

Abstract
c-Myc (Myc) is highly expressed in developing embryos where it regulates body size by controlling proliferation but not cell size. However, Myc is also induced in many postmitotic tissues, including adult myocardium, in response to stress where the predominant form of growth is an increase in cell size (hypertrophy) and not number. The function of Myc induction in this setting is unproven. Therefore, to explore Myc's role in hypertrophic growth, we created mice where Myc can be inducibly inactivated, specifically in adult myocardium. Myc-deficient hearts demonstrated attenuated stress-induced hypertrophic growth, secondary to a reduction in cell growth of individual myocytes. To explore the dependence of Myc-induced cell growth on CycD2, we created bigenic mice where Myc can be selectively activated in CycD2-null adult myocardium. Myc-dependent hypertrophic growth and cell cycle reentry is blocked in CycD2-deficient hearts. However, in contrast to Myc-induced DNA synthesis, hypertrophic growth is independent of CycD2-induced Cdk2 activity. These data suggest that Myc is required for a normal hypertrophic response and that its growth-promoting effects are also mediated through a CycD2-dependent pathway.
Keywords: cardiac growth, cardiac muscle, cell cycle, hypertrophy, Myc
Introduction
During development, cell cycle progression is normally tightly coupled to the accumulation of cell mass (cell growth) (Neufeld and Edgar, 1998); however, in some adult postmitotic tissues, cell growth can become uncoupled from proliferation resulting in hypertrophic growth (Dorn and Force, 2005). The molecular mechanisms that regulate hypertrophy and the means whereby proliferation and cell growth are normally coupled are poorly understood, but the fact that they are coupled during development suggests common regulatory mechanisms. One molecule that has been implicated in mediating both forms of growth in many tissues, including the heart, is c-Myc (Myc).
Myc is highly expressed in fetal, proliferating cardiac myocytes. However, soon after birth the myocytes cease to divide corresponding with the downregulation of Myc. Transgenic mice that overexpress Myc in the fetal myocardium develop ventricular enlargement secondary to myocyte hyperplasia, suggesting that Myc is sufficient to induce proliferative growth in the heart as well (Jackson et al, 1990). Although Myc is not expressed in the adult heart under normal physiological conditions, it is upregulated rapidly in response to virtually all hypertrophic stimuli (Izumo et al, 1988) but the growth response is limited to hypertrophy and not hyperplasia (Soonpaa and Field, 1997). The importance of Myc in mediating this hypertrophic growth in postmitotic myocytes is controversial but several lines of evidence support the concept that Myc can mediate cellular growth in the absence of cell division. In Drosophila, decreased expression of dMyc, the ortholog of mammalian Myc, in wing imaginal disc cells reduced cell proliferation and cell size (Johnston et al, 1999). In contrast, dMyc overexpression resulted in increased cell size without affecting cell division. In mammalian cells, deleting Myc in B cells and hepatocytes reduced cell size (Iritani and Eisenman, 1999; Baena et al, 2005). Conversely, previous studies from our lab demonstrated that activation of Myc specifically in adult myocardium was sufficient to induce hypertrophic growth and this growth was accompanied by cell cycle reentry (Xiao et al, 2001). Similarly, overexpression of Myc in B lymphocytes both in vitro (Schuhmacher et al, 1999) and in vivo (Iritani and Eisenman, 1999) is also associated with an increase in cell size, independent of cell cycle progression.
The mechanisms whereby Myc regulates hypertrophic growth are less clear but it is interesting to note that genes responsible for Myc's ability to promote cell cycling have also been implicated in regulating cell size in certain contexts. Myc activation in the heart is accompanied by the upregulation of Cyclin D2 (CycD2) and cyclin-dependent kinase (Cdk) -2 and -4 activities (Xiao et al, 2001). The link between this Myc-induced Cdk activity and Myc-induced proliferation has been well established (Amati et al, 1998). Myc stimulated Cdk2 kinase activity, which is critical for cell cycle progression (Amati et al, 1998), occurs in large part by antagonizing the association of CdkIs, p27 and p21, with Cdk2 (Bouchard et al, 1999; Perez-Roger et al, 1999). Upregulation of Myc target genes Cdk4 and CycD2 (Coller et al, 2000) leads to the rapid sequestration of p21 and p27, liberating Cdk2. Consistent with this model, cells from CycD2−/− mice, unlike wild-type controls, do not reenter the cell cycle in response to Myc (Bouchard et al, 1999). The role of CycD2 in mediating Myc-induced hypertrophic growth was not addressed in these studies.
Interestingly, CycD and Cdk4 have also been implicated in regulating cell size in Drosophila but, similar to Myc, the effect of CycD–Cdk4 overexpression varied according to cell type (Datar et al, 2000). In undifferentiated proliferating cells, CycD/Cdk4 overexpression caused accelerated cell division (hyperplasia) without affecting cell size (Datar et al, 2000). However, in differentiated cells, CycD/Cdk4 caused cell enlargement (hypertrophy), potentially through Rb-independent pathways. More recent studies have suggested that CycD/Cdk4 stimulates cell growth through regulation of mitochondrial activity (Frei et al, 2005). In mammals, numerous studies have implicated CycD/Cdk4 in regulating cardiac hypertrophy; however, this connection is, so far, based primarily on cell culture experiments. Hypertrophic signals upregulate CycD2 expression and CycD-dependent kinase activity in the cardiac myocytes (Li et al, 1998; Busk et al, 2002). Although forced expression of CycD2 in cardiac myocytes provoked cell division and not hypertrophy (Busk et al, 2005; Pasumarthi et al, 2005), CycD2 was overexpressed at a developmental time point where the myocytes were still capable of reentering the cell cycle. Overexpression of CycD2 in adult, postmitotic cardiac myocytes has not been reported. However, inhibiting G1-Cyc/Cdk activity in adult myocytes blocks hypertrophic growth (Nozato et al, 2001).
To determine the role of Myc in regulating hypertrophic growth specifically in adult, postmitotic myocardium and whether its growth effects are mediated through CycD2-dependent pathways in the heart, we developed inducible, cardiac-restricted Myc-deficient mice and a model of inducible, myocardial-specific Myc expression in a CycD2-null background. Deletion of Myc attenuated hypertrophic growth in response to both hemodynamic and pharmacologic hypertrophic stimuli and resulted in an increase in cardiac apoptosis. Myc-induced hypertrophic growth was dependent on the presence of CycD2 similar to what has been reported for Myc-induced cell cycle reentry, but was independent of Cdk2 activity.
Results
Creation of inducible, cardiac-specific Myc-deficient mice
The role of Myc in cell cycle control and cellular proliferation has been well studied and Myc has been shown to be critical for the regulation of mammalian body size during development by controlling cell number (Trumpp et al, 2001). However, Myc is also expressed in many adult tissues in pathological conditions, including cardiac hypertrophy, where cell growth but not cell division occurs. Myc's role in regulating normal physiology in these adult tissues remains unknown. To explore the role of Myc in cardiac hypertrophy in vivo, we created inducible Myc-deficient mice where Myc can be specifically deleted in adult myocardium with a tamoxifen-regulated Cre, MerCreMer (MCM). When Cre is activated in these mice (MCM;Mycfl/fl) with 4-hydroxytamoxifen (4-OHT), the resulting recombination excises Myc coding exons 2 and 3 (Figure 1A). This deletion can be identified with ΔMycfl primers, which give a ∼600 bp product when recombination has occurred (de Alboran et al, 2001). PCR performed on total ventricular DNA from control (MCM;Myc+/+) or Myc-null (MCM;Mycfl/fl) mice with ΔMycfl primers demonstrated no spontaneous recombination in the absence of ligand. However, 4-OHT treatment resulted in recombination in the ventricles of MCM;Mycfl/fl mice (Figure 1B). This recombination only occurred in the hearts of the 4-OHT-treated, MCM;Mycfl/fl mice and was not seen in other tissues (Figure 1C). As Myc is expressed at very low levels in adult myocardium at baseline, its expression was induced by subjecting mice to hemodynamic or pharmacological hypertrophic stimuli in vivo. MCM;Mycfl/fl mice with or without 4-OHT treatment underwent Sham or transverse aortic constriction (TAC) operation to induce cardiac hypertrophy. TAC induced a 9.7-fold increase in Myc protein expression in vehicle-treated MCM;Mycfl/fl mice when compared to Sham-operated mice. Pretreatment of MCM:Mycfl/fl mice with 4-OHT reduced the expected increase in Myc protein after hemodynamic load by 66.5% (9.67±0.44- versus 3.24±2.11-fold, P<0.005; Figure 1D). This degree of recombination is consistent with previous reports using the MCM mice (Sohal et al, 2001) and likely reflects in part, that the heart consists of a number of different cell types although the majority of the protein arises from authentic cardiac myocytes. To confirm this finding, we analyzed Myc expression in total ventricular RNA prepared from treated or untreated MCM;Mycfl/fl hearts after stimulation with a pharmacologic hypertrophic agonist, isoproterenol (ISO). Myc expression was dramatically attenuated in response to ISO stimulation in 4-OHT treated MCM:Mycfl/fl hearts in vivo (Figure 1E).
Figure 1.
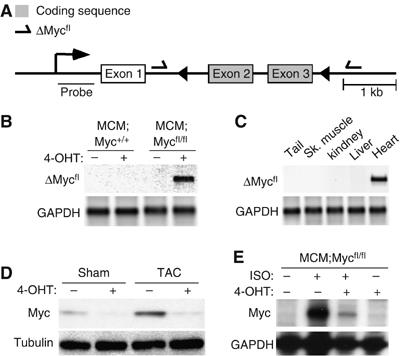
Creation of inducible cardiac-specific Myc-deficient mice. To create inducible, cardiac-restricted Myc-deficient mice, we bred mice with a floxed Myc allele to mice expressing a tamoxifen-inducible Cre only in the heart. (A) Schematic diagram depicting the floxed Myc allele (⇀=ΔMycfl primers; ◂=LoxP sites). (B, C) PCR utilizing ΔMycfl primers was performed on genomic DNA from the indicated tissues and genotypes. (D) Myc protein expression in ventricular lysates from vehicle- and 4-OHT-treated MCM;Mycfl/fl mice after Sham or TAC surgery was determined by Western blotting. (E) RPA analysis on total ventricular RNA from MCM;Mycfl/fl mice after ISO stimulation.
Cardiac hypertrophy is attenuated in Myc-deficient mice after hemodynamic stress
MCM;Mycfl/fl mice were born with expected Mendelian numbers and appeared phenotypically normal. Baseline left ventricular (LV) size, function and histology were normal in MCM;Mycfl/fl mice after 4-OHT treatment to delete Myc (Figure 2A and Supplementary Table 1). To determine the role of Myc in hypertrophic growth, we subjected adult mice to a hemodynamic stress induced by TAC surgery and measured the increase in heart weight normalized to body weight (HW/BW) after 2 weeks. Vehicle-treated MCM;Mycfl/fl mice demonstrated the expected enhanced LV wall thickness (Figure 2A) and concentric hypertrophy after TAC, while 4-OHT-treated MCM;Mycfl/fl in the absence of Myc did not. When compared to Sham-operated animals, MCM:Myc+/+ (4.74±0.01 versus 7.62±0.98 mg/g, P<0.001; Figure 2B) or vehicle-treated MCM;Mycfl/fl (5.05±0.16 versus 7.42±0.24 mg/g, P<0.001; Figure 2B) with intact Myc expression demonstrated the expected increase in HW/BW ratio after TAC. In contrast, the hypertrophic response in Myc-deficient mice was attenuated. Although 4-OHT-treated MCM;Mycfl/fl mice tolerated TAC hemodynamically over the period studied and did not develop left ventricular dysfunction (Supplementary Table 1), they developed less hypertrophy (4.85±0.17 versus 6.01±0.29 mg/g, P<0.01). While TAC induced a 46.9% increase in heart weight in vehicle-treated MCM;Mycfl/fl mice compared to Sham-operated mice, heart weight only increased 23.9% in 4-OHT-treated MCM:Mycfl/fl mice after TAC (P<0.001).
Figure 2.
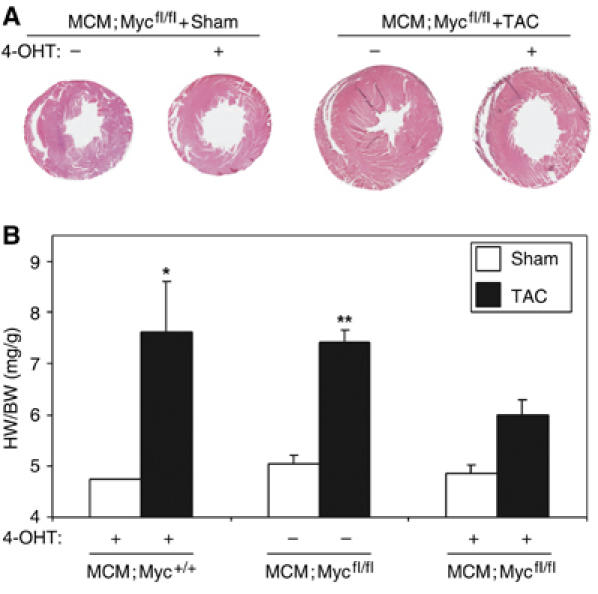
Myc-null mice decrease cardiac mass and cardiac myocyte size. MCM;Myc+/+ and MCM;Mycfl/fl mice were treated with vehicle or 4-OHT for 5 days to induce recombination and then subjected to Sham or TAC surgery and followed for 2 weeks. (A) H&E-stained, perfusion-fixed MCM;Mycfl/fl hearts demonstrating development of concentric LV hypertrophy in vehicle-treated MCM;Mycfl/fl mice. (B) To determine if myocardial hypertrophy had occurred, heart weights (mg) normalized to body weight (g) were analyzed. *P<0.05 for 4-OHT-treated MCM;Myc+/+ mice after TAC versus MCM;Myc+/+ Sham animals. **P<0.05 for vehicle-treated MCM;Mycfl/fl mice+TAC versus Sham animals or 4-OHT-treated MCM;Mycfl/fl mice+TAC (n=6 in each group).
To confirm that the reduced HW/BW ratio in Myc-deficient hearts after pressure-overload represented a reduction in cell growth, we measured cardiac myocyte fiber width on wheat germ agglutinin-stained myocardial sections in these animals (Figure 3A). Cardiac myocyte width was 45% greater in control vehicle-treated MCM;Mycfl/fl subjected to TAC compared with 4-OHT-treated MCM;Mycfl/fl mice (17.58±0.33 versus 12.08±0.28 μm, P<0.01; Figure 3B). Consistent with this reduction in cardiac hypertrophy, Myc-deficient mice also showed attenuated upregulation of hypertrophic marker genes such as atrial natriuretic factor (ANF) and β-myosin heavy chain (βMHC) mRNA compared with untreated MCM:Mycfl/fl mice after TAC (Figure 3C).
Figure 3.
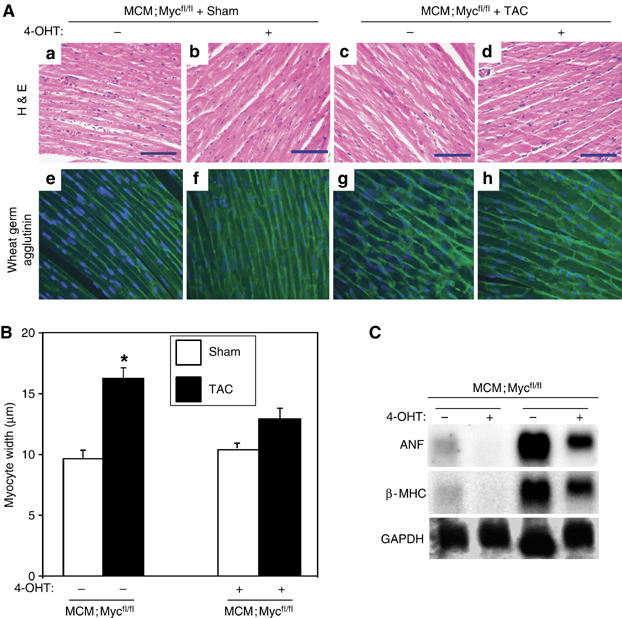
Myc-null mice decrease cardiac mass and cardiac myocyte size. MCM;Mycfl/fl mice with 5 days of 4-OHT or vehicle treatment underwent Sham or TAC surgery. Hearts were harvested after 2 weeks and histological analysis was performed. (A) H&E (a–d) and wheat germ agglutinin (e–h)-stained myocardial sections from Sham-operated MCM;Mycfl/fl mice treated with vehicle (a and e) or 4-OHT (b, f) versus TAC-operated MCM;Mycfl/fl treated with vehicle (c, g) or 4-OHT (d, h). Scale bar=70 μm. (B) Fiber width of wheat germ agglutinin-stained hearts from Sham and TAC mice were quantified (n=4 in each group). *P<0.01 for MCM;Mycfl/fl with vehicle+TAC versus MCM;Mycfl/fl with 4-OHT+TAC. (C) Total ventricular RNA (10 μg) from 4-OHT-treated mice with indicated genotypes was probed with specific 32P-labelled probes for atrial natriuretic factor (ANF), β-myosin heavy chain (βMHC), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Interestingly, there was a significant increase in interstitial fibrosis as measured by picrosirius red staining of ventricular sections from 4-OHT treated MCM;Mycfl/fl mice subjected to Sham versus TAC (1.00±0.04 versus 1.48±0.09-fold, P<0.01; Figure 4A and B). To determine if this was in response to cell loss, we assessed apoptosis by TdT-mediated dUTP-biotin nick-end labeling (TUNEL) staining. Apoptotic cardiomyocytes were rarely detected in MCM;Mycfl/fl mice with or without 4-OHT treatment mice that underwent sham operation. However, apoptosis increased 12-fold in 4-OHT-treated MCM;Mycfl/fl mice subjected to TAC compared to untreated Sham animals (P<0.001; Figure 4C), suggesting Myc was also necessary for cardiac myocyte survival with hypertrophic stimuli.
Figure 4.
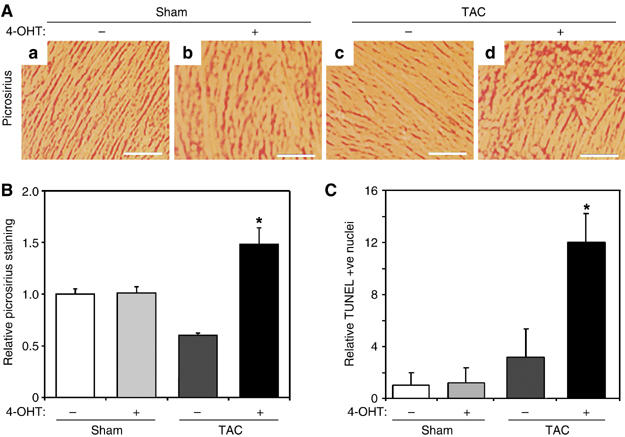
Myc-null hearts demonstrate increased fibrosis and apoptosis after TAC. (A) Representative picrosirius-stained myocardial sections from vehicle- or 4-OHT-treated MCM;Mycfl/fl mice subjected to Sham or TAC surgery. Scale bar=70 μm. (B) The amount of ventricular fibrosis was quantified and displayed relative to vehicle-treated MCM;Mycfl/fl+Sham mice (n=4 in each group). *P<0.01 for 4-OHT-treated MCM;Mycfl/fl+TAC versus 4-OHT-treated MCM;Mycfl/fl+Sham or vehicle-treated MCM;Mycfl/fl+TAC. (C) Results of TUNEL staining were quantified and the numbers of TUNEL –+ve nuclei expressed relative to vehicle-treated MCM:Mycfl/fl+Sham mice. *P<0.01 for 4-OHT-treated MCM;Mycfl/fl+TAC versus 4-OHT-treated MCM;Mycfl/fl+Sham or vehicle-treated MCM;Mycfl/fl+TAC.
Myc-deficient mice display reduced cardiac growth and increased apoptosis with chronic isoproterenol infusion
To determine if this attenuated hypertrophy and fibrosis was a general response to hypertrophic stimuli, we treated MCM:Myc+/+ or MCM;Mycfl/fl mice with 4-OHT for 5 days and then subjected the mice to an infusion of ISO or vehicle for 1 week. ISO stimulation resulted in a 30.4% increase in HW/BW ratio in 4-OHT-treated MCM:Myc+/+ mice compared to only a 17.4% increase in ISO-treated, Myc-deficient MCM;Mycfl/fl mice (P<0.05; Figure 5A). ISO stimulation increased heart-to-body weight ratio in 4-OHT-treated control MCM:Myc+/+ mice (5.17±0.12 versus 6.74±0.40 mg/g, P<0.01; Figure 5A). In contrast, MCM;Mycfl/fl mice demonstrated no significant increase (4.83±0.62 versus 5.67±0.33 mg/g, P=NS). This difference in HW/BW ratio was paralleled by a 19.4% increase in cardiac myocyte fiber width in ISO-stimulated MCM:Myc+/+ mice when compared to fiber width in vehicle-treated MCM:Myc+/+ or MCM;Mycfl/fl hearts (13.79±0.56 versus 11.55±0.30 or 11.04±0.14 μm, P<0.01; Figure 5B). There was no significant increase in cardiac myocyte fiber width in ISO-treated MCM;Mycfl/fl hearts compared to vehicle-treated MCM;Mycfl/fl hearts, suggesting the reduced HW/BW was a result of attenuated myocyte hypertrophy. Similar to the results in the TAC model, ISO treatment in MCM;Mycfl/fl mice was also accompanied by increased interstitial fibrosis as seen on H&E-stained myocardial sections (Figure 5C-b). To further explore the cause for this, we quantified TUNEL +ve nuclei on sections of ventricular tissue. As shown, ISO-treated MCM;Mycfl/fl ventricles displayed a 6.3-fold increase in TUNEL +ve nuclei (P<0.01; Figure 5D), suggesting that Myc was also necessary for cardiac myocyte survival with hypertrophic stimuli.
Figure 5.
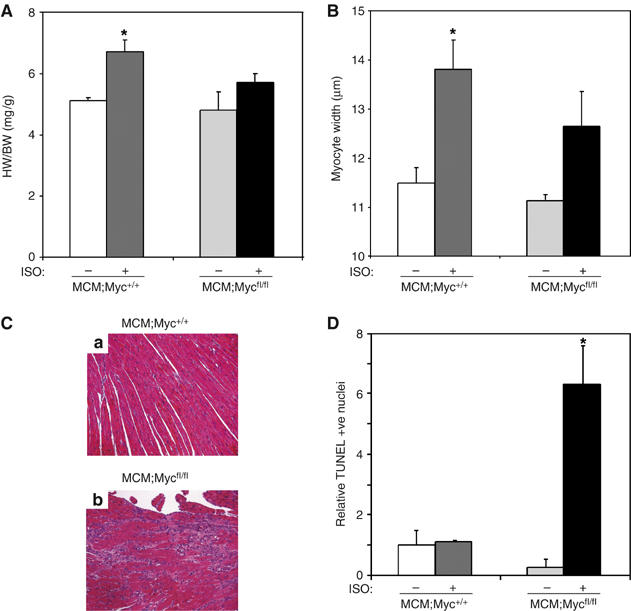
Hypertrophy is attenuated in ISO-stimulated, Myc-deficient hearts. To pharmacologically induce hypertrophy, MCM;Myc+/+ and MCM;Mycfl/fl mice were stimulated with ISO for 1 week after 5 days of 4-OHT treatment. (A) To assess the hypertrophic response, HW/BW ratios were analyzed. *P<0.01 for MCM;Myc+/++ISO versus MCM;Myc+/++vehicle and P<0.05 versus MCM;Mycfl/fl+ISO (n=5 per group). (B) To assess cardiac myocyte hypertrophy, cross-sectional myocyte fiber width in hearts were quantified. *P<0.01 for ISO-stimulated MCM:Myc+/+ versus vehicle-treated MCM:Myc+/+ or MCM;Mycfl/fl hearts. (C) H&E-stained myocardial sections from ISO-stimulated MCM:Myc+/+ (Ca) or MCM;Mycfl/fl (Cb) mice. (D) TUNEL +ve cardiac nuclei were quantified and the results expressed relative to vehicle-treated MCM:Myc+/+ mice. *P<0.05 for ISO-stimulated MCM;Mycfl/fl versus vehicle-treated MCM;Mycfl/fl or ISO-stimulated MCM:Myc+/+ hearts.
Inhibition of Myc in vitro blocks ET-1-stimulated hypertrophic growth and CycD2 upregulation
To explore the mechanisms underlying the growth effects of Myc in cardiac myocytes, we utilized an in vitro model of hypertrophic growth by stimulating neonatal rat ventricular myocytes (NRVMs) with endothelin-1 (ET-1). NRVMs were serum starved for 48 h and then exposed to ET-1, which upregulates Myc expression (Figure 6A) and induces hypertrophic growth (Figure 6Ba versus b) without increasing cell number (data not shown). To confirm our in vivo results, we blocked ET-1-induced Myc expression with an adenovirus overexpressing an siRNA to Myc (Ad-siMyc). Overexpressing a control protein (Ad-LacZ) or scrambled siRNA sequence (data not shown) had no effect on Myc induction.
Figure 6.
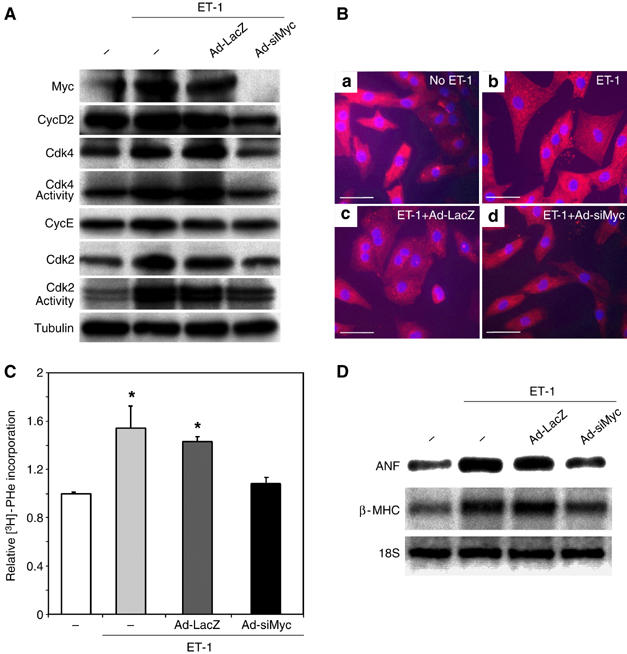
Inhibition of Myc blocks ET-1-dependent hypertrophy in vitro. (A) NRVMs were infected with no virus (−), AdLacZ or AdsiMyc. Myocytes were stimulated with 100 nM ET-1 for 8 h and protein lysates probed for the indicated proteins or Cdk kinase activities determined and autoradiographs shown. (B) To assess the role of Myc on cardiac myocyte cell size, NRVMs were not infected with virus (a, b) or infected with Ad-LacZ (c) or Ad-siMyc (d). NRVMs (b–d) were stimulated with ET-1 for 40 h and then fixed and immunostained with MF20 (red) and DAPI (blue). Scale bar=50 μm. (C) To determine relative protein synthesis, NRVMs were incubated with or without ET-1 for 16 h in the presence of no virus (−), Ad-LacZ or Ad-siMyc and [3H]Phenylalanine incorporation was determined. Results were normalized to uninfected, vehicle-treated cells. *P<0.01 for ET-1-stimulated, uninfected or Ad-LacZ-infected NRVMs versus unstimulated or ET-1-stimulated Ad-siMyc cultures. Each experiment was repeated three times. (D) Total NRVM RNA (2 μg) from the indicated treatments was probed with specific Dig-labeled probes for atrial natriuretic factor (ANF), β-myosin heavy chain (βMHC) and ribosomal 18S RNA.
Myc is well known to regulate the expression of a number of G1 cyclins and Cdk activities. As these proteins are also regulated in cardiac myocytes after hypertrophic agonists, we examined the expression of a panel of cell cycle activators (Figure 6A). Although stimulation of NRVMs with ET-1 upregulated CycD2 leading to increased Cdk2 and 4 activity, these changes were blocked by inhibiting Myc expression (Figure 6A). Inhibiting ET-1-induced Myc expression also prevented the expected increase in myocyte size (Figure 6Bb versus d) presumably though the inhibition of Myc-dependent protein synthesis seen in control myocytes after ET-1 stimulation (1.00±0.05 versus 1.54±0.18, P<0.01; Figure 6C). Although ET-1 induced both ANF and β-MHC, the expression of these hypertrophic markers was attenuated by inhibiting Myc expression, similar to the in vivo results (Figure 6D). It is well known that Cdk2 and 4 activity cooperate for Myc-induced cell cycle progression but the role of these proteins in mediating Myc-induced hypertrophic growth is unknown.
CycD2 is necessary for Myc-induced cardiac hypertrophy
Our previous studies had demonstrated that cardiac-specific activation of Myc in adult α-MHC-MycER mice with 4-OHT could stimulate both cardiac myocyte hypertrophy and cell cycle reentry but not proliferation when compared to similarly treated nontransgenic (NTg) littermates (Xiao et al, 2001). To determine if Myc-induced hypertrophic growth was CycD2 dependent, as has been described for Myc-induced cell cycle entry (Bouchard et al, 1999; Perez-Roger et al, 1999), we created MycER mice deficient for CycD2. Although Myc activation induced CycD2 expression and Cdk2 and 4 activity in MycER myocardium, no upregulation was seen in similarly treated CycD2-null mice (Figure 7A). Treatment of MycER;CycD2+/+ mice with 4-OHT for 1 week resulted in a 26.6% increase in HW/BW ratio (4.36±0.17 versus 5.52±0.19, P<0.01; Figure 7B). However, activation of Myc in CycD2-null mice did not result in a significant change in cardiac mass (4.51±0.11 versus 4.53±0.19, P=NS; Figure 7B). Likewise, the 85-fold increase in BrdU-positive nuclei in MycER;CycD2+/+ seen after 4-OHT activation of Myc (0.03±0.03 versus 2.54±0.8; P<0.01) was abolished in MycER;CycD2−/− mice (0.04±0.03 versus 0.35±0.11, P=n.s.; Figure 7C and D).
Figure 7.
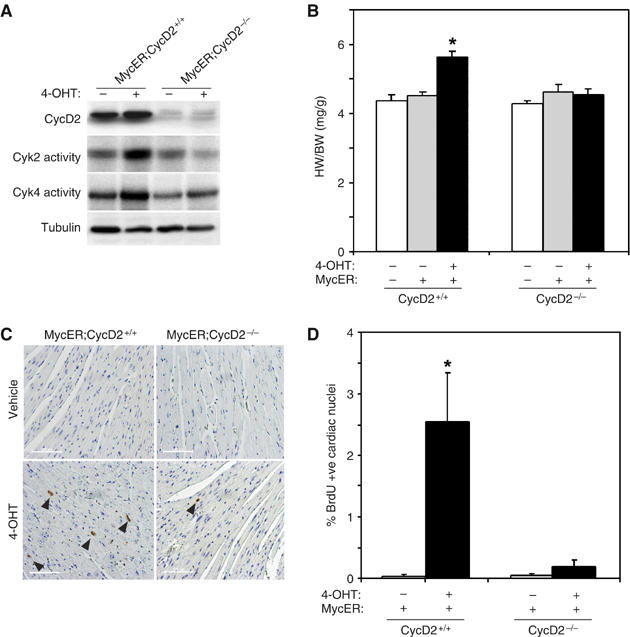
Cyclin D2 is necessary for Myc-induced cell cycle re-entry and hypertrophic growth in the heart. To determine the relative importance of Cyclin D2 to Myc's ability to induce cell cycle re-entry and/or hypertrophic growth in the heart, bigenic MycER;CycD2−/− mice were created. (A) Expression of CycD2 determined by Western blotting and Myc-induced Cdk kinase activity in 4-OHT-treated MycER;CycD2+/+ versus MycER;CycD2−/− hearts is shown. (B) The ratio of HW/BW after 7 days of Myc activation in control (MycER;CycD2+/+) or CycD2-null mice (MycERCycD2−/−) was determined. *P<0.01 for 4-OHT-treated MycER;CycD2+/+ versus MycERCycD2−/− hearts. (C, D) Myocardial sections from 4-OHT-treated MycER;CycD2+/+ or MycER;CycD2−/− were stained for BrdU incorporation (arrowheads identify BrdU+ve cardiac nuclei). The percentage of BrdU +ve nuclei was quantified (n=5 per group). *P<0.05 for 4-OHT-treated MycER;CycD2+/+ versus MycERCycD2−/− hearts. Scale bar=100 μm.
Myc-induced cell cycle reentry but not hypertrophic growth is Cdk2 depend-
To clarify if the lack of hypertrophic growth in MycER;CycD2−/− mice was due to a lack of CycD2 and potentially Cdk4 activity versus the inability to upregulate Cdk2 activity in these hearts, we examined the role of Cdk2 and Cdk4 in Myc-induced hypertrophic growth and protein synthesis in vitro. NRVMs infected with an adenovirus overexpressing Myc (AdMyc) increased Cdk2 activity and to a lesser extent Cdk4, similar to its effects in vivo (Figure 8A). To block Cdk activity, we coinfected AdMyc cultures with a dominant-negative Cdk2 (Ad-dnCdk2) or Cdk4 (Ad-dnCdk4) virus. These vectors specifically blocked Myc-induced activation Cdk2 or 4 activity (Figure 8A). The effect of these interventions on cardiac myocyte cell size was assessed with forward scatter by flow cytometry. Forced expression of Myc increased NRVM forward scatter from 420.5±103.3 to 474.0±119.8 indicating larger NRVMs. Although inhibition of Cdk2 activity had no effect on Myc-induced hypertrophic growth (470.2±120.7), blocking Cdk4 activity attenuated Myc-induced hypertrophy (439.5±97.7). Similarly, overexpression of Myc induced protein synthesis by 47% as measured by [3H]phenylalanine incorporation, which was not affected by inhibiting Cdk2 activity but was prevented by blocking Cdk4 activity (Figure 8C). In contrast, both Ad-dnCdk2 and Ad-dnCdk4 inhibited the Myc-induced 6.2-fold increase in DNA synthesis (6.23±1.23 versus 1.18±0.4 or 3.64±0.33-fold, P<0.01; Figure 8D), suggesting that while Myc-dependent reactivation of DNA synthesis in cardiac myocytes requires both Cdk2 and 4, Myc-stimulated cell growth is Cdk2 independent.
Figure 8.
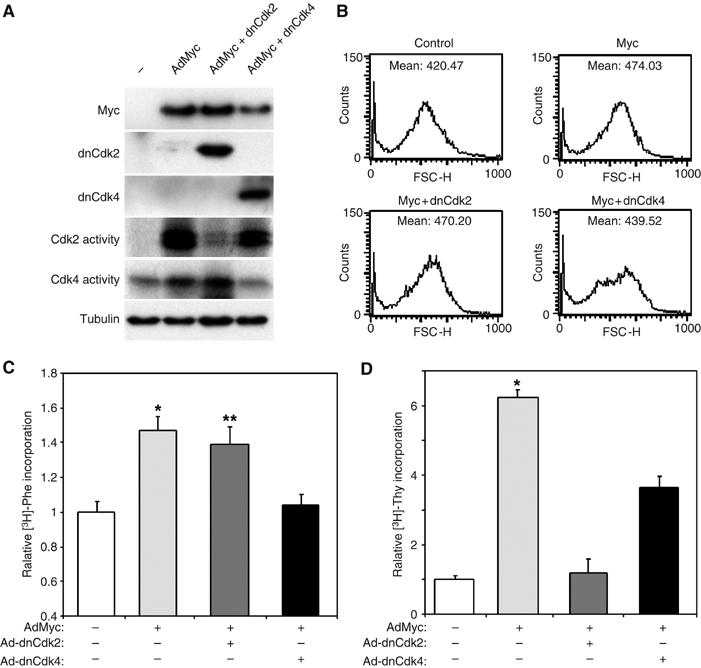
Myc-induced DNA synthesis but not hypertrophy requires Cdk2 activity. To explore the dependence of Myc's growth effects on Cdk2 and 4, we infected NRVM with no virus (−) or AdMyc with or with adenoviruses expressing dominant forms of Cdk2 and Cdk4. (A) Expression of infected proteins was confirmed by Western blotting on lysates prepared from NVRMs infected by the indicated vectors. The ability of the dominant negative constructs to specifically inhibit Myc-induced Cdk kinase activity was determined on parallel lysates. (B) To assess cardiac myocyte cell size, NRVMs were analyzed by flow cytometry and forward scatter (FSC) was measured. Histograms of FSC in a representative experiment of NRVMs infected by the indicated viruses are shown. The mean FSC is shown for each condition. (C) Relative protein synthesis was determined by [3H]Phenylalanine incorporation of myocytes infected with the indicated adenoviral vectors. Results are presented relative to uninfected cultures. *P<0.001 AdMyc versus uninfected cultures or AdMyc+dnCdk4. **P<0.01 for AdMyc+dnCdk2 versus uninfected cultures or AdMyc+dnCdk4. (D) Relative DNA synthesis was determined by [3H]thymidine incorporation of NRVMs infected with the indicated adenoviral vectors. *P<0.0001 for AdMyc versus uninfected cultures, P<0.001 versus AdMyc+dnCdk2 and P<0.05 versus AdMyc+Ad-dnCdk4.
Discussion
We have previously shown that overexpression of Myc is sufficient to induce cardiac hypertrophy in adult myocardium (Xiao et al, 2001). However, whether Myc is also necessary for cell growth and the mechanisms whereby Myc might induce cardiomyocyte hypertrophy remained unknown. In the present study, we investigated Myc's role in cardiac hypertrophy by inactivating Myc both in vivo and in vitro. The results demonstrate that an Myc–Cyclin D2-dependent pathway is required for hypertrophic growth in cardiac myocytes. Deleting Myc in adult myocardium attenuated but did not block cardiac hypertrophy completely. This reduction in cardiac myocyte size was accompanied by a reduction in fetal gene markers normally upregulated in hypertrophic hearts, suggesting that the entire hypertrophic phenotype, not just growth, was suppressed. In contrast, inhibition of Myc in cultured cardiac myocytes completely abolished the hypertrophic response induced by ET-1. One explanation for this divergent response may simply be related to simple technical differences between the two models. ET-1 predominantly stimulates G-protein-coupled, endothelin-A receptors in cardiac myocytes, which activate a downstream signaling cascade mediated by the G(q) heterotrimeric G proteins (Sugden and Clerk, 2005). In contrast, TAC initiates a host of direct and indirect autocrine and paracrine neurohumoral factors, which have been implicated in mediating cardiac growth (Dorn and Force, 2005). It is also possible that the relative role of Myc in the various forms of cardiac hypertrophy explored here varies. Additionally, the time course of the two models differed greatly. The in vitro experiments determined the relative importance of Myc to protein synthesis at an early time point when Myc is highly expressed. In contrast, the in vivo studies compared hypertrophic responses after 1 and 2 weeks of hemodynamic stress. It is possible that Myc is more important to the initial hypertrophic response and that later redundant mechanisms are able to at least partially compensate. This may explain why the observed attenuation of hypertrophy was greater in the shorter, ISO-stimulated model.
The finding that Myc was important for myocyte survival in hypertrophy was unexpected. Hypertrophic agonists led to a small, but significant increase in apoptosis in Myc-null hearts that resulted in increased interstitial fibrosis. One of the paradoxes facing investigators studying Myc function is the observation that both Myc over- and underexpression results in apoptosis. Of the two, Myc-induced apoptosis has been studied in much more detail (Pelengaris et al, 2002). Ectopic expression of Myc sensitizes cells to a wide range of apoptotic stimuli including tumor necrosis factor-alpha (TNFα) (Klefstrom et al, 1994) by inducing cytochrome c (Morrish et al, 2003) release from the mitochondrial intermembrane space into the cytosol where it can promote apoptosis (Iaccarino et al, 2003). The link between Myc deficiency and apoptosis is less clear, but may also be mediated through a mitochondrial pathway. A large proportion of the nuclear-regulated mitochondrial genes are direct Myc targets (Morrish et al, 2003), and are induced in cells upon growth stimulation. Ensuring that the cell maintains the correct stoichiometry of respiratory complex subunits and their correct assembly in the mitochondria is critical for adapting to the increased metabolic demands associated with cell growth (Poyton and McEwen, 1996). Disruption of this balance can have detrimental effects, and examples exist of mitochondrial dysfunction that result from both reduced (Schapira and Cock, 1999) or increased expression of genes involved in mitochondrial function, such as adenine nucleotide translocase-1 (Bauer et al, 1999) and the mitochondrial hinge protein (Okazaki et al, 1998). Therefore, Myc underexpression could lead to mitochondrial dysfunction and apoptosis by deregulating genes involved in mitochondrial function.
A major finding of this study is that a CycD2-dependent pathway mediates Myc-induced hypertrophic growth, although the end effectors remain unclear. It is generally accepted that Cdk2 kinase activation is an essential step in Myc-induced G1-exit and that induction of CycD2 is a critical preliminary step in this process, as the newly formed CycD/Cdk4 complexes sequester Cdk inhibitors p27 and p21 (Bouchard et al, 1999; Perez-Roger et al, 1999). We found that both Myc-induced cardiac cell cycle entry and cellular growth was blocked in CycD2-null mice. However, in contrast to its effects on cell cycle progression, Myc-induced hypertrophic growth was independent of Cdk2 activity. These results are consistent with the preferential role of Cdk2 in cardiac hyperplasia not hypertrophy, shown by forced expression in vivo (Liao et al, 2001). Given that Cdk2 is critical for Myc's proliferative effects, whether CycD2 leads to proliferation or hypertrophy might be dependent on whether excess Cdk inhibitors are present. Thus, the differential growth effect of CycD2 in proliferative versus postmitotic cells may be attributable to a developmental increase in Cdk inhibitors, as is seen in the postnatal heart (Poolman et al, 1998). This report represents the first evidence that Myc-dependent hypertrophic growth is also CycD2 dependent and that the pathways controlling cell cycle progression and cellular growth diverge at CycD.
Several lines of evidence suggest that G1 cyclins and Cdks might be critical for the control of cell growth. In Drosophila, CycD/Cdk4 stimulates and controls cell growth in postmitotic cells (Datar et al, 2000). This CycD/Cdk4-induced cell growth was dependent on a gene encoding the mitochondrial ribosomal protein, mRpL12 (Frei et al, 2005). In the absence of mRpL12, cells demonstrated reduced growth and mitochondrial activity suggesting that CycD/Cdk4 controls cell growth via a mitochondrial-dependent pathway. A number of reports have implicated CycD/Cdk4 in regulating cardiac hypertrophy in mammalian cells as well, although the downstream effectors have not been identified (Busk et al, 2002; Tamamori-Adachi et al, 2002). Likewise, several reports have documented that inhibiting G1-Cyclin/Cdk activity in adult, postmitotic cardiac myocytes can attenuate hypertrophic growth (Tamamori et al, 1998; Nozato et al, 2001). We recently demonstrated that cardiac hypertrophy is accompanied by increased RNA polymerase (pol) III transcription, which is related to changes in both the activity and level of the RNA pol III-specific transcription factor TFIIIB (Goodfellow et al, 2006). Myc can potentiate TFIIIB activity directly by binding and activating transcription (Gomez-Roman et al, 2003) or potentially indirectly, by removing the inhibiting effects of the retinoblastoma gene product (Rb). Hypophosphorylated, active Rb binds TFIIIB and prevents its interactions with TFIIIC or RNA pol III (Larminie et al, 1997). Myc-induced Cdk activity could potentially remove this inhibitory effect. Given the fundamental role Myc plays in regulating cell growth, it will be critical in future studies to determine the role and mechanisms whereby CycD/Cdk4 might induce cardiac hypertrophy.
Materials and methods
Transgenic mice and animal studies
The inducible α-myosin heavy chain (α-MHC)-MycER transgenic mice have been described (Xiao et al, 2001). Myc-floxed mice (Mycfl/fl) were provided by Dr F Alt and genotyped as described (de Alboran et al, 2001). The tamoxifen-inducible MerCreMer (MCM) mice were generated by Dr J Molkentin under the control of the α-MHC promoter (Sohal et al, 2001). To activate Myc or Cre in these transgenic mice, 1 mg of 4-OHT (Sigma) dispersed in peanut oil by sonication was injected intraperitoneally daily. Control littermates were injected with peanut oil alone. To induce excisional recombination of Myc, mice were treated with 4-OHT for 5 days. To identify recombination, PCR conditions were chosen for ΔMycfl primers that do not amplify the >2.5 kb fragment in wild-type mice and no product is obtained. CycD2-deficient mice were a kind gift from Dr P Sicinski (Sicinski et al, 1996). All mice were maintained on a C57 (MycER, CycD2) or FVB (MCM;Mycfl/fl) background. Animals were handled in accordance with institutional guidelines.
For TAC, a fixed pressure overload was obtained by surgically constricting the transverse aorta, as has been previously described (Rockman et al, 1994). Age-matched Sham-operated animals underwent the identical surgical procedure, except that the aortic constriction was not placed. For the ISO infusion model, 7-day osmotic minipumps (Alzet, model 2001) loaded with 0.2 ml of ISO (28 μg/ml per 25 g body weight) were implanted into the subcutaneous space of 10-week-old mice via a small intrascapular incision.
Cell culture and adenovirus preparation
NRVM were prepared as previously described (MacLellan et al, 2000). For all cell culture experiments, NRVM were serum-starved in serum-free DMEM for 36 h before use. Construction of AdMyc (Mitchell and El-Deiry, 1999), dnCdk2 and dnCdk4 (Ferguson et al, 2000) has been previously described. Viruses were propagated and titered according to established protocols (MacLellan et al, 2000).
To create an siRNA adenovirus to Myc, a shuttle plasmid was constructed by inserting a 21-mer RNA oligonucleotide directed against the rat and mouse Myc sequence (5′-aagaggcggacacacaacgtc-3′) into the unique ApaI and HindIII sites of pDC.silencer(U6), which was kindly supplied by Dr Abdellatif (Yue et al, 2004). Adenoviruses were propagated and titered according to published protocols (MacLellan et al, 2000). For cell culture experiments, NRVM were infected with adenoviruses 36 h before treatment.
Protein analysis
Western blots were performed on protein extracts from whole ventricles, according to established protocols (MacLellan et al, 2000). Antibodies were obtained from Santa Cruz Biotechnology, Inc. unless otherwise noted. Protein expression was visualized using horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence reagents (Amersham Biosciences). Immune complex kinase assays for Cdk2 and Cdk4 activity were performed on 500 μg of ventricular lysates (Li et al, 1998). The immunocomplexed pellet was incubated in 30 μl kinase buffer with 1 μg of Rb (Santa Cruz Biotech.) or histone H1 (Upstate Biotech, Inc.) substrate, 5 μCi of [γ-32P]ATP, 1 mM DTT and 5 μM ATP and then electrophoresed through 10% acrylamide gels.
Histology and morphometric analysis
Hearts were fixed overnight in 4% paraformaldehyde buffered with PBS and routinely processed. BrdU labeling was achieved by injecting 50 mg of BrdU per gram of body weight intraperitoneally. To identify DNA synthesis, paraffin-embedded sections were probed with antibodies against BrdU (Zymed). Antibody against sarcomeric myosin heavy chains (MF20; Developmental Hybridoma Studies Bank) and diamidinophenolindole (DAPI) were used according to the manufacturer's instructions. Apoptosis was determined using ApopTag fluorescein in situ apoptosis detection kit (Chemicon) or TACS™ 2 TdT blue label in situ apoptosis detection kit (Trevigen). Apoptotic cells were detected with fluorescein conjugate or visualized with an enzymatic reaction using the TUNEL method. The apoptosis rates were determined by examining >2500 cardiac nuclei per heart sections. Secondary antibodies were purchased from Molecular Probes. Myocyte fiber widths were measured on perfusion-fixed myocardial sections using a computerized morphometric system (SigmaScan, Systat Inc.). All myocytes were measured at the same magnification with the observer blinded to the genotype of the animals.
Northern blot analysis
Total RNA was isolated from ventricles using RNA STAT 60 Kit (Tel-Test, Inc.). Northerns blots were performed according to established protocols using radioactive (MacLellan et al, 2000) or digoxigenin (DIG)-labeled probes (MacLellan et al, 2000). The oligonucleotide and cDNA probes used have been reported (Ross et al, 1998).
Measurement of myocyte cell number, cell size, DNA and protein synthesis
To determine myocyte cell number, DNA and protein synthesis, serum-starved myocytes were infected with the indicated adenoviral vectors (50 PFU/myocyte). To determine cell number, myocytes were trypsinized and total cell counts determined using a Coulter Counter (Becton-Dickinson). To estimate protein synthesis, myocytes were labeled with 5 μCi/ml of [3H]phenylalanine (Amersham Corp.) for 4 h after which cell precipitates were solubilized and counted. To determine the relative DNA synthesis, myocytes were cultured in media containing 5 μCi/ml of [3H]thymidine (ICN) for 6 h and thymidine incorporation quantified. Flow cytometry to assess cell size was performed at the UCLA Flow Cytometry Laboratory based on forward scatter as previously described (Gylys et al, 2004). Aliquots of 105 cells in 500 μl 1 × PBS were dispensed into small conical tubes (Falcon). At least 10 000 events were collected before analysis. All samples were analyzed using a Becton Dickinson FACScan analytic flow cytometer (Becton Dickinson) with FCS Express software (DeNovo).
Statistical analysis
All data are presented as mean±s.e.m. except results of forward scatter, which are presented as mean±s.d. Results were compared by analysis of variance and Fisher's PLSD tests, using significance at a P-value <0.05.
Supplementary Material
Supplement Information – Table 1
Acknowledgments
We thank K Hayakawa, L Hong and S Chan for their technical assistance. We are also grateful to P Sicinski, F Alt, SC Pang, J Molkentin and Michael Schneider for their helpful discussions and reagents. This work was supported by gifts from the Laubisch Fund and the Piansky Family Endowment as well as NIH R01 HL 70748 and AHA 0340087N grants to WRM.
References
- Amati B, Alevizopoulos K, Vlach J (1998) Myc and the cell cycle. Front Biosci 3: D250–D268 [DOI] [PubMed] [Google Scholar]
- Baena E, Gandarillas A, Vallespinos M, Zanet J, Bachs O, Redondo C, Fabregat I, Martinez A, de Alboran IM (2005) c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci USA 102: 7286–7291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer MK, Schubert A, Rocks O, Grimm S (1999) Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol 147: 1493–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J 18: 5321–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk PK, Bartkova J, Strom CC, Wulf-Andersen L, Hinrichsen R, Christoffersen TE, Latella L, Bartek J, Haunso S, Sheikh SP (2002) Involvement of cyclin D activity in left ventricle hypertrophy in vivo and in vitro. Cardiovasc Res 56: 64–75 [DOI] [PubMed] [Google Scholar]
- Busk PK, Hinrichsen R, Bartkova J, Hansen AH, Christoffersen TE, Bartek J, Haunso S (2005) Cyclin D2 induces proliferation of cardiac myocytes and represses hypertrophy. Exp Cell Res 304: 149–161 [DOI] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR (2000) Expression analysis with oligonucleotide microarrays reveals that Myc regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA 97: 3260–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datar SA, Jacobs HW, de la Cruz AF, Lehner CF, Edgar BA (2000) The Drosophila cyclin D–Cdk4 complex promotes cellular growth. EMBO J 19: 4543–4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alboran IM, O'Hagan RC, Gartner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, Alt FW (2001) Analysis of c-Myc function in normal cells via conditional gene-targeted mutation. Immunity 14: 45–55 [DOI] [PubMed] [Google Scholar]
- Dorn GW II, Force T (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115: 527–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KL, Callaghan SM, O'Hare MJ, Park DS, Slack RS (2000) The Rb-Cdk4/6 signaling pathway is critical in neural precursor cell cycle regulation. J Biol Chem 275: 33593–33600 [DOI] [PubMed] [Google Scholar]
- Frei C, Galloni M, Hafen E, Edgar BA (2005) The Drosophila mitochondrial ribosomal protein mRpL12 is required for Cyclin D/Cdk4-driven growth. EMBO J 24: 623–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421: 290–294 [DOI] [PubMed] [Google Scholar]
- Goodfellow SJ, Innes F, Derblay LE, MacLellan WR, Scott PH, White RJ (2006) Regulation of RNA polymerase III transcription during hypertrophic growth. EMBO J 25: 1522–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Cole GM (2004) Enrichment of presynaptic and postsynaptic markers by size-based gating analysis of synaptosome preparations from rat and human cortex. Cytometry A 60: 90–96 [DOI] [PubMed] [Google Scholar]
- Iaccarino I, Hancock D, Evan G, Downward J (2003) c-Myc induces cytochrome c release in Rat1 fibroblasts by increasing outer mitochondrial membrane permeability in a Bid-dependent manner. Cell Death Differ 10: 599–608 [DOI] [PubMed] [Google Scholar]
- Iritani BM, Eisenman RN (1999) c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci USA 96: 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumo S, Nadal-Ginard B, Mahdavi V (1988) Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci USA 85: 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson T, Allard MF, Sreenan CM, Doss LK, Bishop SP, Swain JL (1990) The c-Myc proto-oncogene regulates cardiac development in transgenic mice. Mol Cell Biol 10: 3709–3716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P (1999) Drosophila Myc regulates cellular growth during development. Cell 98: 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klefstrom J, Vastrik I, Saksela E, Valle J, Eilers M, Alitalo K (1994) c-Myc induces cellular susceptibility to the cytotoxic action of TNF-alpha. EMBO J 13: 5442–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larminie CG, Cairns CA, Mital R, Martin K, Kouzarides T, Jackson SP, White RJ (1997) Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J 16: 2061–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM, Poolman RA, Brooks G (1998) Role of G1 phase cyclins and cyclin-dependent kinases during cardiomyocyte hypertrophic growth in rats. Am J Physiol 275: H814–H822 [DOI] [PubMed] [Google Scholar]
- Liao HS, Kang PM, Nagashima H, Yamasaki N, Usheva A, Ding B, Lorell BH, Izumo S (2001) Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res 88: 443–450 [DOI] [PubMed] [Google Scholar]
- MacLellan WR, Xiao G, Abdellatif M, Schneider MD (2000) A novel Rb- and p300-binding protein inhibits transactivation by MyoD. Mol Cell Biol 20: 8903–8915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KO, El-Deiry WS (1999) Overexpression of c-Myc inhibits p21WAF1/CIP1 expression and induces S-phase entry in 12-O-tetradecanoylphorbol-13-acetate (TPA)-sensitive human cancer cells. Cell Growth Differ 10: 223–230 [PubMed] [Google Scholar]
- Morrish F, Giedt C, Hockenbery D (2003) c-Myc apoptotic function is mediated by NRF-1 target genes. Genes Dev 17: 240–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld TP, Edgar BA (1998) Connections between growth and the cell cycle. Curr Opin Cell Biol 10: 784–790 [DOI] [PubMed] [Google Scholar]
- Nozato T, Ito H, Watanabe M, Ono Y, Adachi S, Tanaka H, Hiroe M, Sunamori M, Marum F (2001) Overexpression of Cdk Inhibitor p16INK4a by adenovirus vector inhibits cardiac hypertrophy in vitro and in vivo: a novel strategy for the gene therapy of cardiac hypertrophy. J Mol Cell Cardiol 33: 1493–1504 [DOI] [PubMed] [Google Scholar]
- Okazaki M, Ishibashi Y, Asoh S, Ohta S (1998) Overexpressed mitochondrial hinge protein, a cytochrome c-binding protein, accelerates apoptosis by enhancing the release of cytochrome c from mitochondria. Biochem Biophys Res Commun 243: 131–136 [DOI] [PubMed] [Google Scholar]
- Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ (2005) Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res 96: 110–118 [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Khan M, Evan G (2002) c-Myc: more than just a matter of life and death. Nat Rev Cancer 2: 764–776 [DOI] [PubMed] [Google Scholar]
- Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H (1999) Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J 18: 5310–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolman RA, Gilchrist R, Brooks G (1998) Cell cycle profiles and expressions of p21Cip1 and p27Kip1 during myocyte development. Int J Cardiol 67: 133–142 [DOI] [PubMed] [Google Scholar]
- Poyton RO, McEwen JE (1996) Crosstalk between nuclear and mitochondrial genomes. Annu Rev Biochem 65: 563–607 [DOI] [PubMed] [Google Scholar]
- Rockman HA, Wachhorst SP, Mao L, Ross J Jr (1994) ANG II receptor blockade prevents ventricular hypertrophy and ANF gene expression with pressure overload in mice. Am J Physiol 266: H2468–H2475 [DOI] [PubMed] [Google Scholar]
- Ross RS, Pham C, Shai SY, Goldhaber JI, Fenczik C, Glembotski CC, Ginsberg MH, Loftus JC (1998) Beta1 integrins participate in the hypertrophic response of rat ventricular myocytes. Circ Res 82: 1160–1172 [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cock HR (1999) Mitochondrial myopathies and encephalomyopathies. Eur J Clin Invest 29: 886–898 [DOI] [PubMed] [Google Scholar]
- Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, Kohlhuber F (1999) Control of cell growth by c-Myc in the absence of cell division. Curr Biol 9: 1255–1258 [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA (1996) Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384: 470–474 [DOI] [PubMed] [Google Scholar]
- Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ Res 89: 20–25 [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Field LJ (1997) Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol 272: H220–H226 [DOI] [PubMed] [Google Scholar]
- Sugden PH, Clerk A (2005) Endothelin signalling in the cardiac myocyte and its pathophysiological relevance. Curr Vasc Pharmacol 3: 343–351 [DOI] [PubMed] [Google Scholar]
- Tamamori M, Ito H, Hiroe M, Terada Y, Marumo F, Ikeda MA (1998) Essential roles for G1 cyclin-dependent kinase activity in development of cardiomyocyte hypertrophy. Am J Physiol 275: H2036–H2040 [DOI] [PubMed] [Google Scholar]
- Tamamori-Adachi M, Ito H, Nobori K, Hayashida K, Kawauchi J, Adachi S, Ikeda MA, Kitajima S (2002) Expression of cyclin D1 and Cdk4 causes hypertrophic growth of cardiomyocytes in culture: a possible implication for cardiac hypertrophy. Biochem Biophys Res Commun 296: 274–280 [DOI] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773 [DOI] [PubMed] [Google Scholar]
- Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, Fishbein MC, MacLellan WR (2001) Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res 89: 1122–1129 [DOI] [PubMed] [Google Scholar]
- Yue Y, Lypowy J, Hedhli N, Abdellatif M (2004) Ras GTPase-activating protein binds to Akt and is required for its activation. J Biol Chem 279: 12883–12889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Information – Table 1