

Abstract
Mitochondria are present as tubular organelles in neuronal projections. Here, we report that mitochondria undergo profound fission in response to nitric oxide (NO) in cortical neurons of primary cultures. Mitochondrial fission by NO occurs long before neurite injury and neuronal cell death. Furthermore, fission is accompanied by ultrastructural damage of mitochondria, autophagy, ATP decline and generation of free radicals. Fission is occasionally asymmetric and can be reversible. Strikingly, mitochondrial fission is also an early event in ischemic stroke in vivo. Mitofusin 1 (Mfn1) or dominant-negative Dynamin related protein 1 (Drp1K38A) inhibits mitochondrial fission induced by NO, rotenone and Amyloid-β peptide. Conversely, overexpression of Drp1 or Fis1 elicits fission and increases neuronal loss. Importantly, NO-induced neuronal cell death was mitigated by Mfn1 and Drp1K38A. Thus, persistent mitochondrial fission may play a causal role in NO-mediated neurotoxicity.
Keywords: Alzheimer's disease, dynamin related protein 1, mitofusin 1, Parkinson's disease, stroke
Introduction
That mitochondria become injured in the pathogenesis of neurodegenerative disorders is well documented (Beal, 2000; Bossy-Wetzel et al, 2004a). Less clear, however, is what exactly causes mitochondrial damage observed in common, sporadic disorders like stroke, Parkinson's disease (PD) and Alzheimer's disease (AD). Additionally, the precise role that mitochondrial dysfunction plays in the disease process remains uncertain nor is it clear whether mitochondrial defects are cause or consequence of neurodegeneration.
To accommodate high-energy demands a single neuron harbors several hundreds of mitochondria (Bereiter-Hahn and Voth, 1994; Wallace, 2005) that are dynamic, able to migrate, fuse and divide. Mitochondrial fission and fusion is orchestrated by a conserved machinery, involving dynamin related GTPases that exert competing functions (van der Bliek, 2000; Osteryoung and Nunnari, 2003; Chen and Chan, 2004; Okamoto and Shaw, 2005). Fission requires the large GTPase Dynamin-related protein 1 (Drp1) (Smirnova et al, 2001). Drp1 is present in the cytoplasm and binds to the mitochondrial outer membrane via Fis1 (James et al, 2003). Upon activation by an unknown mechanism, Drp1 assembles into large complexes at future scission sites (cut sites) on the inner mitochondrial membrane (Frank et al, 2001). How Drp1 mediates outer membrane scission is unclear, but it has been proposed that, similar to Dynamin, it may act as a mechano-enzyme (Yoon et al, 2001). Conversely, mitochondrial fusion is regulated by mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy 1 (OPA1) (Olichon et al, 2002; Chen et al, 2003). Mitochondrial fission and fusion play a role in cell division and differentiation (Bereiter-Hahn and Voth, 1994; Yaffe, 1999). More recently, mitochondrial fission has been implicated in synaptic and spine plasticity in neurons (Li et al, 2004).
Mitochondrial fission has to be balanced by fusion events. Emerging new evidence shows that tipping the equilibrium toward continuous mitochondrial fission can evoke a neurodegenerative cascade (Bossy-Wetzel et al, 2003). Intriguingly, inherited loss-of-function mutations of MFN2 or OPA1 cause progressive neuropathies in humans. MFN2 mutations cause Charcot-Marie-Tooth Type 2A (CMT-2A), a peripheral neuropathy characterized by motor and sensory neuron loss (Zuchner et al, 2004). OPA1 mutations cause autosomal dominant optic atrophy characterized by retinal ganglion cell and optic nerve degeneration (Delettre et al, 2000). Thus, loss of mitochondrial fusion genes and consequent continuous fission appears to play a causal role in the initiation of neurodegeneration.
Whether excessive mitochondrial fission also underlies common sporadic neurodegenerative disease such as ischemic stroke, PD and AD is unknown. The goal of this study was to start to address this question. Nitric oxide (NO) is a neurotransmitter in the central nervous system required for learning and memory (Holscher, 1997). However, when produced in excess NO reacts with superoxide anion (O2.−) to form highly neurotoxic peroxynitrite (ONOO−). NO/ONOO− is thought to contribute to the pathogenesis of several neurodegenerative disorders, including stroke, PD and AD (Dawson and Dawson, 1998; Boje, 2004).
How NO/ONOO− promotes neurotoxic effects is still debated, but mitochondrial injury appears to be a potential culprit. NO/ONOO− directly inhibits respiratory chain complexes (Brown and Cooper, 1994; Radi et al, 1994; Brorson et al, 1999). Additionally, NO/ONOO− stress can stimulate release of Zn2+ from intracellular stores, leading to mitochondrial damage and caspase-independent, neuronal demise (Bossy-Wetzel et al, 2004b).
The mechanism by which postmitotic primary neurons die in neurodegenerative disease appears to be distinct from the classical rapid apoptotic cell death of cell lines. Both caspase-dependent and independent pathways appear to operate in the neuronal loss in neurodegenerative disorders (Li et al, 2000; Leist and Jaattela, 2001; Friedlander, 2003; Cregan et al, 2004). Since effective treatments are lacking, neurodegenerative diseases remain at present without cure. Therefore, the molecular pathways associated with neurodegenerative disorders are currently under intense investigation and new discoveries are needed to identify the critical events that mark their onset and progression (Bossy-Wetzel et al, 2004a). Here, we show that a number of neurotoxic agents including NO, rotenone and Amyloid-β (Aβ) peptide induce profound mitochondrial fission. Blocking mitochondrial fission by expression of Drp1K38A or wild-type Mfn1 alleviates neuronal cell death by NO.
Results
NO evokes rapid mitochondrial fission
To model NO-mediated neuronal injury we exposed mixed cortical cultures with the NO donor, S-nitrosocysteine (SNOC) (Lei et al, 1992; Bonfoco et al, 1995). SNOC concentrations of 50–200 μM yield approximately ∼1.5–5 μM [NO], respectively, in the first 20 min (Brorson et al, 1999). To track single mitochondria in real-time, primary cortical neuronal cultures were transfected with a vector encoding Mito-DsRed2, a red fluorescent marker specifically labeling the mitochondrial matrix. Fluorescence time lapse microscopy and three-dimensional (3D) image reconstruction revealed that mitochondria of resting neurons take on typical elongated filamentous morphology of variable length in neuronal projections (Figure 1A). Strikingly, NO exposure triggers dramatic fragmentation of single filamentous mitochondria into many, small and round organelles in the presence of 100 μM pan-caspase inhibitor zVAD-fmk methyl ester (Figure 1A). Caspase inhibitors were added to evaluate their effect on mitochondrial morphology. Mitochondrial fission by NO occurs in the presence and absence of caspase inhibitors (Figure 1A and data not shown). Once initiated, mitochondrial fission is fast and synchronous, affecting all mitochondria in a given neuron. As a result, the total number of mitochondria increases (Figure 1B), with a shift from long (sometimes >20 μm) to short (⩽1 μm) organelles (see Supplementary Video 1).
Figure 1.
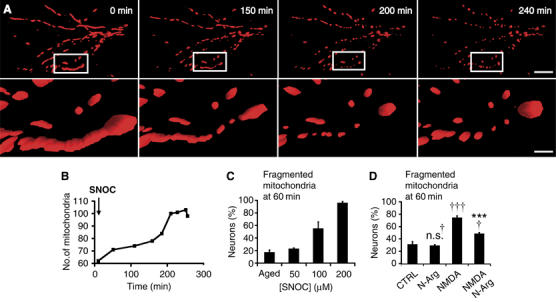
NO triggers mitochondrial fission. (A) 3D time-lapse microscopy of mitochondria undergoing fission in a dendritic arbor of a neuron. Neurons were transfected with Mito-DsRed2, pretreated with the pan-caspase inhibitor zVAD-fmk methyl ester (100 μM), and exposed to SNOC (200 μM). Images were 3D iso-surface rendered. Frames depict representative time points of the movie demonstrating mitochondrial fragmentation within 3 h of NO exposure (upper panels; scale bar, 15 μm) and closeup views (lower panels; scale bar, 3 μm). See Supplementary Video 1. (B) Mitochondrial fragmentation within the field of view shown in (A) results in increased mitochondrial numbers. (C) NO induces dose-dependent mitochondrial fission. Neurons expressing Mito-DsRed2 were exposed to SNOC and fixed at 60 min. The fraction of neurons displaying fragmented mitochondria is shown as the mean±s.e.m. (D) Mito-DsRed2-expressing cortical neurons were pretreated with 1 mM nitro-L-arginine (20 min) and exposed to 25 μM NMDA. The fraction of neurons exhibiting fragmented mitochondria at 60 min after NMDA treatment is shown as the mean±s.e.m. of quintuplicate samples from a representative experiment (†††significance at P<0.001 or n.s.†, not significant compared to control; ***significance at P<0.001 as compared to NMDA treatment).
Next, we determined the lowest concentration of SNOC required to initiate mitochondrial fission. Aged SNOC, from which all NO has been liberated, was used as a negative control. Incubation of cortical neurons with SNOC for 1 h induced dose-dependent mitochondrial fission (Figure 1C). Remarkably, 100 μM SNOC causes mitochondrial fragmentation in 50% of neurons. This event was exacerbated at SNOC concentrations up to 200 μM, which results in ∼80% neuronal cell death at 16 h (see below, Figure 7B).
Figure 7.
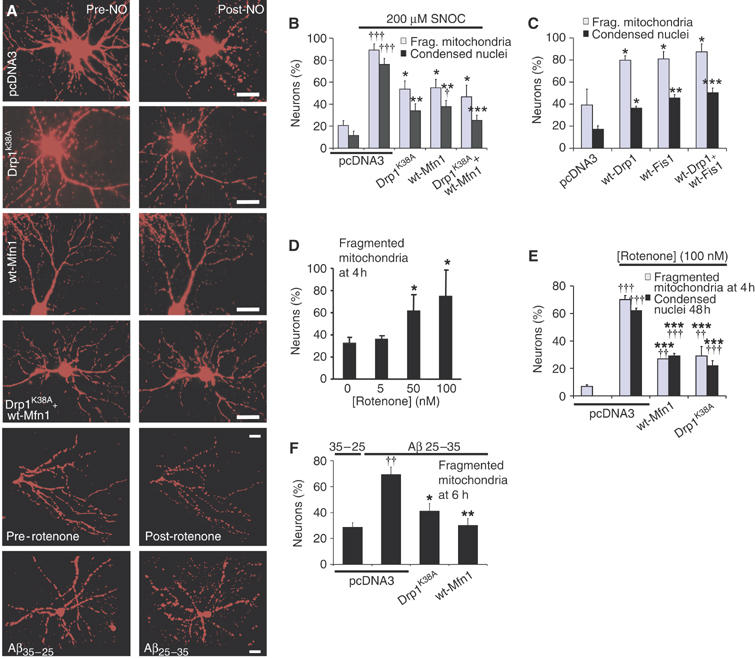
Mitochondrial fission is required for neuronal cell death and mediated by Drp1 and Mfn1. Neurons were co-transfected with Mito-DsRed2 plus the plasmids encoding the indicated protein(s). (A) Representative fluorescence micrographs of mitochondrial morphology before and after SNOC (175 μM; 7 h) or rotenone (30 nM; 2 h) treatments and a comparison of the effects of Aβ25–35 versus Aβ35–25 (10 μM, 6 h), as indicated. Scale bar, 20 μm. (B) Percentage of mitochondrial fission and cell death at 18 h († and ††† significance at P<0.05 and 0.001 as compared to control pcDNA3 transfection; *,** and *** significance at P<0.05 , 0.01 and 0.001, respectively, compared to SNOC treated, pcDNA3 transfected neurons; n=3 independent experiments). Dying neurons were recognized by their shrunken and condensed nuclei after Hoechst 33342 staining. (C) Enforced Drp1 or Fis1 expression (60 h) evokes fission and neuronal cell death (*,** and *** significance at P<0.05, 0.01 and 0.001, respectively, compared to pcDNA3 transfected neurons; n=3 independent experiments). (D) Rotenone induces dose-dependent mitochondrial fission. The fraction of neurons displaying fissioned mitochondria is shown as the mean ±s.e.m. (*significance at P<0.05, n=4). (E) Mfn1 or Drp1K38A inhibits 100 nM rotenone-induced mitochondrial fission (4 h) and cell death (48 h). Data indicate means±s.e.m. of triplicate measurements (†† and ††† significance at P<0.01 and 0.001 as compared to untreated control; ***significance at P<0.001 as compared to rotenone treatment with pcDNA3 transfection; n=3). (F) Mitochondrial fission by Aβ35–25 or Aβ25–35 exposure. The fraction of neurons exhibiting fragmented mitochondria is shown as the mean±s.e.m. (††significance at P<0.01 compared to pcDNA3 transfected, Aβ35−25 treated control; * and ** significance at P<0.05 and 0.001, respectively, compared to pcDNA3 transfected, Aβ25–35 treated neurons; n=3).
To investigate whether our observations using exogenous NO donors are also true for endogenously generated NO, we exposed primary neurons to NMDA and assessed their mitochondrial morphology. Significantly, 25 μM NMDA elicits mitochondrial fission (Figure 1D). Importantly, nitro-L-arginine, an NOS inhibitor, significantly attenuates NMDA-induced mitochondrial fission, suggesting that endogenous NO, in part, mediates mitochondrial fission downstream of NMDAR activation (Figure 1D).
Mitochondrial fission precedes neuronal cell death
To establish the temporal relationship between mitochondrial fission and events linked to neurodegeneration, neuronal cultures were co-transfected with Mito-DsRed2 to trace mitochondrial dynamics, plus MyrPalm-mCFP to observe changes in neurite morphology and apoptosis inducing factor (AIF)-GFP to determine potential release of AIF from mitochondria (Susin et al, 1999). Neurons grown on a two-chambered glass coverslips were exposed to either fresh or aged SNOC (200 μM), and both conditions were imaged simultaneously by revisiting 8–10 selected neurons at 20 min intervals for 12 h (Figure 2).
Figure 2.
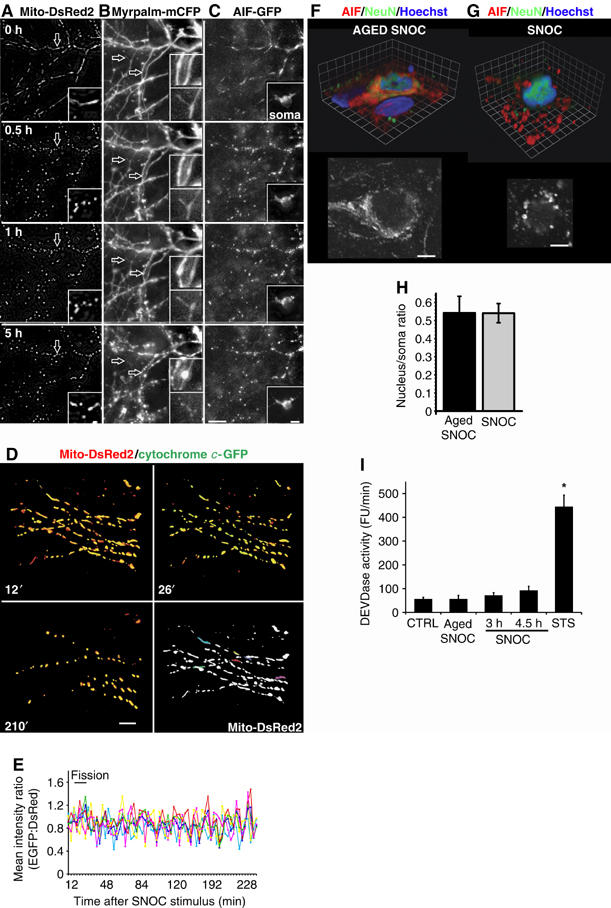
Mitochondrial fission occurs before dendritic injury and without AIF or cytochrome c release from mitochondria. Neurons were co-transfected with vectors encoding (A) Mito-DsRed2 and (B) MyrPalm-mCFP plus (C) AIF-GFP and the effects of SNOC were recorded using 3D time-lapse imaging. Projection images zoomed in onto the dendritic arbor of one neuron (representative of n=42). Arrows indicate dendritic spines. The inset shows the soma at appropriate grayscale levels. The apparent brightening of AIF-GFP fluorescence in the nucleus at 5 h was due to nuclear and cell body shrinkage rather than nuclear translocation of AIF. Scale bars, 10 μm. See also Supplementary Video 2. (D–E) Neurons transfected with Mito-DsRed2- plus cytochrome c-GFP were exposed to 300 μM SNOC. 3D time-lapse images were captured by fluorescence deconvolution microscopy and analyzed using Volocity software. (D) 3D time-lapse image reconstructions of mitochondria and cytochrome c in a dendritic arbor at indicated time points following SNOC addition. Scale bar, 10 μm. Five mitochondria were selected and motion tracked throughout the imaging series. (E) Traces show the ratio of mean GFP to DsRed2 emission intensity from each mitochondrion. Each trace color corresponds to the same colored mitochondrion. (representative of n=9) (F–G). Immunocytochemistry for staining anti-AIF antibodies (red) anti-NeuN antibodies (green) and nuclei labeled with Hoechst 33342 dye (blue) after (F) aged SNOC or (G) SNOC (150 μM; 4 h) treatment. Volume rendered 3D reconstruction of confocal image stacks are shown above, and a single confocal plane in grayscale below. Grid, 3.3 μm for (F) and 1.9 μm for (G), scale bars, 5 μm. (H) Ratio of nuclear to somal AIF signal was measured as mean fluorescence intensity in the center of nuclei divided by the mean fluorescence intensity in the cell body, outlined by the NeuN staining (excluding the nucleus). The bar diagram summarizes the mean±s.e.m. of 282 and 262 neurons measured in n=19 and 20 image stacks in three independent experiments for aged SNOC and SNOC, respectively (P∼0.99 not significant by Student's t-test). (I) DEVDase activity. Purified cortical neurons were exposed to either 200 μM SNOC or 1 μM staurosporine, as positive control. The rate of zDEVD-AMC caspase substrate cleavage per minute was monitored and expressed in arbitrary fluorescence units. Data show mean±s.e.m. for four independent experiments (*significance at P<0.01 by ANOVA).
Interestingly, mitochondrial fission occurs before any overt neurite damage. Typically, SNOC exposure leads to mitochondrial fission within 30 min, followed by focal neurite swellings (varicosities) at 1 h and further swelling at 5 h visualized by MyrPalm-mCFP fluorescence. However, neurites remained connected until 5 h following SNOC exposure, based on their joint movements in time-lapse recordings (Figure 2B). Additionally, nuclear shrinkage was apparent only at 5 h after SNOC exposure (Figure 2C) (see Supplementary Video 2a). Control neurons treated with aged SNOC retained baseline morphology during the course of the time-lapse experiment (see Supplementary Video 2b). During the experiment AIF-GFP remained localized within mitochondria, and its intensity in the nucleus did not increase, suggesting that AIF-GFP was not released from mitochondria (Figure 2C).
We then tested whether cytochrome c would be released from mitochondria in cytochrome c-GFP plus Mito-DsRed2 transfected neurons (Figure 2D). Following exposure to SNOC, fission of tubular mitochondria began at around 25 min. In most (8 of 9) observed neurons, however, cytochrome c-GFP remained in fragmented mitochondria, as evidenced by the overlap of both signals at 210 min. To quantify cytochrome c distribution, individual mitochondria were randomly selected and the mean fluorescence intensity ratio of GFP to DsRed2 signal was plotted over time (Figure 2E). The fluorescence ratio remained constant during the entire imaging period, indicating that cytochrome c was not released from fragmented mitochondria (Figure 2E).
To verify our results with GFP fusion proteins we evaluated the distribution of the endogenous AIF protein by immunocytochemistry (Figure 2F–H). 3D reconstructions derived of confocal image z-Stacks show, using an independent approach, that AIF remains in mitochondria in control and SNOC exposed neurons (Figure 2F–G). Statistical analysis of the ratio of nuclear over soma AIF fluorescence intensity showed no significant difference between control and SNOC treated samples (Figure 2H).
Consistent with a lack of cytochrome c release from mitochondria, 200 μM SNOC did not evoke a robust effector caspase enzyme activity in purified cortical neuronal cultures (Figure 2I), which is in agreement with previous findings that caspases can be inactivated by NO (Melino et al, 1997, 2000). In contrast, 1 μM staurosporine, a kinase inhibitor that typically evokes apoptotic responses in a large variety of cell types triggered significant effector caspase activity.
Thus NO-mediated mitochondrial fission precedes neurite injury and does not result in AIF or cytochrome c release from mitochondria. Furthermore, caspases were not activated here. Thus, cell death pathways activated in primary neurons by patho-physiological triggers like NO might be distinct from classical apoptotic pathways.
Mitochondrial fission occurs in ischemic stroke in vivo
We next asked whether mitochondrial fission initiated in cultured neurons in vitro is of significance in ischemic stroke in vivo. Mice were subjected to 2 h of ischemia using middle cerebral artery occlusion (MCAO), followed by a 3 h reperfusion protocol (Gu et al, 2002). At this early time point ischemic–reperfusion damage was expected at the ultrastructural level, preceding cell death in the penumbral region (Solenski et al, 2002). Intriguingly, in the penumbral region no filamentous mitochondria in EM micrographs were observed. Instead many small, spherical mitochondria were evident when compared to the unaffected, contralateral hemisphere (Figure 3A). Ultrastructural damage in the ischemic brain region is characterized by twisted membranes along the periphery of mitochondria and neurites (Figure 3A). The distribution of measured mitochondrial cross-sectional lengths in the ischemic and contralateral hemisphere is shown in Figure 3B. The left side of the histogram shows no statistical difference between the hemispheres, when short mitochondrial cross sections are compared. However, a statistically significant absence of cross-sections longer than 2 μm was observed in the ischemic hemisphere (Figure 3B; right side of the histogram). Furthermore, mitochondria in the ischemic hemisphere were significantly more frequent in close proximity to one another when compared to the control hemisphere (Figure 3C). Thus, these results suggest that mitochondrial fission occurs in ischemic stroke in vivo prior to neuronal demise.
Figure 3.
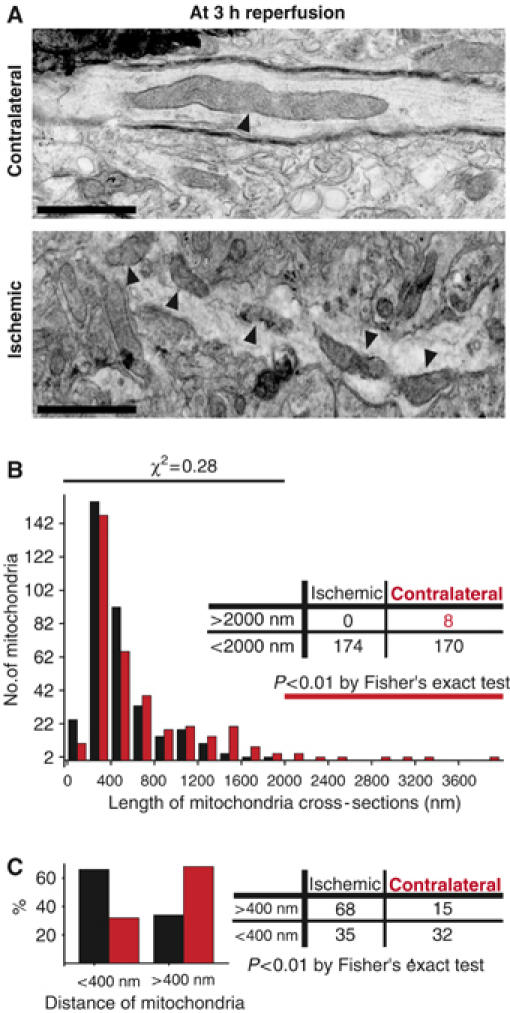
Mitochondrial fission in ischemic stroke in vivo. Thin-section electron micrographs from the ischemic penumbral or contralateral brain region of a mouse subjected for 2 h to middle cerebral artery occlusion, followed by 3 h reperfusion. (A) Elongated mitochondrion (arrow) in neuronal process of the unaffected hemisphere (top). Small mitochondrial segments (arrows) are in close proximity to each other in a neuronal process of the ischemic brain region (bottom). Images are representative of three analyzed mice. Scale bars, 500 nm. (B) Histogram comparing mitochondrial cross-section length of contralateral, control (red) and ischemic (black) hemispheres. The lower bins of the histogram (<2000 nm) were compared with χ2-test and were not different. The table shows the presence of long mitochondrial cross-sections (>2000 nm) in the control hemisphere and their absence in the ischemic hemisphere (P<0.01; Fisher's exact test). (C) Proximities of mitochondria comparing the contralateral (red) and ischemic (black) hemispheres. Bar diagram shows percents of mitochondrial proximities found to be smaller or larger than 400 nm. The same data set is summarized as a contingency table on the right, (P<0.001; two-way Fisher's exact test).
NO causes ultrastructural damage to mitochondria
There is evidence that mitochondrial ultrastructure dictates their function (Hackenbrock, 1966; Perkins et al, 2003; Mannella, 2006). We therefore asked whether fragmented mitochondria are structurally abnormal, which may reflect perturbed bioenergetic functionality. To do so, we analyzed the mitochondrial ultrastructure in neuronal processes using EM tomography. We found that mitochondria in control neurons resemble long narrow tubules, some as long as 25 μm (Figure 4A–D). Intriguingly, mitochondria in neurons exposed to NO were shorter and more globular in morphology (Figure 4E–H). The average length of mitochondria in NO-treated neurons was 830±750 nm, significantly shorter than the length of 2950±1560 nm (mean±s.d.; P<0.00005; Student's t-test) observed in control-treated neurons. Additionally, the mitochondrial matrix was often swollen following NO exposure. Figure 4E–H shows a snapshot of a fission event induced by NO. Remarkably, the lower mitochondrion shows clear damage, including an outer membrane tear and abnormal inner membrane cristae, while the upper mitochondrion exhibits normal ultrastructure. This suggests that mitochondrial fission initiated by NO can be asymmetrical, perhaps resembling bacterial sporulation events (Frank et al, 2003). In addition, the number of cristae per unit of mitochondrial volume is much larger in NO-exposed samples (Figure 4I–L), and occasionally large vesicles inside mitochondria are present, suggesting that uncontrolled inner membrane fission may occur (see Supplementary Video 3, 4 and 5). Thus, NO-induced mitochondrial fission is linked to ultrastructural damage of mitochondria.
Figure 4.
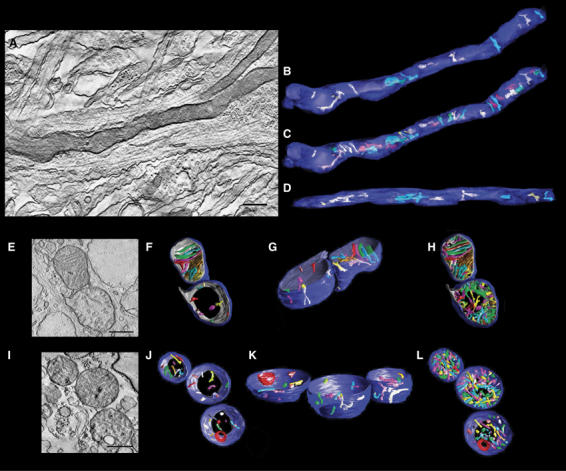
Fission is associated with ultrastructural changes in mitochondria. EM tomography of mitochondria in cultured cortical neurons demonstrates injury associated with mitochondrial fission. Cortical cultures were exposed to aged SNOC control (A–D) or 200 μM SNOC (E–L) for 6 h in the presence of 80 μM zVAD-fmk methyl ester. A slice from each tilt series is shown (A, E and I), followed by orthogonal views of 3D reconstructions after segmentation of the OMM (blue) and IMM (gray), with cristae (various arbitrary colors) (B–D, F–H and J–L). For clarity, 10 representative cristae are shown in (B, D, F, G, J and K), while all cristae are displayed in (C, H and L). The control mitochondrion (A–D) is elongated with a dark matrix, indicating no swelling, and 34 cristae. The NO-exposed mitochondrion in (E–H) is dividing, and the lower mitochondrion of the couplet shows severe damage, recognized by an outer membrane rupture, a regionally confined inner membrane blowout, slight matrix swelling and cristae fragmentation. Each of the daughter mitochondria is shorter and rounder than controls. Unlike the upper daughter, few of the cristae extend throughout the lower mitochondrion. Additionally, cristae are considerably smaller and regionally confined. The lower mitochondrion of the couplet contains 82 cristae, while the upper contains only 39, indicating cristae fragmentation in the former. The NO-exposed mitochondrial triplet in (I–L) displays further damage, including membrane degradation, accumulation of an electron dense mass, greater cristae fragmentation, and apparent mitochondrial fragmentation. All cristae are smaller than controls, with 73 in the top mitochondrion, 112 in the middle, and 47 in the bottom. Scale bars, 400 nm. See Supplementary Video 3, 4 and 5 of tomographic reconstruction.
Mitochondrial fission can be reversible and associated with autophagy
Using time-lapse fluorescence microscopy, we observed that mitochondrial fission can be reversible when young neuronal cultures (8–10 days in in vitro (DIV)) are exposed to 50–200 μM SNOC (Figure 5A–D). Mitochondrial filaments fragmented rapidly as evidenced by a decrease in mean length (Figure 5B) and an increase in mitochondrial number (Figure 5C). However, at 1–2 h mitochondria regained their tubular morphology (26 of 55 neurons) (Figure 5B, C and D; see also Supplementary Video 6). In the remaining group mitochondrial fission was irreversible. Additionally, we observed autophagosomes, recognized by their characteristic double membranes, engulfing injured mitochondria next to intact mitochondria in EM micrographs of SNOC exposed neurons (Figure 5E).
Figure 5.
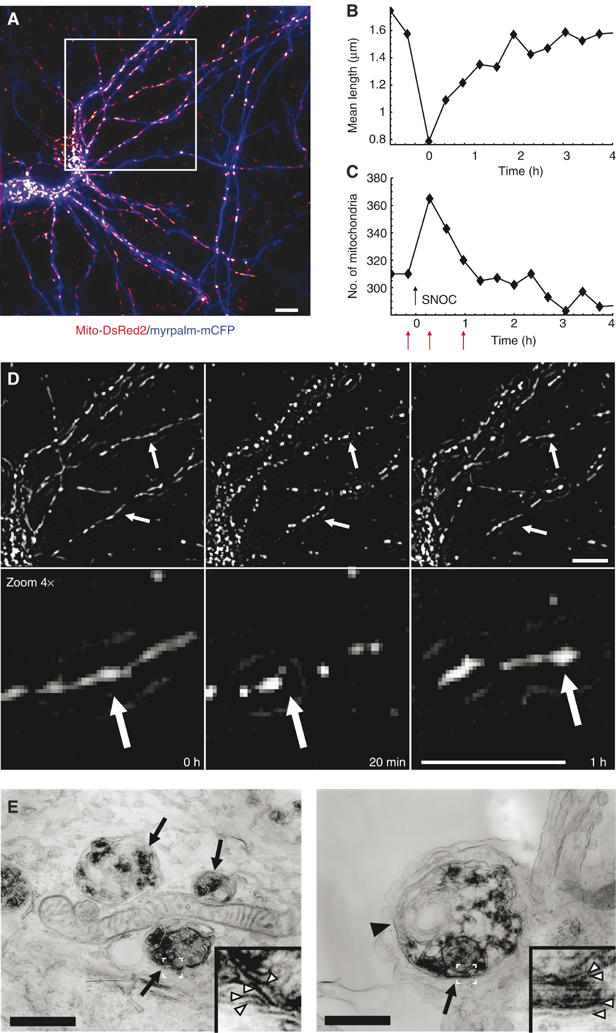
Reversible mitochondrial fission and autophagy. Time lapse fluorescence microscopy of mitochondrial dynamics in response to SNOC (200 μM) of young neurons (DIV 8–10). (A) Projection image of two neurons expressing Mito-DsRed2 (red) and MyrPalm-CFP (blue). Images were high-pass filtered and z-stacks were projected over maximum intensity. (B) Changes in mitochondrial dynamics were evaluated as mean skeletal length of mitochondria or (C) as mitochondrial numbers. (D) Magnified representative frames of the time-lapse recording of the area within the white rectangle of (A) indicate mitochondrial morphology at time 0, 20 min and 1 h. These time points are depicted in (B) and (C) by red arrows. Data are representative of 26 neurons, which exhibited reversible mitochondrial fission from a total of 55 analyzed neurons (six experiments). Scale bar, 10 μm. See Supplementary Video 6. (E) Autophagy in neurites at 6 h after SNOC exposure. Arrows indicate autophagosomes engulfing injured mitochondria next to an intact mitochondrion in the left image. The arrowhead in the right panel indicates membrane wrappers characteristic of autophagosomes. White arrowheads in the insets show the double membranes of the autophagosome and those of mitochondria within the autophagosome. Scale bars, 500 nm.
NO induces bioenergetic failure and increased reactive oxygen species (ROS) production
To test the effects of NO on bioenergetic parameters, we determined the ATP concentration and the ATP/ADP ratio in purified cortical neurons. As shown in Figure 6A, SNOC exposure resulted in a dramatic ATP decline at 20 min. This observation is consistent with previously reported results (Brorson et al, 1999). The ATP level recovered partially at 3 h, only when not more than 50 μM SNOC was used (gray bars), a concentration that typically does not cause cell death. Energy depletion was also indicated by a decrease in the ATP/ADP ratio in neurons exposed to SNOC (Figure 6B). While ATP and the ATP/ADP ratio declined dramatically within the first 20 min of SNOC exposure, cell viability measured as calcein-AM hydrolysis decreased by only ∼20% at 200 μM SNOC. Thus, the major decrease in ATP levels in response to SNOC is not due to a loss in cell viability.
Figure 6.
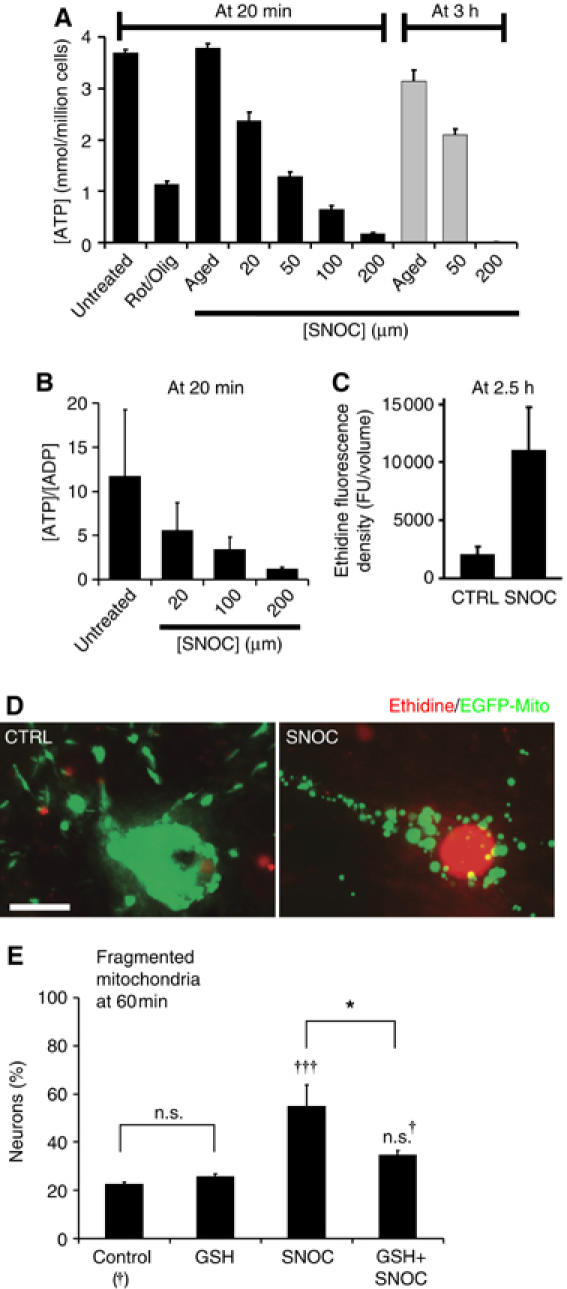
Fission is associated with bioenergetic failure and free radicals. (A) Mitochondrial fission is linked to a drop in ATP. Purified cortical neurons were exposed to increasing SNOC concentrations or mitochondrial inhibitors (2 μM rotenone plus 2 μg/ml oligomycin; Rot/Olig). ATP concentrations are shown as the mean±s.e.m. normalized to the plating density of neurons (n=6) (B) [ATP]/[ADP] ratios in purified cortical neurons exposed to increasing SNOC concentrations (n=4). (C) The bar graph depicts ethidine fluorescence density in arbitrary units of aged and fresh SNOC treated neurons at 2.5 h. (D) Cortical neurons expressing Mito-GFP (green) were exposed to aged or fresh 200 μM SNOC solution. After 2.5 h cultures were loaded with hydroethidine. Images are representative of more that 20 neurons analyzed (in D) for each condition from at least three independent experiments. Scale bar, 10 μm. (E) Reduced glutathione (GSH) partially blocks mitochondrial fission. Neurons were transfected with Mito-DsRed2 pretreated for 2 h with 2 mM GSH monoethyl ester, washed once and then exposed to 35 μM SNOC. Mitochondrial fission was scored after 1 h. Data are means±s.e.m. of quintuplicate samples from one representative experiment of a total of three (†††significance at P<0.001 or n.s., †not significant compared to control; *significance at P<0.05).
Next, we tested whether NO exposure and mitochondrial fission would be associated with an increase in ROS. Cortical neurons, expressing Mito-GFP, were loaded with the ROS-sensitive fluorescent probe, hydroethidine (HEt), which fluoresces red upon oxidation to ethidine (Bossy-Wetzel et al, 2004b). SNOC exposure resulted in an approximately fivefold increase after 2.5 h (Figure 6C). Figure 6D shows a neuron exposed to SNOC with fragmented mitochondria and bright red nuclear ethidine fluorescence, in contrast to a control neuron.
Next, we asked whether ROS is implicated in the mitochondrial fission process. Cortical neurons transfected with Mito-DsRed2 were pretreated with 2 mM reduced glutathione (GSH) monoethyl ester and then exposed to SNOC (35 μM SNOC). As shown in Figure 6E, GSH blunted significantly NO-mediated mitochondrial fission. Thus, mitochondrial fission might be partially triggered by nitrosative and oxidative stress.
Dynamin-related GTPases are required for NO-induced fission and cell death
To determine whether mitochondrial fission plays a causal role in neuronal cell death, first, we followed various cell death related parameters by time lapse microscopy, during SNOC-triggered injury of neurons in primary cortical cultures. Nuclear condensation, but not nuclear fragmentation, always followed the SNOC-triggered, irreversible fission of mitochondria. The uptake of PO-PRO-1 fluorescent probe apoptosis marker and the redistribution of microtubule associated protein 2c (MAP2c)-GFP preceded nuclear condensation. Additionally, nuclear condensation preceded propidium iodide uptake and the complete loss of MAP2c-GFP (see Supplementary Figure 1 and Videos 7 and 8). Therefore, cell death was assessed by counting Mito-DsRed2 expressing neurons with condensed nuclei visualizaed by Hoechst 33342 staining in the following experiments.
To test whether mitochondrial fission plays a causal role in NO-mediated neurotoxicity, the expression levels of Drp1, Fis1 and Mfn1 in primary neurons were manipulated. The expression of Drp1K38A and Mfn1 were confirmed by immunocytochemistry (Supplementary Figure 2). Expression of either dominant-negative Drp1K38A harboring an inactive GTPase domain, wild-type Mfn1, or both, prevents mitochondrial fission in response to NO (Figure 7A and B). Most importantly, overexpression of Drp1K38A, Mfn1, or both, protects neurons from NO-induced cell death (Figure 7B). Surviving neurons did not develop focal neurite varicosities or Bax foci on mitochondria (H Yuan unpublished results). We have never observed dead neurons without fragmented mitochondria. Of note is that, Drp1K38A and Mfn1 did not block NMDA-induced mitochondrial fission and cell death (Supplementary Figure 3).
Forced expression of wild-type Drp1, Fis1 or both, increased the rate of mitochondrial fission and sporadic cell death (Figure 7C). However, in a portion of neurons (20–40%), mitochondrial fission occurred without signs of cell death. Thus, mitochondrial fission in neurons is mediated by the relative activities of fission and fusion factors, including Drp1, Fis1 and Mfn1. Collectively, these results indicate that mitochondrial fission is required but not sufficient for neuronal cell death.
Mitochondrial fission occurs in response to rotenone and A peptide
To examine whether mitochondrial fission might also generally occur with other neurotoxic agents, we tested the effects of the pesticide rotenone and of Aβ peptide. Environmental toxins, such as the pesticide rotenone, can evoke PD-like symptoms (Betarbet et al, 2000). Following exposure to 30 nM rotenone, mitochondria undergo rapid fission in neurons within 2 h, prior to other cellular changes linked to neurodegeneration or cell death (Figure 7A). This effect of rotenone is dose-dependent, with the percentage of neurons displaying fragmented mitochondria reaching a maximum of 80% at 100 nM (Figure 7D). Similarly, rotenone and 1-methyl-4-phenylpyridinium (MPP+), another complex I inhibitor linked with PD, induces mitochondrial fission in the rat dopaminergic cell line N27 (Supplementary Figure 4). Expression of Mfn1 or Drp1K38A prevented significantly mitochondrial fission as well as neuronal cell death of cortical neurons (Figure 7E). These findings suggest that mitochondrial fission might be implicated in neurotoxicity mediated by complex I inhibition.
Next, we tested the effects of Aβ peptide (amino acids 25–35) on mitochondrial morpholgy. Aβ, produced from amyloid precursor protein by proteolytic processing, is a key mediator of AD (Bossy-Wetzel et al, 2004a). Exposure of cortical neurons to 10 μM preaggregated Aβ25–35, but not to the reverse peptide sequence. Aβ35–25 induces striking mitochondrial fragmentation at 6 h (Figure 7A). This fragmentation was inhibited by Mfn1 or Drp1K38A expression (Figure 7F).
Discussion
Here, we provide evidence that mitochondrial fission is an early and required event in NO-induced cell death of primary neurons. We used 3D time-lapse fluorescence imaging to clearly demonstrate that single mitochondrial filaments fragment into multiple isolated organelles, as opposed to simply altering their morphology, as previously suggested for glutamate receptor activation (Rintoul et al, 2003). Mitochondrial fission was accompanied by an increase in mitochondrial number and decrease in their size. Once initiated, fission occurs rapidly within 10 min, and affecting all mitochondria in a single neuron, suggesting that the fission machinery is present and ready to be activated, independent of de novo protein synthesis. Furthermore, fission correlates with an arrest in mitochondrial movement. Intriguingly, multiple neuronal stressors including NO, rotenone, NMDA and Aβ peptide evoke profound mitochondrial fission in primary neurons, indicating that diverse signals may converge on a common pathway to activate fission. Our finding, therefore, might be of broad significance, providing a novel mechanistic explanation for the well-documented mitochondrial defects associated with many neurodegenerative disorders.
We provide several lines of evidence indicating that mitochondrial fission is an early, upstream event in neurodegeneration. Using time-lapse imaging of primary neurons in vitro we show that mitochondrial fission occurs prior to local dendritic swelling, spine loss and neuronal demise. Most importantly, using an MCAO model of ischemia in mice, we demonstrate that fission occurs also in ischemic stroke in vivo, at 3 h reperfusion, many hours before neuronal loss. We used an acute rather than a chronic, progressive model of neurodegeneration because of the relative synchronous entry into neurodegenerative pathways. This allowed us to establish that fission is an upstream and early event in neuronal cell death in vivo.
A central question to be addressed is whether fragmented mitochondria are bioenergetically functional. To investigate this problem we determined the mitochondrial ultrastructure by EM tomography. Using this advanced technique we find that mitochondria in control neurons form long filaments. After fission, mitochondria are more globular with clear ultrastructural abnormalities. Remarkably, we noticed cristae vesiculation and an increase in their number, suggesting enhanced mitochondrial inner membrane division. Mitochondria with this ultrastructure are predicted to be bioenergetically impaired (Hackenbrock, 1966; Perkins et al, 2003; Mannella, 2006).
Another striking and unexpected result was that fission can be asymmetric, where one mitochondrion maintains normal ultrastructure while its fission partner exhibits profound damage. Thus, fission may represent an ancient stress response, similar to bacterial sporulation, to accommodate deficits in metabolic and energy supplies, thereby promoting survival (Frank et al, 2003). Interestingly, fission is accompanied by autophagosomes engulfing injured mitochondria. One role of fission in response to stress might be to excise and eliminate damaged organelles. Along these lines, we observe that fission in immature neurons or at sublethal doses of SNOC can be reversible. However, when stress insult is of intense magnitude fission can be irreversible, leading to neuronal demise.
Here, we show that mitochondrial fission plays a causal role in neuronal demise. Blocking fission by dominant-negative mutant Drp1K38A or wild-type Mfn1 prevents mitochondrial fragmentation and rescues neurons from degeneration and cell death evoked by NO or rotenone. It has been suggested that dynamin-related GTPases may have additional functions including regulation of bioenergetics and ion homeostasis (Pich et al, 2005; Yu et al, 2006). Therefore, it cannot be ruled out that the neuroprotective properties of Mfn1, for example, might due to other functions besides its role in mitochondrial fusion. Here, we find also that Drp1 and Fis1 expression induce fission and an increase in baseline cell death. However, many neurons that exhibit mitochondrial fragmentation remain alive. Mitochondrial fission is therefore required, but does not per se lead to neuronal death. Recent observations in transformed cell lines and Caenorhabditis elegans showed that mitochondrial fission is required for apoptotic cell death (Bossy-Wetzel et al, 2003; Frank et al, 2003; Jagasia et al, 2005; Perfettini et al, 2005; Youle and Karbowski, 2005).
Caspase-dependent and -independent pathways have been implicated in the neuronal demise during neurodegeneration (Li et al, 2000; Leist and Jaattela, 2001; Friedlander, 2003; Cregan et al, 2004). Various forms of cell death are mutually not exclusive and may operate in the same system and at the same time. In our study, AIF and cytochrome c remain localized to mitochondria, even when mitochondria became fragmented and dendrites were injured. Furthermore, we show that SNOC does not induce caspase enzyme activity. Along this line, previous reports indicated that caspases can be inactivated by S-nitrosylation (Melino et al, 1997, 2000). Furthermore we recently showed that NO-induced cell death is poorly blocked by caspase inhibitors (Bossy-Wetzel et al, 2004b). Therefore, the predominant form of cell death here is caspase-independent and perhaps involves type II, autophagic cell death (Levine and Yuan, 2005; Lum et al, 2005). The rate of fission and autophagy might be critical in determining neuronal survival or death (Shintani and Klionsky, 2004; Levine and Yuan, 2005).
Lack of mitochondrial fusion and persistent fission may have a negative impact on neuronal function. Chronic fission may lead to the manifestation of mtDNA mutations that otherwise would remain silent in fused mitochondria (Nakada et al, 2001; Ono et al, 2001). Furthermore, continuous fission might lead to severe mitochondrial dysfunction and energy deficits, which in turn, impair ion homeostasis, synaptic transmission, channel and pump activity, or axonal/dendritic transport. Our results suggest that mitochondrial fission may not lead immediately to cell death and can be reversed, dependent on the age of the neuron and the magnitude of the insult. Fission might therefore perturb neuronal function without leading immediately to cell death. An increase in mitochondrial fission rates induced by internal or external stressors might explain the slow progressive nature of many chronic neurodegenerative disorders. Thus, observations made here may offer new opportunities for drug developments to combat neuronal demise in several neurodegenerative diseases.
Materials and methods
Neuronal cultures, transfection, drug treatments
Mixed cortical neuronal cultures were derived from embryonic day E17 rats and maintained at 37°C in 5% CO2 (Lei et al, 1992). For all experiments except those using Aβ peptide, cultures were transfected at 14–16 DIV using Lipofectamine 2000 (Invitrogen) and experiments were performed after 2–5 days. SNOC was prepared as described previously (Lei et al, 1992). The caspase inhibitor zVAD-fmk methyl ester (BIOMOL) was dissolved in DMSO (Sigma). NMDA (Sigma) exposure was for 20 min in EBSS (Bonfoco et al, 1995) followed by replacement of the buffer with the saved conditioned medium. Rotenone (Calbiochem) was dissolved in ethanol and applied to neurons in growth medium. For the AD model, cultures were transfected with Lipofectamine 2000 at 7 DIV. The Aβ peptide (Sigma) was dissolved in sterile H2O and allowed to aggregate at room temperature for 24 h prior to use. Peptide exposure was performed 72 h after transfection.
Image acquisition and analyses
For live-cell imaging, growth medium was replaced with Hibernate-ELF (BrainBits). Cultures were maintained at 37°C either on a heated microscope stage for time-lapse imaging or in a humidified-atmosphere incubator between repeated imaging.
Images (for Figures 1 and 7) were acquired using an Axiovert Zeiss 100 M microscope equipped with Plan Apochromat 100 × 1.4 NA oil or Plan-Neofluar 40 × 0.8 NA air objectives, an LB-LS (Sutter Instruments) Xe-arc illumination unit, a Sensicam QE CCD camera (PCO AG, Germany) and Slidebook software (Intelligent Imaging Innovations, Inc.). Deconvolution was performed using the constrained iterative algorithm in Slidebook. z-Stacks were 3D-rendered using Volocity software (Improvision). The change in number and length of mitochondria was quantified using fluorescent signal classifiers and object measurements in Volocity.
Long-term time lapse experiments (for Figures 2 and 5) were carried out on an Olympus IX81 inverted microscope equipped with a Plan APO 60 × 1.4 NA oil objective, a DG-4 Xe-arc illumination unit, a Cascade 512B cooled CCD camera (Roper Scientific) and Metamorph 6.2 software (Molecular Devices). Cultures were grown on Lab-Tek two-chambered coverslips and mounted onto an MS-2000 linear encoded motorized stage (ASI Inc.). After exposure to 200 μM SNOC or aged SNOC, 3D images were acquired simultaneously by revisiting 8–10 cells in both wells at time intervals of 20 min. z-Stacks (0.3 × 0.3 × 1.5 μm3 voxel size; 9–11 planes) were de-hazed by applying a high pass spatial filter designed on an empirical basis (Gerencser and Adam-Vizi, 2001) and z-projected using a custom version of Metafluor Analyst (Universal Imaging Corp.). Representative images are shown at elevated (1.3–1.5) gamma value. To analyze changes of mitochondrial length and number in time-lapse experiments, image stacks were processed by a custom algorithm written in Mathematica 5.2 (Wolfram Research) and Delphi 6.0 (Borland). The above projection images were thresholded and skeletonized, followed by determination of skeletal length and the number of skeletons for each frame of the time-lapse series.
To evaluate mitochondrial fragmentation and cell death of a large number of neurons, manual scoring was carried out of neurons grown on glass coverslips using conventional fluorescence microscopy. Cultures were fixed at 4°C with 0.5% glutaraldehyde (Ted Pella) in PBS for 30 min and treated with 1% sodium borohydride (Sigma) in H2O for 30 min to quench autofluorescence. Neurons transfected with Mito-DsRed2 showed elongated tubular mitochondria in neurites were scored as unfragmented. Neurons that revealed 90% of small, round mitochondria were scored as neurons with fragmented mitochondria. Nuclear morphology was evaluated by co-staining with Hoechst 33342 (2 μg/ml) in PBS. Mito-DsRed2 expressing neurons were scored as dead if the nucleus was shrunken (∼half size of the healthy), condensed and spherical.
Immunocytochemistry and confocal microscopy
For immunocytochemistry mixed cortical cultures grown on glass coverslips were fixed in 3.7% formaldehyde (Sigma), 5% sucrose containing KCl-based HEPES buffer (pH 7.0; 15 min; 37°C) and then permeabilized with 4% Triton X-100 in TRIS-buffered salt solution (pH 7.4; 30 min; 37°C), and blocked with 2% BSA, 0.05% Tween-20 plus 0.1% NaN3 in TRIS buffer for 30 min. Anti-AIF antibodies (Cell Signaling; #4642) and anti-NeuN antibodies (Chemicon; MAB377) were used at 1:20 dilution (4°C; overnight). Alexa555- and Alexa488-conjugated secondary antibodies (Molecular Probes) were used for fluorescence labeling at a dilution of 1:200 (37°C, 90 min). Confocal imaging of immunostained samples was carried out on an Olympus Fluoview-1000 laser scanning confocal microscope. Using a Plan Apo N 60 × 1.42 NA oil lens, image stacks of 1024 × 1024 × approximately 30 pixels (0.13 × 0.13 × 0.5 μm3 voxel size) were recorded. Representative image stacks were volume rendered using Volocity 2.6 software. Statistical analysis was done by custom written Mathematica 5.2 and Delphi 6.0 algorithms. For statistical analysis of Hoechst 33342, image stacks were segmented in 3D, and anti-AIF-Alexa555 fluorescence intensity was determined in the center of nuclei. Only NeuN stained nuclei were analyzed. Anti-AIF-Alexa555 fluorescence in the soma was determined by the segmentation of NeuN image stacks.
Stroke model
Focal cerebral ischemia was induced in male mice (C57BL/6) by middle cerebral artery occlusion using the intraluminal filament technique as previously described (Gu et al, 2002). Perfusion was done using physiological buffer, followed by fixation buffer for EM. Brain sections (100-μm-thick) were prepared using a Vibratome and postfixed and embedded for EM analysis.
EM and tomography
Sample preparation and conventional EM were carried out as described before (Ricci et al, 2004). EM tomography was also performed as described previously (Perkins et al, 2003). For comparison of mitochondrial length, EM micrographs of thin sections were evaluated. By modeling the distribution of cross-sections diameter of tubular mitochondria, assuming that mitochondria are randomly oriented in the brain section, one expects to see not more than 6% of cross-sections longer than 10 × of their diameter. Fisher's one-sided exact test was therefore used to determine whether the small number of long mitochondrial cross-sections in the ischemic cross-section was of statistical significance.
ATP measurements
Purified cortical neurons from E17 rats were used for ATP measurements. Neurons were seeded at 5 × 104 cells/well on Biocoat poly-D-lysine-coated black 96-well plates (Becton Dickinson) and maintained in Neurobasal medium (Invitrogen) plus 0.5 mM L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and B27 supplement (Invitrogen). At 4 DIV, the plating medium was diluted with an equal volume of fresh medium lacking L-glutamine containing 1% GlutaMax (Invitrogen). Cultures were then fed every 2–3 days. At 9 DIV, ATP levels were determined using a luciferase-based CellTiter-Glo assay kit (Promega) with a PolarStar plate reader (BMG). Data were collected from multiple replicate wells in each of six experiments (n=6) and plotted as the mean±s.e.m. The ATP/ADP ratio was measured using an ATP regenerating system consisting of pyruvate kinase plus phosphoenol pyruvate, as described previously (Brorson et al, 1999). The regenerating system converts ADP to ATP and thereby allows for measurement of total ATP plus ADP content.
ROS detection
Cortical neurons expressing pEGFP-Mito were loaded with 750 nM hydroethidine (Molecular Probes) in imaging buffer for 20 min at 37°C and washed once. Mitochondrial morphology and ethidine staining were analyzed using a fluorescence deconvolution microscope. Fluorescence density was calculated by Volocity.
Caspase enzyme assay
Caspase activity was measured using the fluorogenic substrate z-DEVD-AMC (Molecular Probes). Neurons were plated in six-well plates in Neurobasal medium and were treated at 9–12 DIV with either 200 μM SNOC or 1 μM staurosporine. At various time points after treatment, neurons were lysed in 50 mM Tris–Cl pH 7.4, 150 mM NaCl, 20 mM EDTA, 0.5% NP40. Protein extract (50 μg) was supplemented with 100 μM DEVD-AMC and the kinetics of enzyme activity was followed at 320 nm excitation and 460 nm emission using a PolarStar plate reader.
Statistics
A Student's t-test (two sided) was used for comparisons of parametric data from cell populations. Multiple comparisons were performed by ANOVA using Tukey's post hoc test. Histograms were compared with χ2-test, while contingency tables were evaluated by Fisher's exact test (one-sided). Statistical analyses were undertaken using Prism 4.0 software (Graphpad) or Mathematica 5.2.
Supplementary Material
Supplementary Figure 1A
Supplementary Figure 1B
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends
Supplementary Video 1a
Supplementary Video 1b
Supplementary Video 2a
Supplementary Video 2b
Supplementary Video 3
Supplementary Video 4
Supplementary Video 5
Supplementary Video 6
Supplementary Video 7
Supplementary Video 8
Acknowledgments
We thank Dr MP Yaffe for helpful discussions and Drs C Freed and KN Prasad for their N27 cell line. We thank Dr E Monosov for advice in fluorescence microscopy. We are also grateful to Drs G Kroemer, MT Fuller for the pcDNA3EGFP-mAIFfl and pcDNA3-Mfn1 vectors, respectively, to Dr AM van der Bliek for the pcDNA3Drp1 and pcDNA3Drp1K38A vectors, to Dr RY Tsien for the pcDNA3MyrPalm-mCFP and pBAD-mPlum vectors, to Dr DC Chen for the pcDNA3-Mfn1-myc vector, to Dr Changjun Zhu for the Histone H2b-CFP and Histone H2b-mCherry vectors and to Dr S Halpain for the MAP2c-GFP vector. This work was supported by NIH Grants R01 NS44314, R01 NS047456-S3, R01 NS047456, R01 EY016164, P01 AI055789, American Parkinson's Disease Foundation, Inc. (to EB-W), a fellowship from the Philippe foundation and Hereditary Disease Foundation (to GL), P41RR04050 and R01 NS14718 (to MHE) and P01 HD29587, R01 EY05477, R01 EY09024, R01 NS43242, R01 NS044326, R01 NS046994 and R01 NS047973 (to SAL).
References
- Beal MF (2000) Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci 23: 298–304 [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Voth M (1994) Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech 27: 198–219 [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301–1306 [DOI] [PubMed] [Google Scholar]
- Boje KM (2004) Nitric oxide neurotoxicity in neurodegenerative diseases. Front Biosci 9: 763–776 [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA (1995) Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 92: 7162–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA (2003) Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol 15: 706–716 [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004a) Molecular pathways to neurodegeneration. Nat Med 10 (Suppl): S2–S9 [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA (2004b) Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron 41: 351–365 [DOI] [PubMed] [Google Scholar]
- Brorson JR, Schumacker PT, Zhang H (1999) Nitric oxide acutely inhibits neuronal energy production. The Committees on Neurobiology and Cell Physiology. J Neurosci 19: 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Cooper CE (1994) Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 356: 295–298 [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC (2004) Mitochondrial dynamics in mammals. Curr Top Dev Biol 59: 119–144 [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Dawson VL, Slack RS (2004) Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 23: 2785–2796 [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM (1998) Nitric oxide in neurodegeneration. Prog Brain Res 118: 215–229 [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210 [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: 515–525 [DOI] [PubMed] [Google Scholar]
- Frank S, Robert EG, Youle RJ (2003) Scission, spores, and apoptosis: a proposal for the evolutionary origin of mitochondria in cell death induction. Biochem Biophys Res Commun 304: 481–486 [DOI] [PubMed] [Google Scholar]
- Friedlander RM (2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348: 1365–1375 [DOI] [PubMed] [Google Scholar]
- Gerencser AA, Adam-Vizi V (2001) Selective, high-resolution fluorescence imaging of mitochondrial Ca2+ concentration. Cell Calcium 30: 311–321 [DOI] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA (2002) S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297: 1186–1190 [DOI] [PubMed] [Google Scholar]
- Hackenbrock CR (1966) Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol 30: 269–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C (1997) Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci 20: 298–303 [DOI] [PubMed] [Google Scholar]
- Jagasia R, Grote P, Westermann B, Conradt B (2005) DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature 433: 754–760 [DOI] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278: 36373–36379 [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA (1992) Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8: 1087–1099 [DOI] [PubMed] [Google Scholar]
- Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2: 589–598 [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J (2005) Autophagy in cell death: an innocent convict? J Clin Invest 115: 2679–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Ona VO, Guegan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM (2000) Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 288: 335–339 [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M (2004) The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119: 873–887 [DOI] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB (2005) Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 6: 439–448 [DOI] [PubMed] [Google Scholar]
- Mannella CA (2006) The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta 1762: 140–147 [DOI] [PubMed] [Google Scholar]
- Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G, Finazzi-Agro A (1997) S-nitrosylation regulates apoptosis. Nature 388: 432–433 [DOI] [PubMed] [Google Scholar]
- Melino G, Catani MV, Corazzari M, Guerrieri P, Bernassola F (2000) Nitric oxide can inhibit apoptosis or switch it into necrosis. Cell Mol Life Sci 57: 612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto YI, Nonaka I, Hayashi JI (2001) Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7: 934–940 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM (2005) Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet 39: 503–536 [DOI] [PubMed] [Google Scholar]
- Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, Guillou E, Delettre C, Valette A, Hamel CP, Ducommun B, Lenaers G, Belenguer P (2002) The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 523: 171–176 [DOI] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K, Hayashi JI (2001) Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28: 272–275 [DOI] [PubMed] [Google Scholar]
- Osteryoung KW, Nunnari J (2003) The division of endosymbiotic organelles. Science 302: 1698–1704 [DOI] [PubMed] [Google Scholar]
- Perfettini JL, Roumier T, Kroemer G (2005) Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol 15: 179–183 [DOI] [PubMed] [Google Scholar]
- Perkins GA, Ellisman MH, Fox DA (2003) Three-dimensional analysis of mouse rod and cone mitochondrial cristae architecture: bioenergetic and functional implications. Mol Vis 9: 60–73 [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A (2005) The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 14: 1405–1415 [DOI] [PubMed] [Google Scholar]
- Radi R, Rodriguez M, Castro L, Telleri R (1994) Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys 308: 89–95 [DOI] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117: 773–786 [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ (2003) Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23: 7881–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306: 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12: 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solenski NJ, diPierro CG, Trimmer PA, Kwan AL, Helm GA (2002) Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke 33: 816–824 [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446 [DOI] [PubMed] [Google Scholar]
- van der Bliek AM (2000) A mitochondrial division apparatus takes shape. J Cell Biol 151: F1–F4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe MP (1999) The machinery of mitochondrial inheritance and behavior. Science 283: 1493–1497 [DOI] [PubMed] [Google Scholar]
- Yoon Y, Pitts KR, McNiven MA (2001) Mammalian dynamin-like protein DLP1 tubulates membranes. Mol Biol Cell 12: 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Karbowski M (2005) Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 6: 657–663 [DOI] [PubMed] [Google Scholar]
- Yu T, Robotham JL, Yoon Y (2006) Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36: 449–451 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1A
Supplementary Figure 1B
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure Legends
Supplementary Video 1a
Supplementary Video 1b
Supplementary Video 2a
Supplementary Video 2b
Supplementary Video 3
Supplementary Video 4
Supplementary Video 5
Supplementary Video 6
Supplementary Video 7
Supplementary Video 8