

Abstract
The X-linked disorder oculocerebrorenal syndrome of Lowe is caused by mutation of the OCRL1 protein, an inositol polyphosphate 5-phosphatase. OCRL1 is localised to the Golgi apparatus and early endosomes, and can translocate to lamellipodia upon growth factor stimulation. We show here that OCRL1 interacts with several members of the rab family of small GTPases. Strongest interaction is seen with Golgi-associated rab1 and rab6 and endosomal rab5. Point mutants defective in rab binding fail to target to the Golgi apparatus and endosomes, strongly suggesting rab interaction is required for targeting of OCRL1 to these compartments. Membrane recruitment via rab binding is required for changes in Golgi and endosomal dynamics induced by overexpression of catalytically inactive OCRL1. In vitro experiments demonstrate that rab5 and rab6 directly stimulate the 5-phosphatase activity of OCRL1. We conclude that rabs play a dual role in regulation of OCRL1, firstly targeting it to the Golgi apparatus and endosomes, and secondly, directly stimulating the 5-phosphatase activity of OCRL1 after membrane recruitment.
Keywords: endosome, Golgi apparatus, OCRL1, phosphoinositide, rab
Introduction
Phosphoinositides (PIs) are essential phospholipids that regulate a number of processes including intracellular signalling, cytoskeletal dynamics, and membrane traffic (Cantley, 2002; Yin and Janmey, 2003; De Matteis and Godi, 2004; Roth, 2004). A major function of PIs is the membrane recruitment or activation of effector proteins that bind via specific modular binding domains (Cullen et al, 2001; Balla, 2005). As different PIs recruit and/or activate different sets of effector proteins, they represent key determinants in defining the functional identity of a membrane compartment. Consistent with this view, different PIs appear to be enriched in different compartments. For example, PtdIns(4,5)P2 is abundant at the plasma membrane where it recruits a number of proteins including clathrin accessory proteins required for endocytosis (Cremona and De Camilli, 2001; Haucke, 2005), whereas PtdIns(4)P appears to be more abundant at the Golgi apparatus and recruits different effector proteins that mediate trafficking to the cell surface and endosomes (De Matteis et al, 2002; De Matteis and Godi, 2004). In order to maintain this spatial segregation of PIs, it is important that the kinases and phosphatases that modulate the levels of different PIs are correctly targeted within the cell. These enzymes should also be temporally regulated to ensure that individual PI species are produced at the correct time during a particular process.
Oculocerebrorenal syndrome of Lowe is an X-linked disorder characterised by mental retardation, congenital cataracts, and renal Fanconi syndrome (Lowe et al, 1952). Lowe syndrome results from mutation of OCRL1, an inositol polyphosphate 5-phosphatase whose preferred substrate is PtdIns(4,5)P2, although it can also hydrolyse PtdIns(3,4,5)P3 as well as soluble Ins(1,4,5)P3 and Ins(1,3,4,5)P4 (Attree et al, 1992; Lowe, 2005). Skin fibroblasts and kidney proximal tubule cells derived from Lowe syndrome patients have elevated PtsIns(4,5)P2 levels, consistent with a role for OCRL1 in regulating the cellular levels of this lipid (Zhang et al, 1998; Wenk et al, 2003). OCRL1 is a peripheral membrane protein that is localised to the Golgi apparatus and early endosomes (Olivos-Glander et al, 1995; Dressman et al, 2000; Ungewickell et al, 2004; Choudhury et al, 2005). OCRL1 can also be detected in lamellipodia or membrane ruffles when quiescent cells are stimulated with growth factors, suggesting that it translocates to these structures upon growth factor stimulation (Faucherre et al, 2005). This may be mediated by rac1, which has been reported to interact with the C-terminal Rho GAP-like domain of OCRL1 (Faucherre et al, 2003). OCRL1 binds directly to the terminal domain of clathrin heavy chain and the ear domain of the α-adaptin subunit of the endocytic AP2 clathrin adaptor complex (Ungewickell et al, 2004; Choudhury et al, 2005), and is enriched in clathrin-coated vesicles (Ungewickell et al, 2004; Choudhury et al, 2005). Overexpression or depletion of OCRL1 inhibits trafficking of proteins from early endosomes to the TGN, suggesting a role in clathrin-mediated trafficking between these compartments (Choudhury et al, 2005). A role for OCRL1 in other trafficking steps is also possible, with endocytosis a likely candidate given the ability of OCRL1 to bind AP2, although this remains to be shown.
The mechanism by which OCRL1 undergoes targeting to Golgi and endosomal membranes is not known. It has been proposed that rac1 is responsible (Faucherre et al, 2003), but this protein is more abundant at the plasma membrane than internal compartments. We, therefore, reasoned that another small GTPase may be involved in OCRL1 targeting, and decided to investigate rab proteins, as these have been found to recruit a variety of proteins onto membranes, and are intimately involved in membrane traffic (Zerial and McBride, 2001). There are more than 60 rabs in humans, each one with a restricted cellular location (Pereira-Leal and Seabra, 2001; Zerial and McBride, 2001). Rab5 is a key regulator of endocytosis and growth factor signalling (Zerial and McBride, 2001). Rab6, in contrast, is predominantly found at the trans-side of the Golgi apparatus and regulates trafficking from the early endosome to the TGN, and retrograde transport from the Golgi apparatus to the endoplasmic reticulum (Antony et al, 1992; White et al, 1999; Mallard et al, 2002). Other Golgi-associated rabs include rab1, rab2, and rab8, which are enriched in the cis-, medial-, and trans-Golgi, respectively (Zerial and McBride, 2001). Here, we show that OCRL1 binds to several rab proteins, with strongest binding to rab1, rab5, and rab6. Rab binding is required for targeting of OCRL1 to the Golgi apparatus and endosomes, and directly stimulates the 5-phosphatase activity of OCRL1. This suggests a dual mode of regulation by rab proteins to ensure OCRL1 activity is directed to the correct cellular membrane compartments.
Results
OCRL1 interacts directly with Golgi and endosomal rab proteins
To determine whether OCRL1 can interact with Golgi or endosomal rab proteins, we performed a directed yeast two-hybrid experiment. Full-length OCRL1 was cotransformed into yeast cells with GTP-locked versions of rab proteins that have been localised to the Golgi apparatus or endosomes, and interaction was detected by growth on high selection medium. OCRL1 interacted with GTP-locked rab1A, rab5A, rab6A (herein referred to as rab1, rab5, and rab6, unless otherwise indicated), rab8, and rab14, but not other rabs that were tested (Figure 1A). Interaction was also detected with rab6A' and rab6B (data not shown). Interaction between OCRL1 and rabs was confirmed in pull-down experiments where GST-tagged, or in the case of rab8 NusA-tagged, versions of the rabs were incubated with HeLa cytosol (Figure 1B). Binding was strongest with GTP-locked rab6 and rab5, followed by rab1, then rab8, and rab14. Rab binding of OCRL1 was dependent upon the nucleotide status of the rab, with binding in the yeast two-hybrid assay only seen between OCRL1 and wild type (WT) or GTP-locked, but not GDP-locked rabs (Figure 1C). Similar results were seen in pull-down experiments in which WT rabs were preloaded with either GDP or the nonhydrolysable GTP analogue GMP-PNP before incubation with HeLa cell extracts. OCRL1 bound only to GMP-PNP-loaded rab1, rab5, rab6, and rab8, but not the GDP-loaded forms of these rabs (Figure 1D). Together, these results indicate that OCRL1 binds only to the active forms of the rab proteins. To determine whether OCRL1 binds directly to the rabs, purified recombinant OCRL1 was incubated with purified GST-tagged GTP-locked versions of the rab proteins and binding was detected by Coomassie blue staining and Western blotting. As shown in Figure 1E, purified OCRL1 bound strongly to GTP-locked rab1, rab5, and rab6, and to a lesser extent to rab14, indicating that OCRL1 binds directly to these rabs. Direct binding could also be monitored using a solid-phase binding assay in which OCRL1 was immobilised on plastic before incubation with increasing amounts of each rab protein. This indicated that OCRL1 preferentially binds directly to rab6, followed by rab1 and rab5, then rab14 and rab8 (Figure 1F). Solid-phase binding also confirmed that direct binding only occurs to GTP-locked rab1, rab5, and rab6, and not to the GDP-locked forms of these proteins (Supplementary Figure S1).
Figure 1.
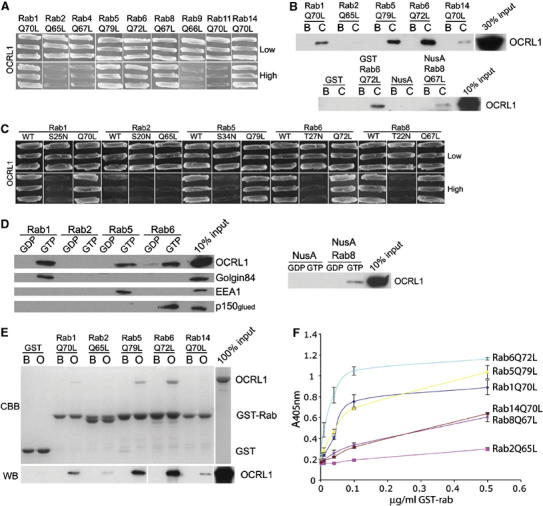
Interaction of OCRL1 with Golgi and endosomal rab proteins. (A) Full-length OCRL1 and GTP-restricted versions of the indicated rab proteins were tested for interaction in the yeast two-hybrid system. Interaction results in growth on high selection. (B) Beads containing GST alone, NusA alone, or GST or NusA-tagged GTP-restricted versions of the indicated rab proteins were incubated with buffer (B) or HeLa cytosol (C) in the presence of 100 μM GMP-PNP and bound proteins eluted and analysed by Western blotting with antibodies to OCRL1. In the top panel, all rabs were GST-tagged. (C) Full-length OCRL1 was tested for interaction with either WT, GDP-restricted (S or T to N) or GTP-restricted (Q to L) versions of the indicated rab proteins. Interaction results in growth on high selection. (D) Beads containing NusA alone, or GST or NusA-tagged WT rab proteins loaded with GDP or GMP-PNP (GTP) were incubated with HeLa cell extract in the presence of 100 μM GDP or 100 μM GMP-PNP (GTP) as indicated. Bound proteins were eluted and analysed by Western blotting with the indicated antibodies. (E) Beads containing GST alone or GST-tagged GTP-restricted versions of the indicated rabs were incubated with buffer alone (B) recombinant full-length OCRL1 (O) in the presence of 100 μM GMP-PNP and bound proteins eluted and analysed by Coomassie blue staining (CBB) or Western blotting (WB) with antibodies to OCRL1. (F) Recombinant full-length OCRL1 was immobilised on plastic and incubated with the indicated amounts of GST-tagged rab proteins. Binding was measured using HRP-conjugated antibodies and colorimetric detection at 405 nm. Results are indicated as the mean±s.d. of two experiments carried out in duplicate.
Mapping of the rab-binding site in OCRL1
We next wanted to identify the region of OCRL1 responsible for rab binding. Truncated versions of OCRL1 were tested for binding to GTP-locked rab5 or rab6 in the yeast two-hybrid system. A construct comprising the N-terminus and 5-phosphatase domain of OCRL1 (amino acids 1–539) did not bind to either rab, whereas a construct containing the region of OCRL1 C-terminal to the 5-phosphatase domain (amino acids 540–893) exhibited strong binding to both rab5Q79L and rab6Q72L (Figure 2A). Deletion of the C-terminal Rho GAP-like domain (amino acids 727–886) had no effect on binding to either rab, suggesting that the rab-binding site lies within the linker region between the 5-phosphatase and Rho GAP-like domains of OCRL1 (amino acids 540–726). This was confirmed when this region was found to be sufficient for binding to both rab5Q79L and rab6Q72L. To further verify these findings, we performed binding experiments with fragments of OCRL1 generated by limited tryptic digestion of the recombinant protein. A ∼57 kDa tryptic fragment of OCRL1 bound strongly to rab6Q72L (Figure 2B, arrow). N-terminal sequencing of this fragment indicated that it starts at amino acid 200 (data not shown). Western blotting revealed the fragment contains the CRAE peptide antibody epitope (residues 616–631), but not that of the KINS (residues 163–177) or 11 520 (residues 878–893) antibodies (Figure 2B). These data confirm the fragment contains the 5-phosphatase and part of the linker domain of OCRL, encompassing residues 200 to at least 631. However, the fragment likely extends to around residue 700, which would fit better with molecular mass predictions from the primary sequence. Thus, in biochemical experiments, this region of OCRL1 is sufficient for rab binding, consistent with the presence of the rab-binding site in the linker domain.
Figure 2.
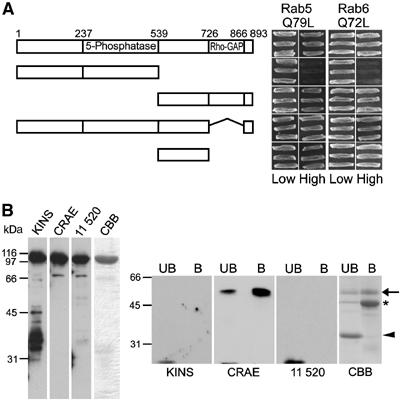
Mapping of the rab binding site in OCRL1. (A) Full-length and truncated OCRL1 constructs were tested for interaction with rab5Q79L or rab6Q72L in the yeast two-hybrid system. Interaction results in growth on high selection. (B) Full-length recombinant OCRL1 (left panel) was digested with trypsin and resulting fragments were incubated with GST-rab6Q72L (right panel). Intact OCRL1 (left) and bound (B) or unbound (UB) proteins (right) were analysed by Coomassie blue staining (CBB) or Western blotting with antibodies to the KINS (residues 163–177), CRAE (residues 616–631), or 11 520 (residues 878–893) epitopes of OCRL1. The rab6-binding fragment of OCRL1 is indicated with an arrow. A smaller fragment that fails to bind rab6 or CRAE antibody is indicated with an arrowhead. The position of GST-rab6Q72L is indicated with an asterisk.
Rab interaction targets OCRL1 to the Golgi apparatus and endosomes
To assess whether rab binding was required for OCRL1 targeting, we first expressed GTP-locked rabs in cells and analysed OCRL1 localisation by immunofluorescence microscopy. Expression of myc-rab5Q79L resulted in enlarged early endosomal structures as has been reported previously (Stenmark et al, 1994). OCRL1 was redistributed onto the limiting membrane of these structures, consistent with a role for rab5 in recruiting OCRL1 onto early endosomes (Figure 3). The extent of OCRL1 recruitment was similar to that seen with EEA1, which is known to undergo rab5-dependent recruitment to early endosomes (data not shown) (Simonsen et al, 1998). Endosomal recruitment of OCRL1 could also be seen upon expression of GFP-tagged WT rab5 (Supplementary Figure S2). Myc-rab6Q72L localised to the Golgi apparatus, which retained its normal perinuclear ribbon-like appearance, but no effect was seen upon the overall distribution of OCRL1 (Figure 3). This was also true for WT myc-rab6 and the GDP-locked T27N mutant (data not shown). However, we also failed to notice any effects of these constructs on Golgi recruitment of the golgin TATA modulatory factor (TMF), which is known to bind via rab6 to the Golgi apparatus (data not shown) (Fridmann-Sirkis et al, 2004).
Figure 3.
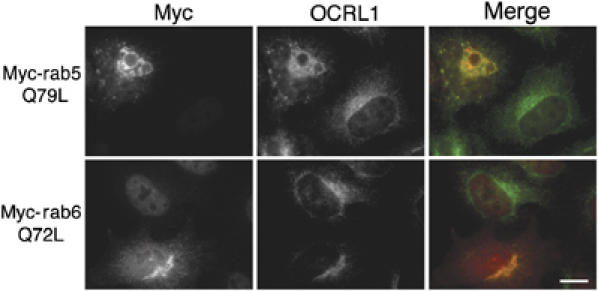
Effects of rab mutant expression on OCRL1 targeting. HeLa cells were transfected with myc-tagged rab5Q79L or myc-tagged rab6Q72L and analysed by immunofluorescence microscopy with antibodies to myc and OCRL1. Bar, 10 μm.
To further investigate the role of rab binding in OCRL1 targeting to the Golgi apparatus, we performed RNA interference to deplete cellular rab1 or rab6 and studied OCRL1 localisation by fluorescence microscopy. Although rab1A was efficiently depleted by our rab1 small interfering RNA (siRNA), there was no depletion of rab1B; in fact, rab1B levels were slightly elevated by this treatment (Figure 4A). Nevertheless, rab1A depletion was sufficient to cause Golgi fragmentation in the majority of cells (Figure 4B). OCRL1 was clearly present on these fragments, indicating that depletion of rab1A alone does not prevent Golgi targeting of OCRL1. Rab6 depletion was efficient, and specific for both expressed rab6 isoforms, rab6A and rab6A', which are recognised by our rab6 antibody (Figure 4A), but had no obvious effects on Golgi morphology or upon OCRL1 targeting to this organelle (Figure 4B). We next performed a double depletion of rab1A and rab6, as binding to one rab may allow targeting in the absence of the other. Codepletion of rab1A and rab6 was efficient (Figure 4A), and resulted in Golgi fragmentation, as expected (Figure 4B). OCRL1 levels on these fragments were dramatically reduced compared to control cells or the single knockdowns, as observed in double (Figure 4B) and triple labelling (Figure 4C) experiments. These results suggest rab1A and rab6 are important for recruitment of OCRL1 to the Golgi apparatus.
Figure 4.
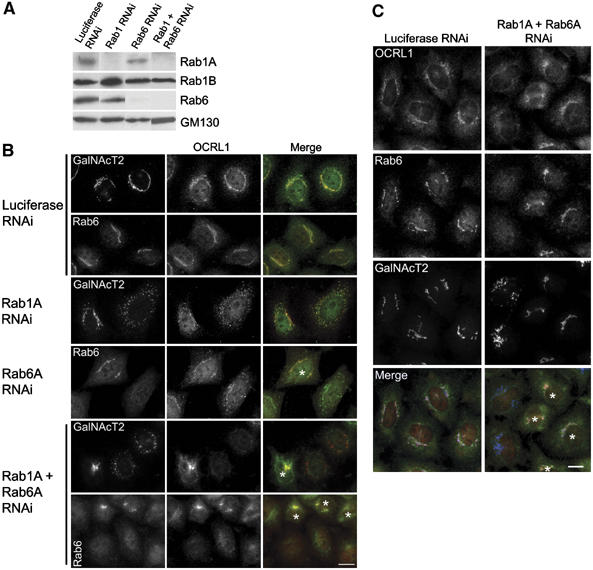
Effects of rab depletion on OCRL1 targeting. (A) HeLa cells were transfected with control siRNA (luciferase) or siRNAs targeting rab1A alone, rab6 alone, or rab1A and rab6 together for 72 h and analysed by Western blotting with the indicated antibodies. (B) SiRNA treated cells were analysed by immunofluorescence microscopy with antibodies to OCRL1 and either GalNacT2 or rab6 as indicated. Nontransfected cells are marked with an asterisk. Depletion of rab1A is indicated by Golgi fragmentation. (C) SiRNA-treated cells were analysed by immunofluorescence microscopy following triple staining with antibodies to OCRL1, rab6, and GalNacT2. Nontransfected cells are marked with an asterisk. Bar, 10 μm.
An alternative approach was devised to further analyse the role of rabs in OCRL1 targeting. This was based upon using random mutagenesis to generate point mutants defective in rab binding, and then studying the targeting of these mutants in cells. A region encompassing the OCRL1 linker region, which contains the rab-binding site, was subjected to error-prone PCR and resulting full-length mutant clones assessed for binding to rab6Q72L in the yeast two-hybrid system. Clones were screened in parallel against clathrin heavy chain terminal domain (CTD), which also binds to the OCRL1 linker region (Choudhury et al, 2005), to eliminate nonsense and frameshift mutations, and missense mutations that unfold the protein. Using this approach, we identified five clones that were unable to bind rab6Q72L but retained an interaction with clathrin. Sequencing revealed each clone contained two point mutations (data not shown). Selected corresponding single mutants were generated in GFP-OCRL1 and binding to rab proteins and targeting to the Golgi apparatus and endosomes were analysed (results are summarised in Table I). As shown in Figure 5A, GFP-tagged WT OCRL1 bound to both clathrin and rab6Q72L, as expected. The D555E, S564P, and G664D mutants were able to bind clathrin to the same extent as WT OCRL1, but severely compromised in rab6 binding (Figure 5A). The P526H, S568G, and N606K mutants retained binding to both clathrin and rab6, with the P526H and N606K mutants binding more strongly than WT OCRL1. To determine whether the D555E, S564P, and G664D mutants were also deficient in binding to other rabs, binding experiments were performed with rab1Q70L and rab5Q79L alongside rab6Q72L. Each mutant was unable to bind rab1, and exhibited much-reduced binding to both rab5 and rab6, with the effects on rab5 interaction the least pronounced (Figure 5B). The S564P mutation had the strongest effect on binding to all rabs. The S568G mutant also bound more strongly than WT OCRL1 to rab1 and rab5 in addition to rab6 (Figure 5B). These results suggest the rabs share a common binding site, with subtle differences in the mode of binding between individual rabs.
Table 1.
Summary of the OCRL1 point mutant rab binding and localisation results
| GFP-OCRL1 | Rab6Q72L interaction | Rab5Q79L interaction | Golgi targeting | Endosomal targeting |
|---|---|---|---|---|
| WT | +++ | +++ | +++ | +++ |
| P526H | +++++ | ++++ | +++ | ND |
| D555E | +/− | +/− | +/− | +/− |
| S564P | − | − | − | − |
| S568G | +++ | ++++ | +++ | ND |
| N606K | ++++ | ++++ | +++ | +++ |
| G664D | +/− | + | +/− | +/− |
| ND, not determined. | ||||
Figure 5.
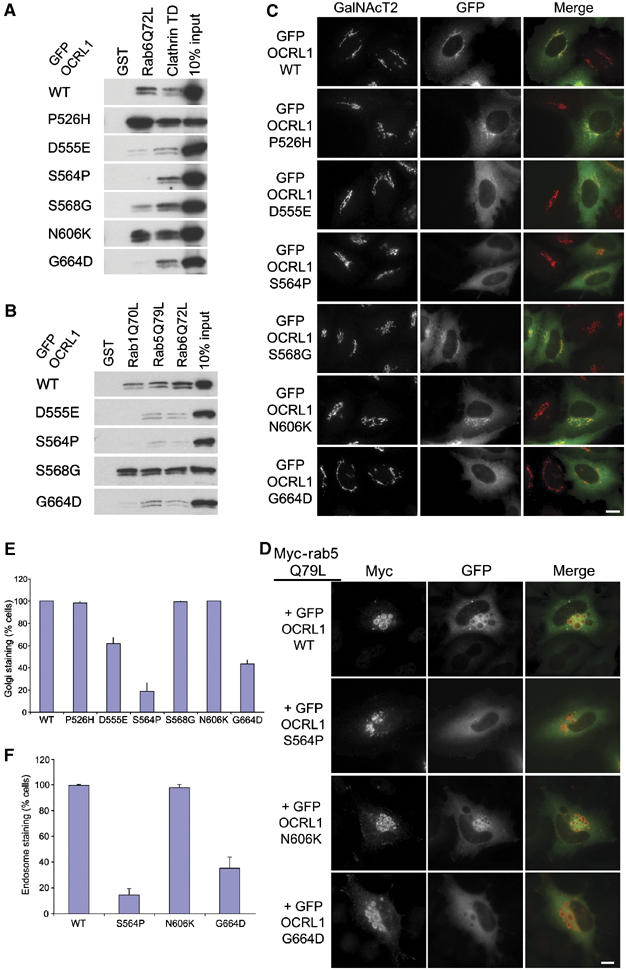
Rab binding-deficient OCRL1 mutants fail to target to the Golgi apparatus and endosomes. (A) Cell lysates prepared from HeLaM cells expressing GFP-tagged full-length OCRL1 or the indicated point mutants were incubated with beads containing either GST alone, GST-clathrin terminal domain (TD), or GST-rab6Q72L, and bound proteins analysed by Western blotting with antibodies to OCRL1. (B) Cell lysates were incubated with beads containing GST alone, GST-rab1Q70L, GST-rab5Q79L, or GST-rab6Q72L and bound proteins analysed by Western blotting with antibodies to OCRL1. (C) HeLaM cells expressing GFP-tagged full-length OCRL1 or the indicated point mutants were analysed by immunofluorescence microscopy with antibodies to GalNacT2. (D) HeLaM cells were cotransfected with myc-rab5Q79L and GFP-tagged full-length OCRL1 or the indicated point mutants were analysed by immunofluorescence microscopy with antibodies to myc. Bar, 10 μm. (E, F) Quantitation of Golgi and endosomal targeting of WT and mutant GFP-OCRL1. Targeting was defined as a discernable increase in GFP fluorescence above background on GalNacT2 (E) or Myc-Rab5Q79L (F) positive structures. The results are expressed as the mean±s.d. from three independent experiments, counting 100 cells per experiment.
As expected, WT GFP-OCRL1 was localised predominantly to the Golgi apparatus and cytoplasm (Figure 5C and E). Mutants deficient in rab binding (S555E, S564P, and G664D) showed reduced targeting to the Golgi apparatus and accumulated in the cytoplasm. In contrast, mutants that retained rab binding (P526H, S568G, and N606K) were efficiently targeted to the Golgi apparatus. To assess whether rab binding is also required for targeting of OCRL1 to endosomes, as suggested by our rab5 overexpression experiments (Figure 3 and Supplementary Figure S2), we studied the localisation of the OCRL1 point mutants in cells coexpressing the dominant rab5Q79L mutant. As expected, WT GFP-OCRL1 was efficiently targeted to the enlarged rab5-positive endosomes (Figure 5D and E). In contrast, rab-binding-deficient mutants were poorly recruited to these structures, suggesting rab binding is also required for targeting of OCRL1 to endosomes. Similar results were seen in A431 cells in the absence of rab5 overexpression. WT GFP-OCRL1 was present on both Golgi and early endosomes in A431 cells, whereas the S564P and G664D mutants were predominantly cytosolic (Supplementary Figure S3 and data not shown).
Rab interaction is required for changes in Golgi and endosomal dynamics induced by overexpression of catalytically inactive OCRL1
We have previously reported that high-level expression of OCRL1 fragments the Golgi apparatus and causes a dramatic enlargement of endosomes concomitant with a block in endosome to Golgi transport (Choudhury et al, 2005). These effects are more pronounced with a deletion construct lacking the 5-phosphatase domain (GFP-OCRL1 Δ237–539). To assess whether rab binding is required for these OCRL1-induced effects, the linker region point mutations described above were introduced into the OCRL1 construct lacking the 5-phosphatase domain, and effects upon Golgi and endosomal markers were analysed by immunofluorescence microscopy. As expected, the parental GFP-OCRL1 Δ237–539 construct induced Golgi fragmentation, and redistribution of the transferrin receptor to large cytoplasmic structures containing the OCRL1 mutant protein, corresponding to enlarged endosomes (Figure 6A, B and D). The cation-independent mannose 6-phosphate receptor (CI-MPR) was also redistributed from the Golgi region to the OCRL1-containing structures, consistent with a block in endosome to Golgi recycling (Figure 6C and D). In contrast, high level expression of GFP-OCRL1 Δ237–539 containing the D555E, S564P, or G664D mutations failed to elicit any changes in Golgi enzyme, transferrin receptor, or CI-MPR distribution (Figure 6A–D, and data not shown), indicating that Golgi and endosome dynamics are unaffected by these mutants.
Figure 6.
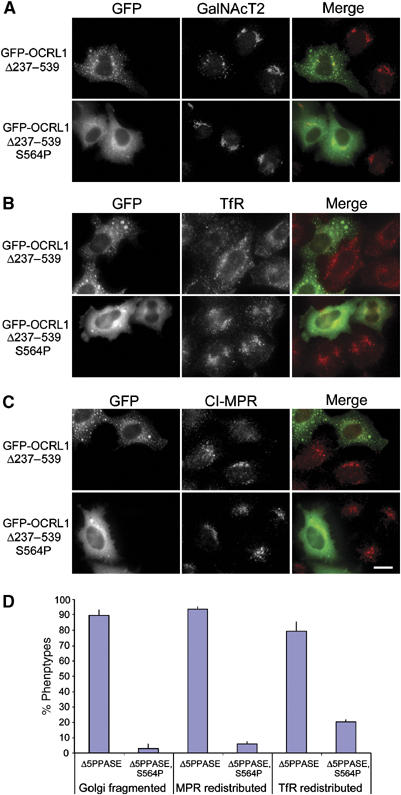
Membrane recruitment of OCRL1 is required for regulation of Golgi and endosomal dynamics. HeLa cells expressing GFP OCRL1Δ237–539 or GFP OCRL1Δ237–539/S564P were analysed by immunofluorescence microscopy with antibodies to GalNacT2 (A), transferrin receptor (TfR) (B), or the cation-independent mannose 6-phosphate receptor (CI-MPR) (C). Bar, 10 μm. (D) Quantitation of the Golgi, CI-MPR, and TrR phenotypes. The results are expressed as the mean±s.d. from three independent experiments, counting 100 cells per experiment.
Endosome to Golgi trafficking was directly analysed in the mutant-expressing cells using the shiga toxin B subunit (STxB), which is internalised from the cell surface before retrograde trafficking from early endosomes to the TGN, which occurs in a rab6-dependent manner (Mallard et al, 2002). Expression of GFP-OCRL1 Δ237–539 inhibited STxB delivery to the TGN, as previously reported (Figure 7; Choudhury et al, 2005). This was also true for the N606K mutant that can bind rabs and target to the membrane (Figure 7). In contrast, STxB trafficking from endosomes to the Golgi was unaffected by the rab-binding-deficient S564P or G664D mutants. Together, these results indicate that interaction with rab proteins is required for OCRL1-induced changes in the both the morphology and trafficking of the Golgi apparatus and endosomes.
Figure 7.
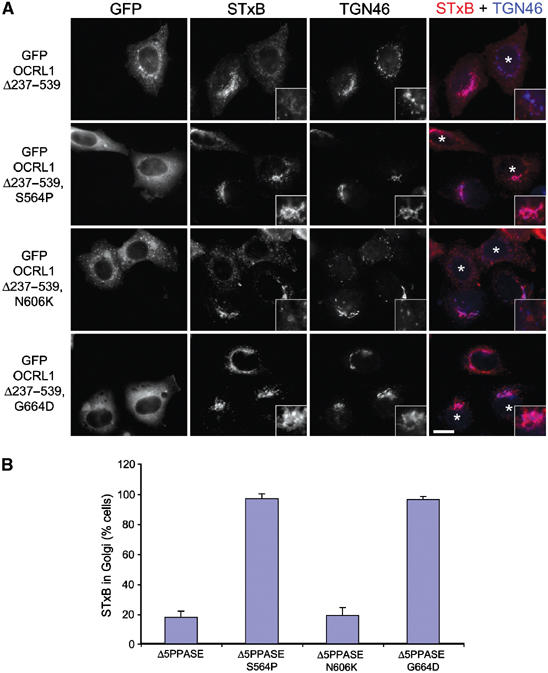
Membrane recruitment of OCRL1 is required for regulation of endosome to Golgi trafficking. (A) HeLa cells expressing GFP OCRL1Δ237–539 or GFP OCRL1Δ237–539 with the indicated point mutations were incubated with cy3-STxB (red) for 45 min at 37°C and processed for immunofluorescence microscopy. Cells were labelled with antibodies to TGN46 (blue). Asterisks indicate transfected cells. Regions of overlap between STxB and TGN46 are pink. Bar, 10 μm. (B) Quantitation of STxB transport to the TGN. The results are expressed as the mean±s.d. from three independent experiments, counting 100 cells per experiment.
Rab interaction stimulates the 5-phosphatase activity of OCRL1
We next wanted to assess whether rab binding affects the catalytic activity of OCRL1. For this purpose, we used an in vitro 5-phosphatase assay in which purified recombinant OCRL1 was incubated with PtdIns(4,5)P2-containing liposomes and conversion to PtdIns(4)P assessed by thin layer chromatography. Recombinant OCRL1 displayed significant 5-phosphatase activity towards PtdIns(4,5)P2 in the absence of additional factors (Figure 8A). Addition of GST-rab5Q79L or GST-rab6Q72L stimulated OCRL1 5-phosphatase activity by 1.5- and 2-fold, respectively. In contrast, no effect was seen with GST-rac1Q61L, which has been reported to bind OCRL1 (Faucherre et al, 2003). The stimulation by rab5 and rab6 was not due to contaminating phosphatase activities, as these protein preparations exhibited no 5-phosphatase when assayed alone. To confirm that the stimulation in OCRL1 activity was due to rab binding, the G664D mutant deficient in rab binding was analysed alongside the WT protein. As shown in Figure 8B, the mutant failed to show a significant stimulation of activity in the presence of rab6Q72L, in contrast to the WT protein which gave a ∼1.8-fold stimulation. We conclude from these experiments that rab5 and rab6 specifically stimulate the 5-phosphatase activity of OCRL1 in vitro.
Figure 8.
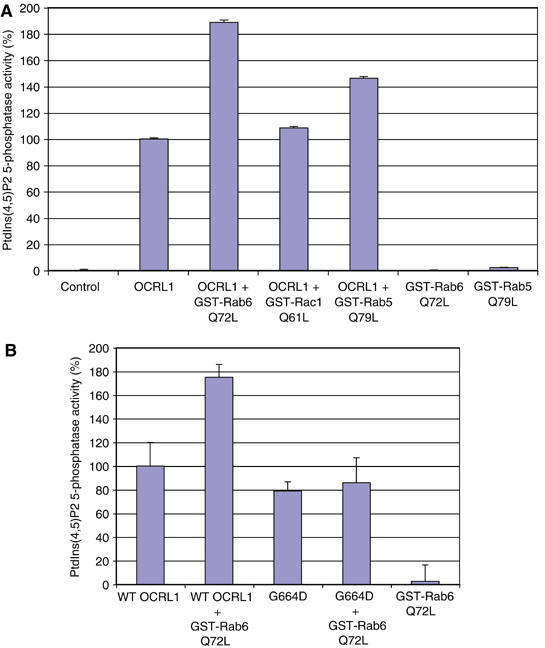
Rabs 5 and 6 stimulate OCRL1 5-phosphatase activity. (A) Incubations were performed in the absence or presence of full-length, WT OCRL1 with or without 2.4 μM GST-tagged rab5Q79L, rab6Q72L, or rac1Q61L as indicated. (B) WT OCRL1 or the G664D mutant were incubated with rab6Q72L as indicated. PtdIns(4,5)P2 5-phosphatase activity was measured as described in the Materials and methods. Results are expressed as the mean±s.d. of three independent experiments.
Discussion
In this study, we have identified the lipid phosphatase OCRL1 as a binding partner for Golgi and endosomal rab proteins. OCRL1 was able to interact with several rabs, with strongest binding to endosomal rab5 and Golgi-associated rab1 and rab6. Rab binding was required for efficient targeting of OCRL1 to endosomes and the Golgi apparatus, and directly stimulated the PtdIns(4,5)P2 5-phosphatase activity of OCRL1.
Simultaneous depletion of both rab1 and rab6 by RNA interference resulted in reduced targeting of OCRL1 to the Golgi apparatus. This suggests that these rabs are required for OCRL1 recruitment to the Golgi apparatus, although it remains possible the effects of rab depletion are indirect. To further test the hypothesis, we generated OCRL1 point mutants that are defective in rab binding. This allowed us to assess the involvement of rab binding in targeting to both the Golgi apparatus and endosomes. We observed a tight correlation between rab binding and Golgi or endosomal targeting. Mutants defective in rab binding were predominantly cytosolic, whereas mutants that retained rab association were efficiently recruited to the Golgi apparatus and early endosomes. Importantly, mutants that were deficient in rab binding retained the ability to bind clathrin heavy chain, which also binds to the OCRL1 linker region (Choudhury et al, 2005), suggesting this part of the protein is correctly folded. To further verify the folding of the point mutants, we performed limited proteolysis. Each mutant had a similar tryptic digestion pattern to WT OCRL1, consistent with these proteins having the same three-dimensional structure (Supplementary Figure S4). The one exception was P526H, which had a slightly altered digestion pattern. Nevertheless, this protein was correctly targeted to the Golgi. The mutagenesis studies suggest different rabs can bind to the same site in OCRL1, albeit with subtle differences in binding between different rab proteins. It will be interesting to resolve the three-dimensional structure of this region to understand how this is brought about, and to determine the mechanism by which rab binding can influence the catalytic domain to stimulate 5-phosphatase activity.
Of the mutated residues characterised in our study, only P526 corresponds to naturally occurring Lowe syndrome mutations (P526T and P526L) (Roschinger et al, 2000). Interestingly, the P526H mutant induced Golgi fragmentation and altered early endosome morphology when expressed at moderate levels in cells, suggesting that it is functionally defective (Figure 5C and data not shown). These effects appear independent of rab binding, as this mutant bound well to rabs and was efficiently targeted to the Golgi and endosomes. They may rather be due to loss of 5-phosphatase activity given the proximity of the mutation to the catalytic site. None of the rab-binding mutants identified in this study have so far been described in Lowe syndrome patients. The vast majority of Lowe missense mutations are in the catalytic domain, but one has been identified in the linker region (L687P) and an in-frame deletion of E585 has also been reported (Monnier et al, 2000). Whether these mutations affect rab binding remains to be shown.
There are many cases of effectors binding to more than a single rab protein, although it is unusual to find binding to quite as many rabs as seen with OCRL1. An obvious question that arises is why does OCRL1 bind to so many rabs? A potential explanation is that not all of the rabs represent physiological binding partners. This is difficult to exclude, but in each case binding appeared specific in that it only occurred to the active, GTP-bound form of the rab. An alternative explanation for multiple rab binding is that it allows targeting to multiple membrane domains. This may be expected for a protein with housekeeping duties, as has been proposed for OCRL1, where it would remove PtdIns(4,5)P2 from internal compartments to spatially restrict this lipid to the plasma membrane (Lowe, 2005). Alternatively, binding to multiple rabs may indicate a role for OCRL1 in several aspects of endosomal or Golgi function, or in regulating different trafficking pathways at these compartments. We have previously shown that OCRL1 can modulate protein trafficking from endosomes to the TGN (Choudhury et al, 2005). Trafficking of STxB from early endosomes to the TGN is regulated by rab6 (Mallard et al, 2002). Thus, OCRL1 may represent one of the rab6 effectors in this pathway. Our data suggest that rab binding is important for OCRL1 function here, as rab-binding-deficient versions of the OCRL1 5-phosphatase deletion mutant fail to exert dominant inhibitory effects on STxB trafficking. CI-MPR redistribution to endosomes in OCRL1-overexpressing cells was also lost in the rab-binding-deficient mutants, although whether this reflects a role for rab6 interaction or that of another rab is unclear. Interaction with rab5 also suggests a role for OCRL1 in other aspects of endosomal function, which would be consistent with the ability of the protein to bind the endocytic clathrin adaptor AP2 (Ungewickell et al, 2004). Redistribution of the transferrin receptor by overexpression of the OCRL1 5-phosphatase deletion mutant was dependent on rab binding. Rab5 may be the relevant rab here, as it known to play a key role in endosomal receptor trafficking. Binding to rab1 and rab8, on the other hand, may indicate a role for OCRL1 in ER to Golgi or intra-Golgi trafficking, or in trafficking from the TGN to the plasma membrane (Zerial and McBride, 2001). Finally, Rab14 has been implicated in trafficking between the TGN and endosomes (Junutula et al, 2004), and a role for OCRL1 in this trafficking step is suggested by its presence in TGN-associated clathrin buds (Choudhury et al, 2005). Given the scope for indirect effects in the overexpression experiments described here, further experiments will be required to determine the precise role of OCRL1 in the individual trafficking steps occurring at the Golgi apparatus and endosomes, and to define the specific rabs involved in each step.
It has previously been reported that OCRL1 binds rac1 via its Rho GAP-like domain, leading to the suggestion that rac1 targets OCRL1 to the TGN (Faucherre et al, 2003). Our data suggest this is not the case, and that, instead, interaction with rab proteins is required for efficient recruitment of OCRL1 to the TGN. Golgi and endosomal rabs are certainly in the right place to recruit OCRL1, in contrast to rac1, which is mostly found at the plasma membrane. Rac1 is particularly abundant in lamellipodia, structures to which OCRL1 translocates upon growth factor stimulation (Faucherre et al, 2005). It was recently proposed that rac1 is also responsible for OCRL1 translocation to these structures (Faucherre et al, 2005). However, rab5 is also abundant in lamellipodia, where it regulates actin dynamics, intracellular signaling, and endocytosis (Zerial and McBride, 2001; Lanzetti et al, 2004; Shin et al, 2005). We propose that rab5 targets OCRL1 to lamellipodia when cells undergo growth factor stimulation. This would be analogous to Inpp5b, a 5-phosphatase closely related to OCRL1, which also binds rab5 and translocates to lamellipodia upon growth factor stimulation (Shin et al, 2005).
Rabs are known to recruit a variety of effectors onto membranes, and this is thought to be important for organising membranes into functional domains. Rab-mediated recruitment of OCRL1 to domains of early endosomes and the Golgi apparatus is likely important for the functional identity of these compartments. Recruitment will ensure a high local concentration of OCRL1 on the membrane, and this will be enhanced by the ability of rabs to directly stimulate the intrinsic rate of 5-phosphate hydrolysis. This will allow for localised consumption of PtdIns(4,5)P2 and production of PtdIns(4)P. An interesting possibility is that through combined interactions with clathrin and rab proteins, OCRL1 may consume or generate significant amounts of PtdIns(4,5)P2 or PtdIns(4)P, respectively, in clathrin buds or vesicles, which in turn would regulate coat protein dynamics in these structures. For example, OCRL1 together with rab6 or rab14 could regulate dynamics of PtdIns(4)P-binding clathrin adaptor proteins at the TGN, whereas rab5 and OCRL1 could regulate PtdIns(4,5)P2-binding adaptors during endocytosis. Alternatively, OCRL1 may hydrolyse PtdIns(3,4,5)P3 at the plasma membrane in a rab5-regulated manner, which could turn off signals generated by this lipid, or alternatively, indirectly lead to enhanced production of PtdIns(3)P, as has recently been suggested for Inpp5b (Shin et al, 2005).
Materials and methods
Materials and antibodies
All materials were from Sigma or Merck unless otherwise stated. Protease inhibitors (cocktail set III, used at 1:250) were from Calbiochem. Cy3-STxB was from Ludger Johannes (CNRS, Paris). Anti-OCRL1 CT peptide antibody 11520 was a gift from Philip Majerus (Washington University School of Medicine, St Louis, MO). Sheep polyclonal antibody against OCRL1 has been reported previously (Choudhury et al, 2005). KINS and CRAE anti-OCRL1 were raised in rabbits against the peptides KINSQNQPTGIHREPC and CRAEPFEGYLEPNETV, respectively. Sheep antibodies to GFP have been described previously (Diao et al, 2003). Sheep antibodies to GST were prepared from serum raised against GST-OCRL1 by affinity purification on GST coupled to glutathione–Sepharose. Mouse antibodies to p150glued and EEA1 were from BD Transduction Laboratories. Rabbit antibodies to rab1A, rab1B, rab6 (recognises both rab6A and rab6A′), and the myc-tag were purchased from Santa Cruz and Cambridge Antibody, respectively. Mouse antibody UH-4 to GalNAcT2 was a kind gift from Dr Henrik Clausen (University of Copenhagen, Denmark). Monoclonal antibodies to CI-MPR and transferrin receptor were from Affinity Bioreagents and Labvison Corp., respectively. MLO7 polyclonal antibodies to GM130 have been described previously (Diao et al, 2003). Fluorophore and HRP-conjugated secondary antibodies were purchased from Molecular Probes and Tago Immunologicals, respectively.
Molecular biology and yeast two-hybrid analysis
Standard molecular biology techniques were used to make all constructs. Full-length and truncated forms of OCRL1 cDNA (isoform b, Acc. No. NP_001578) were cloned into pEGFP-C1 and pGBKT7 vectors (BD Biosciences). Full-length rab6 cDNA was cloned into a modified pcDNA3.1 vector (Stratagene) to generate an amino-terminal myc-tag. Rab8 was cloned into the pET43.1 (Novagen) vector for expression with NusA/6xhis/S-tags to improve solubility of the recombinant protein. Rac1 and rab cDNAs were cloned into pGex-4T2 (Amersham Pharmacia) and pGADT7 vectors (BD Biosciences) except for constitutively active mutants of rab1, rab2, rab5, and rab6 in the pGADT7 vector, which were a gift from Francis Barr (Max-Planck-Institute of Biochemistry, Martinsried, Germany). Point mutations were generated by PCR using the Quikchange method (Stratagene). All constructs were verified by DNA sequencing using the ABI Prism Big Dye Terminator Cycle Sequencing kit (Applied Biosystems). Primer sequences are available upon request. Yeast two-hybrid analysis was performed as previously described according to the BD Biosciences Clontech manual (Choudhury et al, 2005). Myc-tagged rab5Q79L pcDNA3 was provided by Liz Smythe (University of Sheffield, Sheffield, UK). pGADT7-clathrin terminal domain (TD, residues 1–579) was a gift from Harald Stenmark (Norwegian Radium Hospital, Oslo, Norway).
Random mutagenesis
Random mutagenesis of the OCRL1 linker region was performed using a GeneMorph® II EZClone Domain Mutagenesis Kit (Stratagene). Primers were designed to the OCRL1 internal EcoRI (forward) and SacI (reverse) restriction sites, which were used for the first round of PCR with Mutazyme II polymerase to create a ‘megaprimer'. A second round of PCR incorporated the ‘megaprimer' into pGBKT7-OCRL1 using the appropriate EZClone enzyme mix. DNA was transformed into XL1-Gold Escherichia coli cells; all colonies were harvested to extract the ‘mutaOCRL1' DNA. MutaOCRL1 pGBKT7 was coexpressed with Rab6Q72L pGADT7 in the Y2H system as described. Resulting colonies (low selection) were streaked onto both low and high selection plates. Colonies not growing on high selection were harvested from the equivalent low selection plates and grown in 5 ml of low selection medium. Plasmids were harvested by yeast DNA mini-prep and transformed into electro-competent XL1 Blue E. coli cells that were grown on kanamycin plates to select for the mutaOCRL1-pGBKT7 vector.
Cell culture and transfection
Adherent HeLa, HeLaM, and A431 cells were grown at 37°C and 5% CO2 in DMEM containing 10% foetal calf serum (FCS), 2 mM glutamine, 100 μg/ml penicillin G, and 100 μg/ml streptomycin sulphate. Suspension HeLa cells were grown at 37°C and 5% CO2 in RPMI 1640 medium supplemented as DMEM. Adherent cells were transiently transfected with FuGENE 6 (Roche Diagnostics) according to the manufacturer's instructions and incubated for 20 h before fixation or lysis. Metabolic labelling was performed in labelling medium (nine parts met/cys-free DMEM containing 10% dialysed FCS mixed with one part met/cys-containing DMEM) containing 50 μCi/ml 35S-met/cys (NEN Life Sciences) for 18–22 at 37°C.
Shiga toxin trafficking
Shiga toxin trafficking was performed as described previously (Choudhury et al, 2005).
RNA interference
HeLa cells were transfected with siRNA oligos using Oligofectamine (Invitrogen) as described previously (Choudhury et al, 2005). Rab1A and rab6A were targeted with SMARTpool oligonucleotides from Dharmacon Research. Control siRNA pGL2 targeting luciferase was purchased from Eurogentec. Cells were analysed 72 h post-transfection.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as described previously (Choudhury et al, 2005).
Protein preparation
His-tagged WT OCRL1 and the G664D mutant were prepared from insect cells as previously described (Choudhury et al, 2005). Plasmids encoding GST, GST-rab WT, GST-rab constitutively active mutants, GST-rac1Q61L, and GST-clathrin TD were transformed into E. coli BL21 (DE3) cells. Cells were induced with 0.1 mM IPTG for 3 h at 30°C. Cells were lysed in Bugbuster HT (Novagen) containing protease inhibitors, and recombinant proteins were purified on glutathione–Sepharose beads (Amersham Pharmacia). Rab8 WT and constitutively active mutant were prepared with a NusA tag as previously reported (Hattula et al, 2002), except that the culture was induced at 30°C and cells were lysed in Bugbuster HT.
Preparation of cell extracts
Cytosol was prepared from S3 HeLa cells as previously described (Choudhury et al, 2005). HeLa extracts for RNAi analysis were prepared by lysing adherent cells from a 12-well dish in 200 μl HNT (20 mM Hepes, pH 7.4, 0.2 M NaCl, 1% Triton X-100, 1 mM DTT) containing protease inhibitors at RT for 5 min. HeLa extracts for pull-down experiments were prepared by lysing cells from a 10 cm dish in 2 ml HNMT (20 mM Hepes, pH 7.4, 0.1 M NaCl, 5 mM MgCl2, 0.1% Triton X-100, 1 mM DTT) containing protease inhibitors on ice for 5 min. Lysates were clarified by centrifugation at 90 000 g for 15 min at 4°C.
Pull-down experiments
HeLa cytosol was desalted into HNM buffer (20 mM Hepes, pH 7.4, 0.1 M NaCl, 5 mM MgCl2, 1 mM DTT) and clarified by centrifugation at 50 000 r.p.m. for 15 min in a TLA55 rotor. Nucleotide loading onto WT rab-proteins was performed as previously described except that GMP-PNP was used as a GTP analogue (Christoforidis and Zerial, 2000). HeLa cytosol (1 mg) or cell lysate (400 μl) were incubated for 3 h or overnight at 4°C with 100–250 μg of GST-fusion protein coupled to glutathione–Sepharose beads in the presence of 100 μM GDP or GMP-PNP. In some experiments, recombinant 6his-OCRL1 or tryptic digest (10 μg) was incubated with beads coupled to 10 μg GST-fusion protein. Beads were washed three times with HNMT containing 0.25% Triton X-100 supplemented with 10 μM GDP or GMP-PNP. Bound proteins were eluted with SDS–PAGE sample buffer (GFP-OCRL1 lysate and recombinant 6his-OCRL1 pull downs) or by incubating beads in elution buffer (20 mM Hepes, pH 7.4, 1 M NaCl, 20 mM EDTA, 0.25% Triton X-100, 1 mM DTT) for 20 min at RT (pull downs with HeLa cytosol). Eluted protein was TCA precipitated and resuspended into SDS–PAGE sample buffer. Bound and input proteins were subjected to SDS–PAGE and Western blotting or Coomassie blue staining. Pull downs using NusA-Rab8 proteins were performed as above except the proteins were immobilised on S-protein agarose (Novagen).
Solid-phase binding
Binding was performed in 96-well plates (Costar). Wells were coated with 50 μl purified recombinant OCRL1 (50 μg/ml in HNM) overnight at 4°C. After washing twice with 0.2 ml HNMTW (HNM containing 0.05% Tween 20), wells were blocked in HNMTW containing 1% BSA for 1.5 h on ice. Wells were incubated with the 50 μl GST-tagged binding partner diluted in HNMTW for 2 h on ice, washed three times with 0.2 ml HNMTW, before incubation with 50 μl sheep anti-GST antibodies (0.2 μg/ml in HNMTW) for 1 h on ice. Wells were washed again and incubated with 50 μl HRP-conjugated anti-sheep IgG (0.5 μg/ml) for a further 1 h on ice. HRP was detected by incubating with 50 μl/well ABTS substrate at room temperature for 5 min, followed by measuring absorbance at 405 nm in a plate reader.
Limited proteolysis
Purified recombinant OCRL1 (5 μg) was incubated with 125 ng trypsin for 30 min at 30°C in trypsin buffer (20 mM Hepes, pH 7.4, 0.1 M KCl, 5 mM MgCl2). GFP-tagged OCRL1 constructs were immunoprecipitated from metabolically labelled HeLa cells before digestion. Cells expressing GFP-OCRL1 constructs were lysed in HNMT (500 μl per 6 cm dish), and lysates clarified by centrifugation before immunoprecipitation with 2 μg sheep anti-GFP and protein G-Sepharose. After washing with HNMT followed by TB, beads were split into four equal aliquots and incubated for 20 min at 25°C in TB containing 0, 0.01, 0.1, or 1 μg trypsin. Reactions were stopped by addition of SDS sample buffer containing protease inhibitors and boiling before SDS–PAGE.
Lipid phosphatase assays
Lipid vesicles were prepared by sonication in PBS to give a final concentration of 200 μg/ml phosphatidylcholine, 40 μg/ml phosphatidylethanolamine, 8 μg/ml PtdIns(4,5)P2 (Lipid Products, UK), and 0.04 μCi 3H-PtdIns(4,5)P2 (Perkin–Elmer, Wellesley, MA) in the final assay. 6his-OCRL1 (0.04 μM) was allowed to interact with GST-tagged small GTPases (2.4 μM) on ice for 20 min in HNM buffer. The assay was started by addition of vesicles, and after incubation at 30°C for 20 min, stopped by adding acidified choloroform/methanol (1:2). The production of PIP was analysed by thin-layer chromatography (TLC) followed by phosphorimaging (Fuji). Data were processed using AIDA software.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Acknowledgments
We thank our colleagues for generously providing reagents as noted in the text. We also thank Drs Bruno Goud (CNRS, Paris) and Francis Barr (Max Planck Institute for Biochemistry, Martinsried) for sharing unpublished data. Matthew Ball provided excellent technical assistance. We are also grateful to Drs Philip Woodman and Viki Allan for critical reading of the manuscript. This work was supported by an MRC senior research fellowship (G117/494) and a BBSRC project grant (34/C17842) awarded to ML and a Wellcome Trust PhD studentship to NH (068847/Z/02/A). SC was supported by a grant from the UK Lowe Syndrome Trust.
References
- Antony C, Cibert C, Geraud G, Santa Maria A, Maro B, Mayau V, Goud B (1992) The small GTP-binding protein rab6p is distributed from medial Golgi to the trans-Golgi network as determined by a confocal microscopic approach. J Cell Sci 103 (Part 3): 785–796 [DOI] [PubMed] [Google Scholar]
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, McInnes RR, Nussbaum RL (1992) The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 358: 239–242 [DOI] [PubMed] [Google Scholar]
- Balla T (2005) Inositol-lipid binding motifs: signal integrators through protein–lipid and protein–protein interactions. J Cell Sci 118: 2093–2104 [DOI] [PubMed] [Google Scholar]
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655–1657 [DOI] [PubMed] [Google Scholar]
- Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, Konstantakopoulos A, Lucocq J, Johannes L, Rabouille C, Greene LE, Lowe M (2005) Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell 16: 3467–3479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforidis S, Zerial M (2000) Purification and identification of novel Rab effectors using affinity chromatography. Methods 20: 403–410 [DOI] [PubMed] [Google Scholar]
- Cremona O, De Camilli P (2001) Phosphoinositides in membrane traffic at the synapse. J Cell Sci 114: 1041–1052 [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Cozier GE, Banting G, Mellor H (2001) Modular phosphoinositide-binding domains—their role in signalling and membrane trafficking. Curr Biol 11: R882–R893 [DOI] [PubMed] [Google Scholar]
- De Matteis M, Godi A, Corda D (2002) Phosphoinositides and the golgi complex. Curr Opin Cell Biol 14: 434–447 [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Godi A (2004) PI-loting membrane traffic. Nat Cell Biol 6: 487–492 [DOI] [PubMed] [Google Scholar]
- Diao A, Rahman D, Pappin DJ, Lucocq J, Lowe M (2003) The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J Cell Biol 160: 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressman MA, Olivos-Glander IM, Nussbaum RL, Suchy SF (2000) Ocrl1, a PtdIns(4,5)P(2) 5-phosphatase, is localized to the trans-Golgi network of fibroblasts and epithelial cells. J Histochem Cytochem 48: 179–190 [DOI] [PubMed] [Google Scholar]
- Faucherre A, Desbois P, Nagano F, Satre V, Lunardi J, Gacon G, Dorseuil O (2005) Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum Mol Genet 14: 1441–1448 [DOI] [PubMed] [Google Scholar]
- Faucherre A, Desbois P, Satre V, Lunardi J, Dorseuil O, Gacon G (2003) Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. Hum Mol Genet 12: 2449–2456 [DOI] [PubMed] [Google Scholar]
- Fridmann-Sirkis Y, Siniossoglou S, Pelham HR (2004) TMF is a golgin that binds Rab6 and influences Golgi morphology. BMC Cell Biol 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattula K, Furuhjelm J, Arffman A, Peranen J (2002) A Rab8-specific GDP/GTP exchange factor is involved in actin remodeling and polarized membrane transport. Mol Biol Cell 13: 3268–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V (2005) Phosphoinositide regulation of clathrin-mediated endocytosis. Biochem Soc Trans 33: 1285–1289 [DOI] [PubMed] [Google Scholar]
- Junutula JR, De Maziere AM, Peden AA, Ervin KE, Advani RJ, van Dijk SM, Klumperman J, Scheller RH (2004) Rab14 is involved in membrane trafficking between the Golgi complex and endosomes. Mol Biol Cell 15: 2218–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetti L, Palamidessi A, Areces L, Scita G, Di Fiore PP (2004) Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 429: 309–314 [DOI] [PubMed] [Google Scholar]
- Lowe CU, Terrey M, Mac LE (1952) Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child 83: 164–184 [DOI] [PubMed] [Google Scholar]
- Lowe M (2005) Structure and function of the Lowe syndrome protein OCRL1. Traffic 6: 711–719 [DOI] [PubMed] [Google Scholar]
- Mallard F, Tang BL, Galli T, Tenza D, Saint-Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L (2002) Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol 156: 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier N, Satre V, Lerouge E, Berthoin F, Lunardi J (2000) OCRL1 mutation analysis in French Lowe syndrome patients: implications for molecular diagnosis strategy and genetic counseling. Hum Mutat 16: 157–165 [DOI] [PubMed] [Google Scholar]
- Olivos-Glander IM, Janne PA, Nussbaum RL (1995) The oculocerebrorenal syndrome gene product is a 105-kD protein localized to the Golgi complex. Am J Hum Genet 57: 817–823 [PMC free article] [PubMed] [Google Scholar]
- Pereira-Leal JB, Seabra MC (2001) Evolution of the Rab family of small GTP-binding proteins. J Mol Biol 313: 889–901 [DOI] [PubMed] [Google Scholar]
- Roschinger W, Muntau AC, Rudolph G, Roscher AA, Kammerer S (2000) Carrier assessment in families with Lowe oculocerebrorenal syndrome: novel mutations in the OCRL1 gene and correlation of direct DNA diagnosis with ocular examination. Mol Genet Metab 69: 213–222 [DOI] [PubMed] [Google Scholar]
- Roth MG (2004) Phosphoinositides in constitutive membrane traffic. Physiol Rev 84: 699–730 [DOI] [PubMed] [Google Scholar]
- Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, Frankel WN, Solimena M, De Camilli P, Zerial M (2005) An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol 170: 607–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H (1998) EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394: 494–498 [DOI] [PubMed] [Google Scholar]
- Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M (1994) Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J 13: 1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell A, Ward ME, Ungewickell E, Majerus PW (2004) The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci USA 101: 13501–13506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk MR, Lucast L, Di Paolo G, Romanelli AJ, Suchy SF, Nussbaum RL, Cline GW, Shulman GI, McMurray W, De Camilli P (2003) Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat Biotechnol 21: 813–817 [DOI] [PubMed] [Google Scholar]
- White J, Johannes L, Mallard F, Girod A, Grill S, Reinsch S, Keller P, Tzschaschel B, Echard A, Goud B, Stelzer EH (1999) Rab6 coordinates a novel Golgi to ER retrograde transport pathway in live cells. J Cell Biol 147: 743–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HL, Janmey PA (2003) Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol 65: 761–789 [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H (2001) Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2: 107–117 [DOI] [PubMed] [Google Scholar]
- Zhang X, Hartz PA, Philip E, Racusen LC, Majerus PW (1998) Cell lines from kidney proximal tubules of a patient with Lowe syndrome lack OCRL inositol polyphosphate 5-phosphatase and accumulate phosphatidylinositol 4,5-bisphosphate. J Biol Chem 273: 1574–1582 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4