential, positive and rate-limiting regulator of docking. Both functions of Munc18-1 are likely required to obtain this apparently simple relationship between Munc18-1 level and secretion capacity.
Munc18-1 is necessary for high-affinity tethering, but the protein is hydrophilic with no inherent affinity for membranes. The target SNARE syntaxin is present at the membrane in embryonic chromaffin cells (Supplementary Figure S5) and the effect of BoNT-C on NPA identifies this t-SNARE as an important component for these tethering mechanisms. The target SNARE proteins, syntaxin and SNAP25, were originally proposed to account for vesicle docking through complex assembly with the vesicular SNARE (Sollner et al, 1993), but a large number of interference studies have subsequently concluded, on the basis of electron microscopy, that this SNARE complex assembly occurs downstream of morphological docking (Hunt et al, 1994; Broadie et al, 1995; O'Connor et al, 1997; Schoch et al, 2001; Washbourne et al, 2002; Sørensen et al, 2003; Borisovska et al, 2005). Although our data are in agreement with these studies, we identify a step immediately downstream of morphological docking (high-affinity tethering) as being the first instance where SNARE proteins are involved. We propose two alternative working models for tethering principles. First, vesicles may dock on a docking platform formed by the Munc18-1/syntaxin dimer, as we proposed previously (Voets et al, 2001). In this model, the vesicle participant in docking remains elusive. Second, target SNAREs (syntaxin and SNAP25) may form a docking platform that interacts with vesicular synaptotagmin (Rickman et al, 2006), possibly in a redundant combination with vesicular synaptobrevin/VAMP. In this case, Munc18-1 may play a role in setting up the t-SNARE dimer for binding of the vesicular protein.
Materials and methods
Cell culturing and viral constructs
Embryonic mouse chromaffin cells were isolated and cultured as described (Sørensen et al, 2003). Acute expression of heterologous genes was induced using infection with Semliki Forest virus (SFV; Ashery et al, 1999). Genes of interest were expressed from a bicistronic message containing a Poliovirus internal ribosomal entry site and either EGFP or Neuropeptide Y fused to Venus (NPY-Venus; Nagai et al, 2002). Experiments were performed between 4 and 8 h after infection. SFV munc18-1-IRES-egfp and SFV-egfp have been described before (Voets et al, 2001). BoNT-C light chain was a kind gift from T Galli (INSERM, Paris, France). TeNT light chain was a kind gift from T Südhof (Howard Hughes Medical Institute, Dallas, TX, USA). All constructs were verified by DNA sequencing and infection with BoNT-C or TeNT particles resulted in complete loss of high potassium-evoked release after 6 h (not shown).
Electron microscopy
Chromaffin cells from munc18-1-deficient mice, heterozygous or wild type mice (E18) were plated on collagen-coated (32 μg/ml; Sigma) coverslips (Cellocate, Eppendorf, Germany) and infected (DIV2) with SFV-egfp, BoNT-CLC-IRES-egfp, TeNTLC-IRES-egfp or munc18-1-IRES-egfp. Cells were observed under a fluorescence microscope 4–8 h after infection and the location of infected/wild type cells was mapped. In some experiments, munc18-1-deficient chromaffin cells were incubated for 6 min in 1 μM Lat A (Molecular Probes) in culture medium before fixation. After fixation and embedding, cells of interest were selected by observing the flat Epon-embedded cell monolayer (containing the CELLocate print) under the light microscope, and mounted on pre-polymerized Epon blocks for thin sectioning. Ultrathin sections (∼90 nm) were cut parallel to the cell monolayer and collected on single-slot, formvar-coated copper grids, and stained in uranyl acetate and lead citrate. For each condition, the distribution of secretory vesicles was analyzed (in three different grids per animal) in a JEOL 1010 electron microscope. Distances from the granule membrane to the plasma membrane were measured (Voets et al, 2001) on digital images taken at × 20 000 magnification using analySIS software (Soft Imaging System, Germany). Secretory vesicles were recognized by their round, dense core and had a diameter of approximately 90 nm (see Supplementary data for details).
Capacitance measurements and amperometry
Whole-cell patch clamp, membrane capacitance measurements, carbon fiber amperometry, ratiometric intracellular [Ca2+] measurements and flash photolysis of caged Ca2+ were performed as described previously (Nagy et al, 2002). Data are presented as mean±s.e.m. The nonparametric Mann–Whitney test was used to test statistical significance of differences between experimental groups (see Supplementary data for details).
Rhodamine-phalloidin staining
Wild type cells infected with SFV IRES-egfp, or munc18-1-IRES-egfp and non-infected wild type and munc18-1 null mutant cells were fixed in 4% paraformaldehyde. In addition, munc18-1 null mutant cells were incubated for 6 min in 1 μM Lat A in culture medium preceding fixation. Filamentous actin was stained with 0.25 U/ml rhodamine-phalloidin (Molecular Probes) in PBS for 40 min. Images were acquired on a Zeiss LSM510 using identical settings for all genotypes. Intactness of the F-actin cortex was measured in equatorial confocal slices as the percentage of gaps in a circular region covering the F-actin ring with a fixed threshold for all genotypes.
TIRF microscopy
The evanescent field decay constant (1/e depth) was measured with surface-coated fluorescent beads of known geometry (Mattheyses and Axelrod, 2002) and was found to be d=120 nm. Cells were imaged in Ringer solution (in mM: 147 Na, 2.8 K, 5 Ca, 1 Mg, 10 HEPES, 10 glucose). For Latrunculin experiments, 1 μM Lat A (Calbiochem, Schwalbach, Germany) was applied for 10 min. Osmolality of all solutions was 305–310 mOsm, pH 7.3. Image acquisition for lifetime and ΔZ-ACF analyses were performed at 3.3 and 30 Hz respectively (see Supplementary data for details).
Imaging data analysis
Stacks of acquired images were analyzed off-line using custom-written routines in IgorPro 4.0 (Wavemetrics, Lake Oswego, OR, USA). Residency time analysis and vesicle tracking were preceded by band-pass spatial Fourier filtering of image stacks with a Hanning window to eliminate background fluorescence and enhance contrast. To automatically detect single vesicles, filtered stacks of TIRF images were subjected to an à-trous wavelet transformation with iteration level k=3 and detection threshold ld=1.0 (Olivo-Marin, 2002), resulting in a stack of segmented mask images. The á-trous transformation was shown to be effective in automated detection of spots of varying intensity from biological images (Olivo-Marin, 2002). The transform algorithm applied to a single image results in a segmented image in which pixels belonging to detected spots have zero values and non-zero elsewhere, thus forming a set of binary masks for further analysis of automatically identified objects/events. Spots on the mask images, each representing potentially detected vesicles (mask area ⩾4 pixels), were identified and positions of the vesicles determined as the center-of-intensity of all pixels in the mask. Only objects whose centers moved less than 1.5 pixels laterally between adjacent frames and whose time-averaged areas were between 6 and 12 pixels, circularities between 0.7 and 1.2 and intensities over cytosolic background >1.2 were included in the analysis. For lifetime analysis, objects with residency times less than or equal to two frames (τ⩽0.3 s) were excluded, as they mostly represented false detection events due to the noise in images. Vesicles that disappeared for at least one frame were scored as undocked, and those not visible on a preceding frame, as newly docked. Residency times were binned and cell-wise normalized to the footprint area of the membrane and total observation time to reflect hit rates per unit area. Average occurrence of vesicles with particular residency time was estimated from the whole stack of images by first counting the density of vesicles of that residency time at every frame of the stack, averaging obtained densities among all images and finally among the cells. The average occurrence distribution thus provides an expectation value for instantaneous density of vesicles with certain residency time at a random snapshot of the cell's footprint.
For tracking vesicles, spatial vesicle XY positions were followed in time using 2D Gaussian fitting. The relative fluorescence intensity of the vesicles was calculated by normalization to the local cytoplasmic background fluorescence, measured for every granule as the average value of surrounding pixels. Corrected intensity traces were used for the relative Z-position estimation at time points ti according to
![]()
with I0=max (I(ti)) and Z0 an unknown but constant offset for every vesicle. First-order differences of Z(t), calculated as ΔZ(ti)=Z(ti+1)−Z(ti), were used for calculating the ACF 〈ΔZ(τ)ΔZ(0)〉, referenced here as ΔZ-ACF, which is proportional to the velocity ACF given the constant sampling time interval δt=ti+1−ti. Zero (τ=0) and first-order (τ=δt) points were excluded from the analysis, as they reflect the mean of the squared ΔZ values (high positive value) and ‘minus' variance of Z(t) values (high negative value), respectively. ΔZ-ACFs from individual vesicles were noisy (Johns et al, 2001). Therefore, we reduced the original sampling rate to 10 Hz by three-point binning of intensity traces (δt′=3 δt=99 ms). The ensemble average of individual ΔZ-ACFs was four-point binned to minimize noise. The error bars shown are ±s.e.m.
Supplementary Material
Legends to Supplementary Figures
Supplementary Figures
Supplementary Materials and Methods
Acknowledgments
We thank Robbert Zalm, Desiree Schut, Joke Wortel, Angela Sarabdjitsingh and Joost Hoetjes for excellent technical assistance and Drs E Neher and TC Südhof for discussions and advice. This work was supported by grants from the Netherlands Organization for Scientific Research (NWO: GBMW 903-42-023 and ZonMW Veni grant 016-066-101 to RFT, GpD 970-10-036 to MV and HdW, ZonMW Veni grant 916-36-043 to HdW and ZonMW-Pionier grant 900-01-001 to MV), The NeuroBsik Mouse Phenomics Consortium (BSIK03053), by the European Commission (the EUSynapse project, LSHM-CT-2005-019055) and the Deutsche Forschungsgemeinschaft (SFB 406 and GRK 723 to JK and SO 708/1-1 to JBS). OK was a recipient of a fellowship of the Deutsche Forschungsgemeinschaft (GRK 723) and is a student of the International Neurosciences MSc/PhD Program at the Georg-August University, Göttingen.
References
- Ashery U, Betz A, Xu T, Brose N, Rettig J (1999) An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol 78: 525–532 [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Shareef MM, Sharma VM, Shetty AP, Ramamohan Y, Pant HC, Raju TR, Shetty KT (2004) Co-purification and localization of Munc18-1 (p67) and Cdk5 with neuronal cytoskeletal proteins. Neurochem Int 44: 35–44 [DOI] [PubMed] [Google Scholar]
- Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Sudhof T, Bruns D (2005) v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J 24: 2114–2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Prokop A, Bellen HJ, O'Kane CJ, Schulze KL, Sweeney ST (1995) Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron 15: 663–673 [DOI] [PubMed] [Google Scholar]
- Gil A, Gutierrez LM, Carrasco-Serrano C, Alonso MT, Viniegra S, Criado M (2002) Modifications in the C terminus of the synaptosome-associated protein of 25 kDa (SNAP-25) and in the complementary region of synaptobrevin affect the final steps of exocytosis. J Biol Chem 277: 9904–9910 [DOI] [PubMed] [Google Scholar]
- Han X, Wang CT, Bai J, Chapman ER, Jackson MB (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science 304: 289–292 [DOI] [PubMed] [Google Scholar]
- Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Sudhof TC, Niemann H (1994) Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J 13: 5051–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H (1994) A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron 12: 1269–1279 [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112: 519–533 [DOI] [PubMed] [Google Scholar]
- Jahn R, Niemann H (1994) Molecular mechanisms of clostridial neurotoxins. Ann NY Acad Sci 733: 245–255 [DOI] [PubMed] [Google Scholar]
- Johns LM, Levitan ES, Shelden EA, Holz RW, Axelrod D (2001) Restriction of secretory granule motion near the plasma membrane of chromaffin cells. J Cell Biol 153: 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korteweg N, Maia AS, Thompson B, Roubos EW, Burbach JP, Verhage M (2005) The role of Munc18-1 in docking and exocytosis of peptide hormone vesicles in the anterior pituitary. Biol Cell 97: 445–455 [DOI] [PubMed] [Google Scholar]
- Lang T, Wacker I, Steyer J, Kaether C, Wunderlich I, Soldati T, Gerdes HH, Almers W (1997) Ca2+-triggered peptide secretion in single cells imaged with green fluorescent protein and evanescent-wave microscopy. Neuron 18: 857–863 [DOI] [PubMed] [Google Scholar]
- Lang T, Wacker I, Wunderlich I, Rohrbach A, Giese G, Soldati T, Almers W (2000) Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys J 78: 2863–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattheyses A, Axelrod D (2002) Measurement of fluorophore excitation and emission properties near bare and metal coated surfaces. Biophys J 82: 499a [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20: 87–90 [DOI] [PubMed] [Google Scholar]
- Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sørensen JB (2002) Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci 22: 9278–9286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor V, Heuss C, De Bello WM, Dresbach T, Charlton MP, Hunt JH, Pellegrini LL, Hodel A, Burger MM, Betz H, Augustine GJ, Schafer T (1997) Disruption of syntaxin-mediated protein interactions blocks neurotransmitter secretion. Proc Natl Acad Sci USA 94: 12186–12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oheim M, Stuhmer W (2000) Tracking chromaffin granules on their way through the actin cortex. Eur Biophys J 29: 67–89 [DOI] [PubMed] [Google Scholar]
- Olivo-Marin JC (2002) Extraction of spots in biological images using multiscale products. Pattern Recogn 35: 1989–1996 [Google Scholar]
- Qian H, Sheetz MP, Elson EL (1991) Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys J 60: 910–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Jimenez JL, Graham ME, Archer DA, Soloviev M, Burgoyne RD, Davletov B (2006) Conserved prefusion protein assembly in regulated exocytosis. Mol Biol Cell 17: 283–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC (2002) Snares and munc18 in synaptic vesicle fusion. Nat Rev Neurosci 3: 641–653 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deak F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294: 1117–1122 [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM (1987) Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J 52: 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly–disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75: 409–418 [DOI] [PubMed] [Google Scholar]
- Sollner TH, Rothman JE (1996) Molecular machinery mediating vesicle budding, docking and fusion. Experientia 52: 1021–1025 [DOI] [PubMed] [Google Scholar]
- Steyer JA, Almers W (2001) A real-time view of life within 100 nm of the plasma membrane. Nat Rev Mol Cell Biol 2: 268–275 [DOI] [PubMed] [Google Scholar]
- Steyer JA, Horstmann H, Almers W (1997) Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature 388: 474–478 [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114: 75–86 [DOI] [PubMed] [Google Scholar]
- Toonen RF, Verhage M (2003) Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol 13: 177–186 [DOI] [PubMed] [Google Scholar]
- Tsuboi T, Fukuda M (2006) The Slp4-a linker domain controls exocytosis through interaction with Munc18-1.syntaxin-1a complex. Mol Biol Cell 17: 2101–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–869 [DOI] [PubMed] [Google Scholar]
- Vitale ML, Seward EP, Trifaro JM (1995) Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron 14: 353–363 [DOI] [PubMed] [Google Scholar]
- Voets T (2000) Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron 28: 537–545 [DOI] [PubMed] [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M (2001) Munc18-1 promotes large dense-core vesicle docking. Neuron 31: 581–591 [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19–26 [DOI] [PubMed] [Google Scholar]
- Zenisek D, Steyer JA, Almers W (2000) Transport, capture and exocytosis of single synaptic vesicles at active zones. Nature 406: 849–854 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Legends to Supplementary Figures
Supplementary Figures
Supplementary Materials and Methods