

Abstract
The IκB-inducing kinase (IKK) is composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ. IKK-regulated signaling pathways are believed to promote the proliferation of normal cells as well as the aberrant proliferation of cancer cells. The molecular mechanisms linking the IKK signaling pathway components to the cell cycle machinery are not entirely understood. To study the function(s) of the catalytic subunits of the IKK complex, we used pancreatic cancer cells, with constitutive IKK activity. We show that the G1 phase of the cell cycle is specifically regulated by the IKKα subunit, which regulates the stability of the cyclin-dependent kinase inhibitor p27Kip1. Increased p27Kip1 protein levels following the transfection of IKKα-specific siRNAs are a result of the downregulation of the F-box protein S-phase kinase-associated protein 2 (skp2). Additionally, we demonstrate that IKKα signaling regulates the transcription of the skp2 gene by controlling the composition of a RelB-containing NF-κB complex. Together, this work defines a novel IKKα-regulated growth pathway involving the p52/RelB-dependent transcriptional regulation of the skp2 gene.
Keywords: cell cycle, IKK, NF-κB, p27Kip1RelB, skp2
Introduction
The IκB kinase (IKK) complex consists of at least three subunits, IKKα and IKKβ, which function as the catalytic subunits, and IKKγ, which functions as the regulatory subunit (Yamamoto and Gaynor, 2004). Despite the structural and biochemical similarities of the catalytic subunits IKKα and IKKβ, there are clear functional distinctions between them. IKKβ is the dominant kinase involved in transducing pro-inflammatory signals, which lead to the phosphorylation of the IκB proteins (Yamamoto and Gaynor, 2004). In contrast, IKKα provides the dominant kinase activity needed for development of secondary lymphoid organs and B-cell development by regulating the alternative NF-κB activation pathway (Hayden and Ghosh, 2004). In this alternative NF-κB pathway, an IKKα dimer phosphorylates the p52 precursor NF-κB2/p100, leading to p100 processing and translocation of RelB/p52 to the nucleus. In addition, IKKα has distinct nuclear functions. For example, IKKα can modify chromatin by phosphorylating Ser-10 of histone H3, thereby altering gene expression at specific promoters (Anest et al, 2003, 2004; Yamamoto et al, 2003). Furthermore, IKKα can stimulate the exchange of corepressor for coactivator complexes on chromatin (Hoberg et al, 2004).
Both IKK catalytic subunits are linked to the cell cycle machinery and tumorigenesis. IKKα−/− fibroblasts have reduced levels of cyclin D1 (Albanese et al, 2003). Furthermore, cyclin D1 is involved in a pathway including RANKL, RANK, and IKKα that is essential for mammary epithelial cell proliferation (Cao et al, 2001). However, the detailed molecular mechanism(s) by which IKKα controls cell cycle progression of cancer cells is unknown. An important role for IKKβ in inflammation-associated colon carcinogenesis was recently demonstrated (Greten et al, 2004). Additionally, the negative regulation of Foxo3a by the IKK complex contributes to the proliferative control of mammary cancer cells (Hu et al, 2004).
Mammalian cell cycle progression is regulated by the activities of protein complexes composed of cyclins and cyclin-dependent kinases (CDKs). These complexes are tightly controlled by a group of cyclin-dependent kinase inhibitors (CKIs). The CKI p27Kip1 is a central regulator of cell cycle progression. One major target of p27Kip1 is the cyclin-dependent kinase 2 (CDK2) associated with a cyclin coactivator. Overexpression of p27Kip1 leads to a cell cycle arrest during G1 phase, whereas reduced expression of p27Kip1 increases the number of cells in S phase. Unlike other tumor suppressors, the p27Kip1gene is rarely found mutated in tumor cells. Nevertheless, the contribution of p27Kip1 to malignant transformation is well documented (Bloom and Pagano, 2003). Multiple mechanisms control p27Kip1 abundance and function. Although p27Kip1 mRNA expression and translation are regulated, expression levels are controlled primarily by post-translational modifications that affect p27Kip1 protein stability (Reed, 2003; Lin and Diehl, 2004). For example, phosphorylation of p27Kip1 on Thr-187 by the cyclin E–CDK2 complex targets it for ubiquitination and degradation by mediating its recognition by the S-phase kinase-associated protein 2 (skp2), an F-box protein that functions as a receptor component of the skp1/Cul1/F-box (SCF) ubiquitin ligase complex. The variable F-box component of the SCF complex serves as a molecular adaptor between the E3 ligase components (skp1, CUL1, Rbx1) and the target protein substrates. The prototypic SCF complex contains the F-box protein skp2 and targets p27Kip1, p57Kip2, p21WAF1, p130, CDT1, c-myc, SMAD4, and Foxo1 for ubiquitin-dependent degradation (Nakayama and Nakayama, 2005).
To analyze the function of the individual catalytic subunits IKKα and IKKβ in cell cycle regulation, we used pancreatic cancer cell lines that have recently shown to display constitutive IKK activity (Liptay et al, 2003). Using RNA interference, we demonstrate a novel growth control mechanism regulated specifically by IKKα and not IKKβ. IKKα induces skp2 gene transcription by regulating the composition of a RelB-containing NF-κB complex at the proximal skp2 gene promoter. This results in a decrease in p27Kip1 protein stability, thereby contributing to the inactivation of the Rb-dependent G1-phase cell cycle checkpoint.
Results
IKKα regulates G1-phase progression
Pancreatic cancer cells display constitutive IKK activity as indicated by constitutive IκBα phosphorylation (Liptay et al, 2003; Li et al, 2004). To determine if IKKα and IKKβ have different effects on cell growth, we inhibited their expression using RNA interference. As shown in Figure 1A, 48 h after MiaPaCa2 cells were transfected with two specific siRNAs directed against IKKα and two specific siRNAs directed against IKKβ, there is a significant reduction in IKKα and IKKβ protein abundance, respectively. IKKβ expression was not downregulated in IKKα siRNA-treated cells and IKKα expression was not affected by IKKβ siRNA treatment (Figure 1A). The IKK complex has been shown to be involved in cell cycle progression regulation. Therefore, we tested the effect(s) of the knock-down of the IKK catalytic subunits on proliferation as assayed by BrdU incorporation. IKKα knock-down resulted in a significant reduction in BrdU incorporation (59.1 and 58.2% relative to controls), whereas the effect of IKKβ knock-down was negligible (Figure 1B). The IKKα knock-down-mediated reduction in BrdU incorporation was due to a G1-phase cell cycle arrest as determined by FACS of propidium iodide-stained cells. Forty-eight hours post-transfection, MiaPaCa2 cells containing IKKα-specific siRNAs displayed a significant increase in the G1 fraction (55.6% in untransfected cells, 66.3% in IKKα+81 siRNA-transfected cells, and 71.2% in IKKα+129 siRNA-transfected cells). The increase in G1 phase was accompanied by a decrease in S phase (13.8% in untransfected cells, 12.5% in IKKα+81 siRNA-transfected cells, and 6.6% in IKKα+129 siRNA-transfected cells) and G2/M phase (22.5% in untransfected cells, 12.9% in IKKα+81 siRNA-transfected cells, and 11.3% in IKKα+129 siRNA-transfected cells) (Figure 1C). In agreement with the BrdU incorporation assay, no significant change of cell cycle distribution was observed in scramble, IKKβ+81, or IKKβ+591 siRNA-transfected cells. These data demonstrate that IKKα regulates G1-phase progression.
Figure 1.
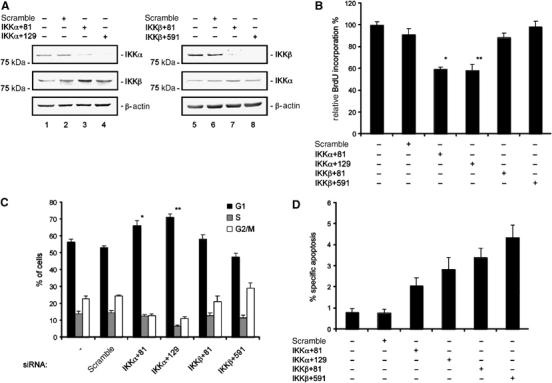
Cell cycle regulation by IKK. (A) Western blots of IKKα and IKKβ protein levels in MiaPaCa2 cells transfected with the indicated siRNAs 48 h post-transfection compared to untransfected or control siRNA-transfected cells. The membrane was stripped and probed for IKKβ (in IKKα siRNA-treated cells) or IKKα (in IKKβ siRNA-treated cells) to control specificity. (B) BrdU incorporation in MiaPaCa2 cells 48 h post-transfection with indicated siRNAs (Student's t-test: *P=0.002 and **P=0.003 versus controls). (C) MiaPaCa2 cells were transfected with the indicated siRNAs. At 48 h post-transfection, cells were stained with PI and cell cycle distribution was analyzed by FACS. The fraction of the cells in G1, S, and G2/M phases is indicated (Student's t-test: *P=0.004 and **P<0.001 versus controls). (D) Apoptotic fraction of MiaPaca2 cells 48 h post-transfection with the indicated siRNAs using fluorescence microscopy following Hoechst stain.
In contrast to the significant effect of IKKα on G1-phase progression, IKKα knock-down resulted in only a slight increase in apoptosis (Figure 1D).
IKKα regulates the G1-phase restriction point independent of cyclin D1 mRNA and protein abundance
We next examined the molecular mechanism(s) responsible for the G1-phase arrest induced by the knock-down of IKKα. In other model systems, cyclin D1 functions downstream of IKKα and NF-κB. As shown in Figure 2A, no significant reduction in cyclin D1 protein levels was observed in either IKKα siRNA or IKKβ siRNA-transfected MiaPaCa2 cells after 48 h. In addition, cyclin D1 mRNA levels were not significantly affected by the knock-down of the IKK catalytic subunits (Figure 2B). The G1-phase Rb-dependent restriction point is functionally inactivated in pancreatic cancer cells (Rozenblum et al, 1997). Rb protein mediates the exit from G1 phase and is regulated by a complex coordination of CDKs and CKIs, such as p27Kip1. This regulation is mediated by changes in the net phosphorylation status of Rb, with the hypophosphorylated form being active. We detected an upregulation of p27Kip1 and cyclin E protein levels in IKKα siRNA-transfected cells, whereas CDK2 and E2F1 abundance was not altered (Figure 2C). Furthermore, Rb activation was observed in those cells transfected with the IKKα-specific siRNAs (Figure 2D). In contrast, we did not detect changes in the phosphorylation status of Rb following transfection of MiaPaCa2 cells with scrambled or IKKβ-specific siRNAs. Similarly, the Rb-related pocket protein p130 was activated specifically in response to the knock-down of IKKα in these cells (Figure 2D). In line with the observed hypophosphorylation of Rb, we found a distinct reduced CDK2 kinase activity after the IKKα knock-down (Figure 2E). Furthermore, p27Kip1 was found to be complexed with the cyclin E/CDK2 kinase only in the case of IKKα siRNA transfection (Figure 2F). Taken together, these data suggest that IKKα controls G1-phase progression via a p27Kip1- and Rb-dependent mechanism.
Figure 2.
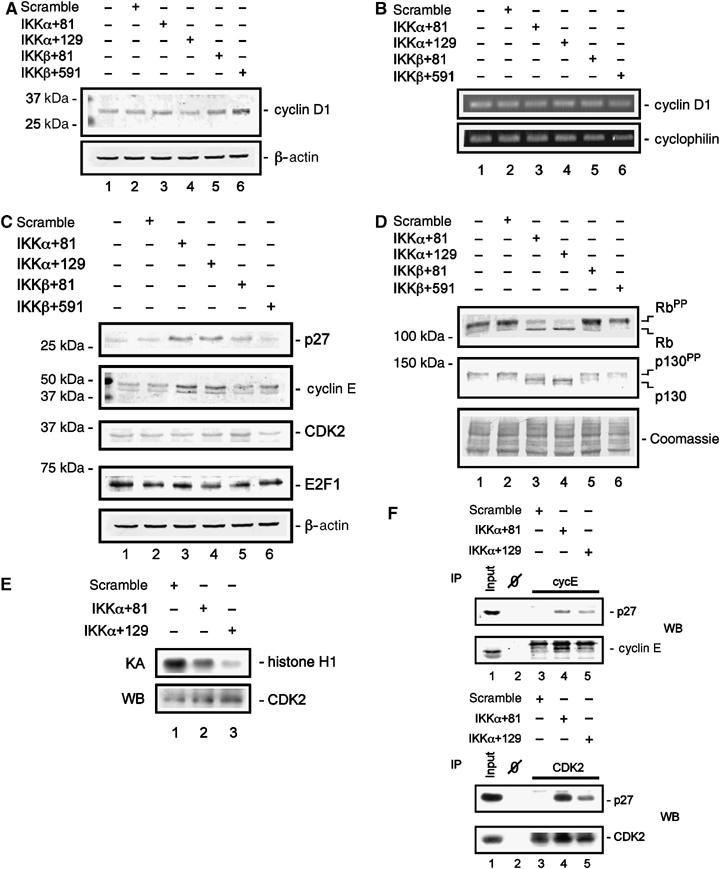
IKKα-mediated regulation of cell cycle proteins. (A) Western blot analysis of cyclin D1 protein levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. (B) Semiquantitative reverse transcriptase–PCR (RT–PCR) of cyclin D1 mRNA levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. Cyclophilin mRNA serves as control for mRNA quantification. (C) Western blot analysis of p27Kip1, cyclin E, CDK2, and E2F1 protein levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. (D) Western blot analysis of Rb and p130 protein levels and phosphorylation status 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. Hyperphosphorylated proteins are denoted by ‘PP'. The Coomassie-stained membrane provides a control for equal protein loading. (E) Immunocomplex CDK2 kinase assay 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. (F) Immunoprecipitation of cyclin E (upper panel) and CDK2 (lower panel) 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. Western blot analysis of p27Kip1 association with the cyclin E/CDK2 complex.
p27Kip1 protein stability is regulated by IKKα
Transfection of IKKα-specific siRNAs does not change p27Kip1 mRNA levels in MiaPaCa2 cells (Figure 3A). To examine p27Kip1 protein stability in IKKα siRNA-transfected cells, we treated MiaPaCa2 cells 48 h post-transfection with the protein synthesis inhibitor cycloheximide. In scrambled siRNA-transfected cells, a rapid decrease in p27Kip1 protein levels was observed, with a 50% reduction between 1 and 2 h of cycloheximide treatment. In contrast, p27Kip1 protein levels were stabilized in MiaPaCa2 cells transfected with IKKα-specific siRNAs (Figure 3B). Cyclin D1 protein levels did not change over the time period investigated. These data suggest that the upregulation of p27Kip1 protein levels following IKKα knock-down is due to increased protein stability.
Figure 3.
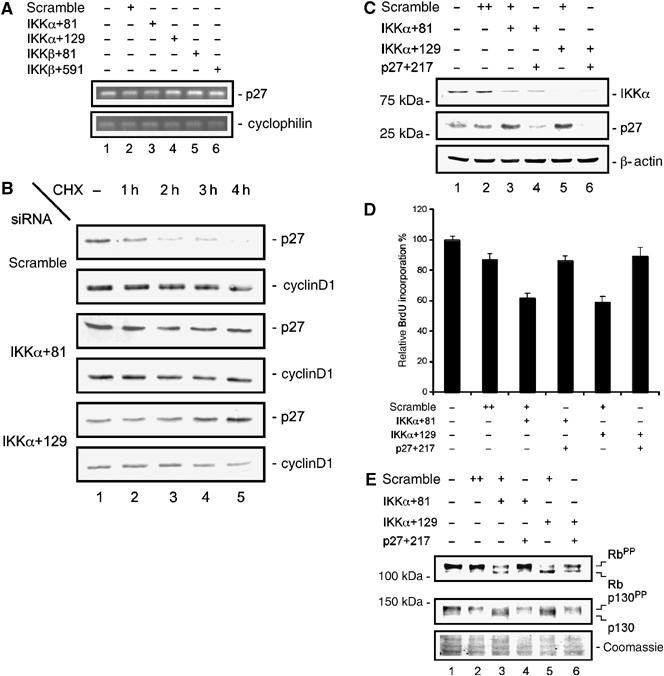
IKKα regulates p27Kip1 stability. (A) Semiquantitative RT–PCR of p27Kip1 mRNA levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. Cyclophilin mRNA serves as an internal control. (B) Determination of p27Kip1 degradation following transfection of MiaPaCa2 cells with the indicated siRNAs. Forty-eight hours after transfection, cells were treated with cycloheximide (CHX) (50 μg/ml) for 4 h. Cyclin D1 was detected on the same membrane and controls loading and specificity. (C) Simultaneous knock-down of IKKα and p27Kip1. Western blot analysis of IKKα and p27Kip1 protein levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. (D) BrdU incorporation after simultaneous knock-down of IKKα and p27Kip1. MiaPaCa2 cells were transfected as indicated. Forty-eight hours later, BrdU incorporation was analyzed. (E) Western blot analysis of Rb and p130 protein levels after the simultaneous knock-down of IKKα and p27Kip1. Hyperphosphorylated proteins are denoted by ‘PP'. The Coomassie-stained membrane serves as a control for equal protein loading.
The increase of p27Kip1 protein levels is essential for impaired proliferation following IKKα knock-down
To examine the consequences of p27Kip1 protein stabilization by IKKα knock-down on the activation of the Rb-dependent G1 checkpoint, we simultaneously transfected IKKα-specific siRNAs and p27Kip1-specific siRNAs into MiaPaCa2 cells. As shown in Figure 3C, the upregulation of p27Kip1 protein following the knock-down of IKKα was completely repressed by transfection of a p27Kip1-specific siRNA (Figure 3C). Also, the simultaneous knock-down of p27Kip1 and IKKα rescued the decreased BrdU incorporation observed in cells transfected only with IKKα-specific siRNAs (Figure 3D). Next, we examined whether the changes in the activation status of Rb and p130 observed in response to IKKα knock-down were also rescued by cotransfection of p27Kip1-specific siRNAs. As shown in Figure 3E, the transfection of the IKKα-specific siRNAs activates Rb as well as p130. When IKKα- and p27Kip1-specific siRNAs were simultaneously transfected, Rb and p130 activation was clearly reduced, a result consistent with the rescue of proliferation as assayed by BrdU incorporation (Figure 3E). These data suggest that the upregulation of p27Kip1 protein levels is essential for the observed growth defect in IKKα siRNA-treated cells.
IKKα regulates skp2 protein and mRNA levels
The upregulation of cyclin E and p27Kip1 observed in MiaPaCa2 cells transfected with IKKα-specific siRNAs is similar to that observed in skp2-deficient cells (Nakayama et al, 2000). Therefore, we examined skp2 protein and mRNA levels in IKKα siRNA-transfected cells. Western blot analysis revealed a significant reduction of skp2 protein in response to IKKα knock-down (Figure 4A), which was not observed in cells transfected with either scramble or IKKβ-specific siRNAs. The protein abundance of cul1 and Rbx1 was not changed after the IKKα knock-down (Figure 4A). Next, using quantitative real-time PCR, we examined skp2 mRNA levels following the knock-down of the catalytic IKK subunits. As shown in Figure 4B, a specific downregulation of skp2 mRNA was observed only in IKKα siRNA-transfected MiaPaCa2 cells. This skp2 mRNA downregulation was not observed in scramble or IKKβ siRNA-transfected cells. To determine whether IKKα-mediated regulation of skp2 was specific to MiaPaCa2 cells, we transfected BxPc3, DanG, and PaTuII pancreatic cancer cells with IKK-specific siRNAs. The knock-down of the catalytic IKK subunits was confirmed by Western blot analysis (Supplementary data 1). In all cell lines tested, a clear downregulation of skp2 protein (Figure 4C) and mRNA (Figure 4D) was detected in IKKα siRNA-treated cells. This effect is specific for IKKα, as scramble or IKKβ-specific siRNAs did not downregulate skp2. Furthermore, in all cell lines tested, transfection of the IKKα-specific siRNAs led to a specific reduction in BrdU incorporation (Figure 4E). The strongest IKKα siRNA-mediated downregulation of skp2 was found in DanG cells, which displayed a reduction in BrdU incorporation to approximately 40% (Figure 4E). No significant reduction of BrdU incorporation was observed in BxPc3 or PaTuII cells transfected with IKKβ-specific siRNAs. In contrast, BrdU incorporation was reduced to approximately 70% in DanG cells following IKKβ knock-down, suggesting a mitogenic role for IKKβ in these cells. Downregulation of the skp2 protein levels following the transfection of IKKα-specific siRNAs was also observed in HeLa cells as well as the stomach adenocarcinoma cell line AGS (data not shown), a finding that further supports the hypothesis that IKKα-mediated regulation of skp2-p27Kip1 involves a general growth regulatory pathway.
Figure 4.
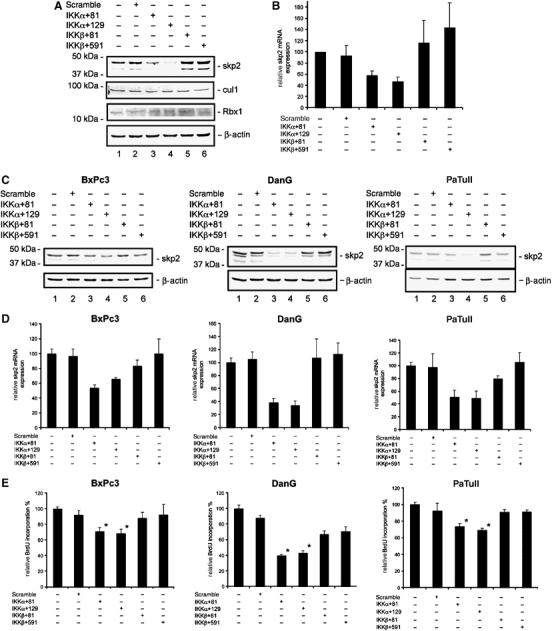
IKKα regulates skp2 protein and mRNA levels. (A) Western blot analysis of skp2, cul1, and Rbx1 protein levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. (B) Quantitative skp2 mRNA expression analysis. Total RNA was prepared 48 h post-transfection with the indicated siRNAs. mRNA levels were quantified using real-time PCR analysis and normalized to cyclophilin expression levels. (C) Expression of skp2 protein after knock-down of the catalytic IKK subunits in BxPc3, DanG, and PaTuII cells. Western blot analysis of skp2 protein levels 48 h post-transfection of BxPc3, DanG, and PaTuII cells with the indicated siRNAs. (D) Quantitative skp2 mRNA expression analysis after knock-down of the catalytic IKK subunits in BxPc3, DanG, and PaTuII cells. Total RNA was prepared 48 h post-transfection with indicated siRNAs. mRNA levels were quantified using real-time PCR analysis and normalized to cyclophilin expression levels. (E) BrdU incorporation of BxPc3, DanG, and PaTuII after knock-down of the catalytic IKK subunits (Student's t-test: *P=<0.001 versus controls).
Skp2 bridges IKKα to p27Kip1
To compare the effects of IKKα and skp2 on cell cycle regulation, we transfected MiaPaCa2 cells with skp2-specific siRNAs and measured p27Kip1 and cyclin E protein levels (Figure 5A). In skp2 siRNA-treated MiaPaCa2 cells, a clear increase in p27Kip1 and cyclin E protein levels comparable to that observed in IKKα siRNA-treated cells was seen (Figure 5A). The upregulation of p27Kip1 in skp2 siRNA-transfected cells led to a reduction in BrdU incorporation (Figure 5B) and induced a G1-phase cell cycle arrest as detected by FACS analysis (data not shown). To determine whether IKKα inhibition-mediated upregulation of p27Kip1 was dependent on skp2, we stably transfected MiaPaCa2 with a murine HA-tagged skp2 expression vector. Transgene expression was detected in the HA-skp2 clones 17 and 20 but not in the untransfected or pcDNA3-transfected clone (Figure 5C). Overexpression of the skp2 transgene resulted in reduced levels of p27Kip1 protein (Figure 5C). Next, we transiently transfected cells stably expressing skp2 with IKKα-specific siRNAs (Figure 5D). Using an antibody that recognizes only human skp2, we detected a downregulation of endogenous skp2 in the pcDNA3 clone 4, the HA-skp2 clone 17, and the HA-skp2 clone 20. However, transgenic murine HA-tagged skp2 protein levels were unaffected following transfection of IKKα siRNAs (Figure 5D). In control cells, transfection of IKKα-specific siRNAs led to an upregulation of p27Kip1, whereas no upregulation of p27Kip1 was detected in the HA-skp2-overexpressing clones 17 and 20 (Figure 5D). These data demonstrate that skp2 mediates the upregulation of p27Kip1 in IKKα siRNA-transfected cells.
Figure 5.
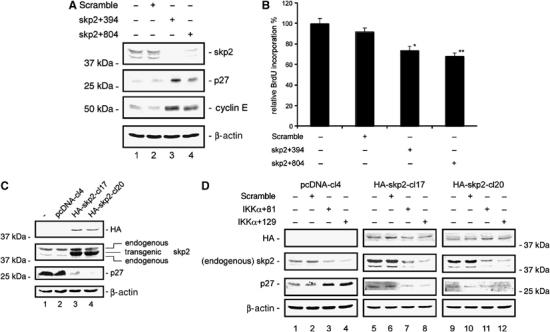
Skp2 mediates upregulation of p27Kip1 after knock-down of IKKα. (A) Western blot analysis of skp2, p27Kip1, and cyclin E protein levels 48 h post-transfection of MiaPaCa2 cells with skp2-specific siRNAs. (B) BrdU incorporation of MiaPaCa2 cells after knock-down of skp2 (Student's t-test: *P=0.004 and **P<0.001 versus controls). (C) Western blot analysis of HA-skp2, skp2 (endogenous and murine overexpressed), and p27Kip1 protein levels in MiaPaCa2 cells stably transfected with murine pcDNA3-HA-skp2 (clones 17 and 20) compared to stably pcDNA3-transfected or untransfected cells. (D) Western blot analysis of HA-skp2, skp2 (endogenous), and p27Kip1 protein levels in stably pcDNA3- or murine pcDNA3-HA-skp2- (clones 17 and 20) transfected MiaPaCa2 cells after knock-down of IKKα.
IKKα regulates transcription of the skp2 gene
To confirm that IKKα regulates transcription of the skp2 gene, we inhibited mRNA synthesis by treating untransfected, control siRNA-transfected, and IKKα siRNA-transfected MiaPaCa2 cells with actinomycin D and measured skp2 mRNA levels over time. As shown in Figure 6A, no differences in skp2 mRNA stability were observed among these cells by 4 h. The half-life of skp2 mRNA in these cells was between 1.5 and 2 h, independent of IKKα expression. These data suggest that IKKα regulates skp2 transcription. To verify the IKKα-mediated transcriptional regulation of the skp2 gene, we performed Chromatin immunoprecipitation (ChIP) assays. As shown in Figure 6B, the presence of RNA polymerase II at the transcriptional start site of the skp2 gene was clearly diminished following knock-down of IKKα. In contrast, RNA polymerase II presence at the transcriptional start of the c-Flip gene did not significantly change following knock-down of IKKα. Furthermore, reduced Ser-10 phosphorylation of histone H3 and decreased acetylation of histone H3 and histone H4 at the skp2 gene was detected following the knock-down of IKKα, whereas the phosphorylation and acetylation status of histones H3 and H4 were not significantly altered at the c-Flip gene (Figure 6B). Together, these data demonstrate that the transcription of the skp2 gene is regulated by IKKα.
Figure 6.
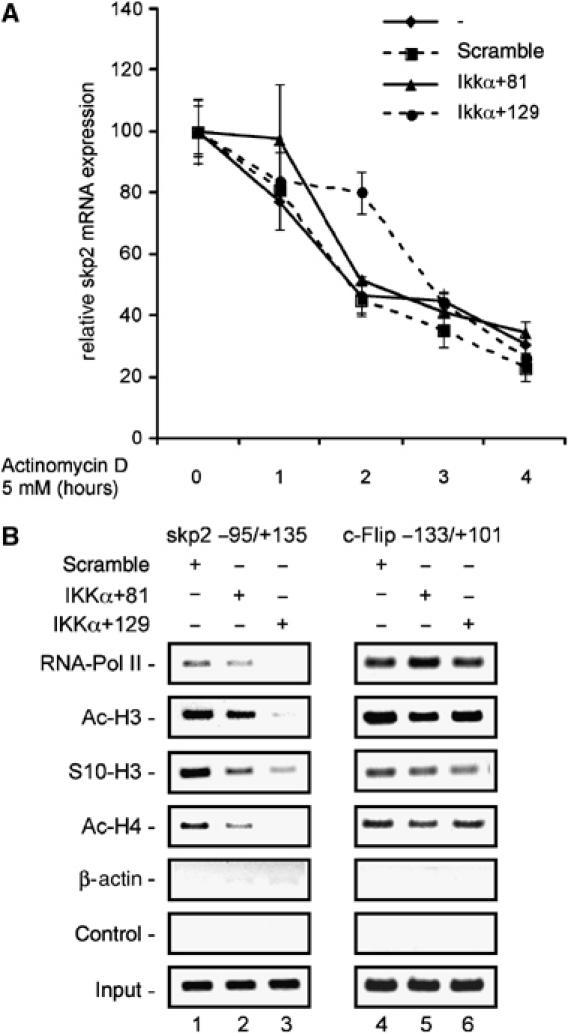
Transcriptional regulation of skp2 by IKKα. (A) Determination of the skp2 mRNA half-life. MiaPaCa2 cells were transfected with the indicated siRNAs or left as an untreated control. Forty-eight hours post-transfection, cells were treated with actinomycin D (5 mM) for 4 h. Total mRNA was prepared every hour and quantified using real-time PCR analysis. Skp2 mRNA levels of untreated controls were set to 100% and compared with the skp2 mRNA levels following actinomycin D treatment. (B) ChIP analysis of the skp2 gene. Chromatin of MiaPaCa2 cells transfected with a control siRNA or two IKKα-specific siRNAs was immunoprecipitated with an RNA polymerase II-specific antibody, an anti-phospho-Ser-10-histone H3, an anti-acetyl-histone H3, an anti-acetyl-histone H4, an β-actin, or no antibody as a negative control. Precipitated DNA or 10% of the chromatin input was amplified with gene-specific primers for skp2 or c-Flip.
RelB binds to the skp2 gene promoter and regulates its transcription
To determine the contribution of NF-κB family members to the regulation of skp2 expression, we transfected specific siRNAs for RelA/p65, c-Rel, and RelB into MiaPaCa2 cells. Western blot analysis was used to demonstrate the specific knock-down of the respective proteins 48 h post-transfection (Figure 7A). No skp2 expression changes were observed after the knock-down of RelA/p65 and c-Rel, whereas a significant reduction in skp2 protein expression was detected after knock-down of RelB. Consistently, p27Kip1 was upregulated following the RelB knock-down but was unaffected by the knock-down of RelA/p65 or c-Rel (Figure 7A). Furthermore, skp2 mRNA expression was specifically reduced (to 50.3%) as compared to untreated controls following RelB knock-down, whereas it was not significantly changed following RelA/p65 or c-Rel knock-down (Figure 7B).
Figure 7.
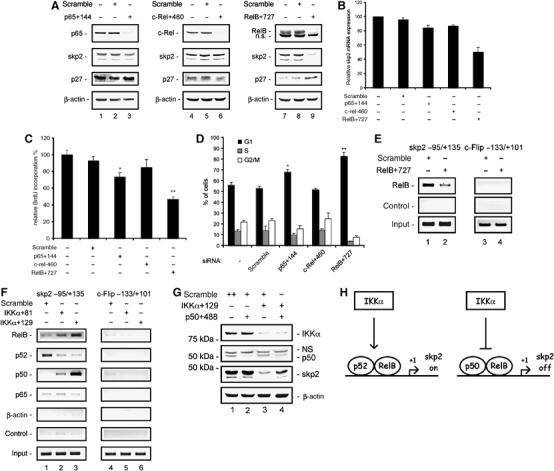
IKKα-controlled RelB complexes bind to and regulate the skp2 gene. (A) Western blot analysis of RelA/p65, c-Rel, RelB, skp2, and p27Kip1 protein levels 48 h post-transfection of MiaPaCa2 cells with RelA/p65-, c-Rel-, or RelB-specific siRNAs. (B) Quantitative skp2 mRNA expression analysis after knock-down of RelA/p65, c-Rel, and RelB in MiaPaCa2 cells. Total RNA was prepared 48 h post-transfection with the indicated siRNAs. mRNA levels were quantified using real-time PCR analysis and normalized to cyclophilin expression levels. (C) BrdU incorporation following the knock-down of RelA/p65, c-Rel, and RelB in MiaPaCa2 cells (Student's t-test: *P=0.001 and **P<0.001 versus controls). (D) FACS analysis of cell cycle distribution. MiaPaCa2 cells were transfected with the indicated siRNAs. Forty-eight hours later, cells were stained with PI and analyzed by FACS. The fraction of the cells in G1, S, and G2/M phases is indicated (Student's t-test: *P=0.005 and **P=0.002 versus controls). (E) ChIP analysis of the skp2 gene. Chromatin of MiaPaCa2 cells transfected with a control siRNA or RelB-specific siRNA was immunoprecipitated with a RelB-specific antibody or no antibody as a negative control. Precipitated DNA or 10% of the chromatin input was amplified with gene-specific primers for skp2 or c-Flip. (F) ChIP analysis of the skp2 gene. Chromatin of MiaPaCa2 cells transfected with a control siRNA or two IKKα-specific siRNAs was immunoprecipitated with RelB-, p52-, p50, or p65-specific antibodies or with a β-actin or no antibody as negative controls. Precipitated DNA or 10% of the chromatin input was amplified with gene-specific primers for skp2 or c-Flip. (G) Simultaneous knock-down of IKKα and p50. Western blot analysis of IKKα, p50, and skp2 protein levels 48 h post-transfection of MiaPaCa2 cells with the indicated siRNAs. (H) Illustration of skp2 transcriptional regulation by IKKα.
To determine if RelB knock-down causes impaired proliferation, we examined BrdU incorporation and cell cycle distribution. Following transfection of RelA/p65 siRNA, c-Rel siRNA, or RelB siRNA, BrdU incorporation was reduced to 73.7, 85, and 46.9%, respectively, relative to untreated control cells (Figure 7C). Additionally, an increase of cells in the G1 phase was observed following the knock-down of RelA/p65 (68.1%) and RelB (82.8%) relative to untreated (56%) or scramble treated (52.9%) controls, whereas no change was observed following the c-Rel knock-down (Figure 7D).
To examine possible direct binding of RelB to the skp2 gene, we used ChIP assays. As shown in Figure 7E, direct binding of RelB was observed at the proximal skp2 gene promoter. In contrast, RelB binding was not observed at the proximal promoter of c-Flip. Furthermore, RelB binding at the skp2 gene promoter was diminished by transfection of RelB siRNA (Figure 7E). Together, these data suggest that RelB-mediated regulation of skp2 expression controls G1-phase progression in pancreatic cancer cells.
IKKα regulates the exchange of p50/RelB for p52/RelB at the skp2 gene promoter
To analyze the contribution of RelB to the downregulation of skp2 following the knock-down of IKKα, we performed ChIP assays. As shown in Figure 7F, we observed increased binding of RelB to the proximal skp2 gene promoter in response to the knock-down of IKKα in MiaPaCa2 cells. No binding of RelB was detected at the proximal promoter of the c-Flip gene. RelB does not form homodimers, but rather only heterodimerizes with p50 and p52 (Dobrzanski et al, 1994). Therefore, skp2 gene promoter binding by p50 and p52 following the knock-down of IKKα was investigated using ChIP assays. A decrease in the binding of p52 to the proximal skp2 gene promoter was observed (Figure 7F). In contrast, we detected a significant increase in p50 binding to the proximal skp2 gene promoter following the knock-down of IKKα (Figure 7F). Faint binding of RelA/p65 was found at the proximal skp2 gene promoter, not regulated by IKKα siRNAs. Neither p52 nor p50 binding was detected at the proximal promoter of the c-Flip gene (Figure 7F). Similar regulation was observed in DanG cells (data not shown). To investigate p50 function after the IKKα knock-down, we simultaneously transfected IKKα-specific siRNAs and p50-specific siRNAs into MiaPaCa2 cells. As shown in Figure 7G, the downregulation of skp2 after the transfection of IKKα-specific siRNAs was rescued by the cotransfection of IKKα- and p50-specific siRNAs. These results suggest that IKKα regulates the exchange of a p50/RelB repressive complex for a p52/RelB activating complex at the skp2 gene promoter.
Discussion
In the present study, we show that the G1 phase of the cell cycle is regulated by IKKα, one of the two catalytic components of the IκB kinase complex. We demonstrate that IKKα regulates the stability of the CKI p27Kip1 protein and thereby controls the Rb-dependent G1-phase checkpoint. Increased levels of p27Kip1 following knock-down of IKKα are a result of the downregulation of the F-box protein skp2. IKKα regulates skp2 at the transcriptional level. Furthermore, we demonstrate that IKKα controls the exchange of p50/RelB for a p52/RelB dimer at the skp2 gene promoter.
Skp2, identified as a component of the cyclin A/CDK2 complex, is currently one of the best-characterized F-box proteins. It functions as a receptor subunit for the SCF ubiquitin E3 ligase. Skp2 controls the turnover of several key cell cycle regulatory proteins, including p27Kip1 (Nakayama and Nakayama, 2005). Skp2 knockout mice have a hypoproliferative phenotype and several severe cellular defects, including nuclear enlargement, polyploidy, and increased numbers of centromeres (Nakayama et al, 2000). Consistent with the skp2 knockout mice phenotypes, we found reduced skp2 expression and increased p27Kip1 and cyclin E expression following knock-down of IKKα. Elevated cyclin E levels in skp2−/− mouse embryonic fibroblasts and hepatocytes do not increase CDK2 kinase activity because of the contemporary accumulation of p27Kip1, a potent inhibitor of CDK2 (Nakayama et al, 2000; Kossatz et al, 2004). Immunoprecipitation of p27Kip1 with cyclin E/CDK2 complexes and CDK2 kinase assay after the IKKα knock-down suggest a similar mechanism compensating for the increased cyclin E expression in our model system. Although biochemical data demonstrate multiple targets of skp2, genetic evidence suggests that p27Kip1 is the major downstream target of skp2, as the skp2 knockout phenotype is almost completely reverted in the p27Kip1/skp2 double knockout mice (Kossatz et al, 2004; Nakayama et al, 2004). Correspondingly, we found a rescue of the proliferative defect and Rb checkpoint activation in the IKKα/p27Kip1 double knock-down cells.
Skp2 protein levels are low or undetectable in G0 and early G1 phase and gradually increase during the progression of the cell cycle into the S phase. This induction is due to transcriptional activation of the skp2 gene and stabilization of the skp2 protein; the contribution of each of these regulatory mechanisms varies between different cell types (Zhang et al, 1995; Wirbelauer et al, 2000; Carrano and Pagano, 2001; Imaki et al, 2003; Bashir et al, 2004; Wei et al, 2004). In the early G1 phase, skp2 protein is degraded by the anaphase-promoting complex/cyclosome containing cdh1 (APC/Ccdh1) (Bashir et al, 2004; Wei et al, 2004). Protein levels of stably transfected HA-tagged skp2 following IKKα knock-down are unchanged. These data suggest that IKKα does not seem to target skp2 protein stability. However, the discrepancy between skp2 protein and mRNA levels observed after the IKKα and RelB knock-down would argue for an additional post-transcriptional mode of regulation.
Transcriptional regulation of the skp2 gene is not entirely understood. The murine skp2 promoter was recently analyzed and demonstrated to bind the transcription factor GA-binding protein in a cell cycle-dependent manner (Imaki et al, 2003). Furthermore, the transcription of skp2 was shown to be regulated by a PI3 kinase-dependent pathway (Mamillapalli et al, 2001). In addition, recent studies have demonstrated that the Notch signaling pathway is linked to skp2 gene transcription (Sarmento et al, 2005). The results presented here demonstrate that skp2 is regulated at the transcriptional level by IKKα. This conclusion is based on the following observations: (1) skp2 protein and mRNA levels are similarly reduced in IKKα siRNA-transfected cells, (2) skp2 mRNA half-life is unchanged following IKKα knock-down, and (3) binding of RNA-Pol II to the transcriptional start site of the skp2 gene is decreased following IKKα knock-down. Moreover, we show the hypoacetylation of histone H3 and H4 at the skp2 gene 5′ regulatory region in cells treated with IKKα siRNAs.
IKKα, an essential component of the NF-κB pathway, influences many aspects of normal and disease physiology (Hayden and Ghosh, 2004). Two signaling pathways activate the NF-κB pathway. The classical pathway, activated by TNFα or IL-1β, leads to the phosphorylation and ubiquitin-dependent degradation of NF-κB inhibitors IκBα and IκBβ via the IKK complex. The resulting free p50/p65 NF-κB subunits translocate into the nucleus and activate transcription. In contrast to classical inducers, lymphotoxin β, BAFF, and CD40L activate the alternative pathway. In this pathway, the IKKα signal results in p100 processing and translocation of p52/RelB to the nucleus. RelB displays a dimerization pattern unique to the NF-κB family. It does not homodimerize or heterodimerize with c-Rel. When complexed with RelA/p65, RelB is unable to bind DNA. It forms dimers with p100, p52, and p50 and does not interact with classical IκB molecules (Ryseck et al, 1992; Dobrzanski et al, 1994; Solan et al, 2002). RelB-containing complexes were shown to act as both activators and repressors of NF-κB-dependent gene transcription (Ruben et al, 1992; Bours et al, 1994; Suhasini and Pilz, 1999; Marienfeld et al, 2003). Importantly, the recruitment of RelB to the IL-12p40 promoter correlates with transcriptional down-regulation, whereas the transcription of ELC and MDC genes was induced by RelB (Saccani et al, 2003). More recently, dimer exchange has been shown to allow for the finely regulated expression of NF-κB responsive genes in dendritic cells. Although RelB is inducible by mitogenic stimulation and B cells of RelB knockout mice are defective in proliferative responses, molecular pathways linking RelB to the cell cycle machinery are not completely understood (Ruben et al, 1992; Snapper et al, 1996). In our cellular model, RelB clearly regulates the transcription of skp2. We detected binding of p52/RelB dimers to the proximal skp2 gene promoter in random cycling cells. Reducing the binding of RelB to the skp2 gene promoter induced a transcriptional downregulation of skp2, suggesting an activating function of the p52/RelB complex. After blocking IKKα signaling using siRNAs, we found an exchange of p52/RelB dimers for p50/RelB dimers at the skp2 gene promoter. Because p50/RelB dimers were shown to possess repressive functions and the IKKα knock-down downregulates skp2 transcription, the p50/RelB dimer likely represses the skp2 gene (Ruben et al, 1992). The incomplete rescue of the skp2 downregulation by the cotransfection of IKKα- and p50-specific siRNA is most likely owing to the incomplete p50 knock-down. The model of IKKα-dependent regulation of skp2 is illustrated in Figure 7H. Consistent with our data, the human skp2 gene promoter was recently cloned and sequence analysis demonstrated NF-κB binding sites in the proximal promoter (Zhang and Wang, 2006). The molecular mechanism of RelB-dependent repression is unclear. Although dimer exchange is exploited by the NF-κB system to finely regulate or even shut down transcription, mechanisms driving dimer exchange are not well understood and require further investigation (Natoli et al, 2005).
Recently, transcriptionally inactive complexes of RelA/p65 and RelB that prevent RelB DNA binding were described (Marienfeld et al, 2003; Jacque et al, 2005). However, this mechanism of regulation of RelB cannot account for the observed regulation of skp2 in our model, as we observed a significant increase of RelB binding to the skp2 gene promoter following the IKKα knock-down. In line with the increased binding of RelB to the skp2 gene promoter, we observed no significant effect regarding p100 processing after the IKKα knock-down in our model system (Supplementary data 2).
In addition to the regulation of the alternative pathway, nuclear functions of IKKα have recently been described. Following TNFα and EGF stimulation, IKKα bound to the promoters of NF-κB dependent genes like IκBα, IL-8, IL-6, or c-Fos phosphorylates Ser-10 of histone H3, an event important for the subsequent Lys-14 acetylation of histone H3 by the histone acetyltransferase CBP (Anest et al, 2003, 2004; Yamamoto et al, 2003). Although we observed reduced phosphorylation of histone H3 Ser-10 at the 5′ regulatory region of the skp2 gene following the knock-down of IKKα, we were unable to detect genomic sequences 1000 bp downstream of the skp2 transcriptional start by ChIP-assays using several different IKKα-specific antibodies (data not shown). This result argues against a direct Ser-10 histone H3 kinase activity for IKKα at the skp2 locus.
Studies of IKKα-deficient fibroblasts and mice with kinase-defective IKKαAA knock-in alleles have demonstrated that IKKα controls cyclin D1 gene expression and G1-phase progression (Cao et al, 2001; Albanese et al, 2003). Additionally, recent studies have demonstrated that the expression of cyclin D1 in mammary gland development and carcinogenesis is regulated by a p52/RelB dimer (Demicco et al, 2005). Furthermore, siRNA-mediated knock-down of IKKα in the breast cancer cell line MCF7 results in a reduction of basal and estradiol-induced cyclin D1 expression (Park et al, 2005). We did not find significant regulation of cyclin D1 in our cellular model. One possible explanation for this discrepancy is that the activities of the remaining IKK complexes following the knock-down of the individual catalytic subunits may be capable of maintaining cyclin D1 expression. The cyclin D1 promoter was shown to bind p52/RelB-, p52/bcl3-, and p50/p65-containing NF-κB complexes (Guttridge et al, 1999; Hinz et al, 1999; Rocha et al, 2003; Demicco et al, 2005). Therefore, the lack of deregulation of cyclin D1 following the knock-down of IKKα may be owing to the compensatory regulation of the cyclin D1 gene by the classical NF-κB pathway. Consistent with this prediction, we detected impaired expression of cyclin D1 following the transfection of RelA/p65 siRNA in our model system (data not shown).
Overexpression of skp2 has been described in a large number of human cancers. Furthermore, ectopic expression of skp2 in mice leads to lymphomagenesis and low-grade prostate cancer, demonstrating an oncogenic function of skp2 (Latres et al, 2001; Shim et al, 2003). Skp2 cooperates with Ras to transform cells in vitro and in vivo (Gstaiger et al, 2001; Latres et al, 2001). Our work serves to connect the oncogenic and cell cycle regulatory functions of skp2 to IKKα and RelB. Given the role of skp2 in tumorigenesis, our work describes how the alternative NF-κB activation pathway may be considered as a potential target for tumor therapies.
Materials and methods
Cell culture and reagents
Pancreatic carcinoma cell lines were cultured as described previously (Schneider et al, 2006). Cycloheximide and actinomycin D (Sigma-Aldrich) were dissolved in ethanol and stored at −20°C.
Stable transfection
For stable transfection, linearized plasmids were transfected using FuGene6 (Roche Applied Science). Twenty-four hours post-transfection, cells were serial diluted and cultured in medium containing 1 mg/ml Geneticin (Invitrogen). After 2 weeks, single clones were picked, propagated, and analyzed for protein expression.
Statistical methods
All data were obtained from at least three independent experiments performed in duplicate, and the results are presented as mean and standard error of the mean (s.e.m.). To demonstrate statistical significance, Student's t-test was used in various experiments. P-values are indicated in the figure legends.
siRNA transfection
Double-stranded siRNAs were transfected at a final concentration of 200 nM using oligofectamine (Invitrogen) essentially as described (Schneider et al, 2006). For the simultaneous transfection of siRNAs directed against two different genes, the total amount of siRNA (400 nM) was kept constant using scramble siRNA. siRNAs were purchased from Ambion. Target sequences of the used siRNAs are shown in Supplementary data 3.
Plasmids
The murine HA-tagged skp2 expression vector was a generous gift of Dr Nakayama (Nakayama et al, 2000).
Protein analysis
Whole-cell lysates were prepared and Western blots were performed as described (Schneider et al, 2006). For Western blot and immunoprecipitation, the following antibodies were used: IKKα, cyclin E, E2F1, cul1, Rbx1, CDK2, p65, RelB, c-Rel, p100, p105, Oct1, α-tubulin (Santa Cruz Biotechnology), IKKβ (Upstate), skp2 (Zymed), p27Kip1, Rb1, Rb2/p130 (BD Pharmingen), cyclin D1 (Oncogene Research Products), and β-actin (Sigma-Aldrich). Proteins recognized by these antibodies were detected by the Odyssey Infrared Imaging System (Licor) using Alexa680-coupled (Molecular Probes) or IRDeye800-coupled (Rockland) secondary antibodies. Immunocomplex kinase assay was carried out essentially as recently described using histone H1 as a substrate (Roche Applied Science) (Liptay et al, 2003).
Semiquantitative and quantitative RT–PCR
Total RNA was isolated from pancreatic carcinoma cell lines using the RNeasy kit (Qiagen) according to the manufacturer's instructions. One hundred nanograms of total RNA was used to perform RT–PCR using the TaqMan Reverse Transcription reagents (Applied Biosystems). Quantitative mRNA analysis was performed using real-time PCR analysis as described previously (Schneider et al, 2002). Primer sequences are available upon request.
BrdU incorporation assay, apoptosis staining, and cell cycle analysis
BrdU incorporation assays, Hoechst 33342 stains, and cell cycle analysis were performed as described (Schneider et al, 2002, 2006).
ChIP assay
Standard protocol was used for ChIP assays (Saccani et al, 2003). An equal amount of chromatin (50–100 μg) was used for each precipitation. The antibodies used were as follows: anti-RNA polymerase II, anti-RelB, anti-p52, anti-p50, anti-p65 (Santa Cruz Biotechnology), anti-acetyl-histone H3, anti-acetyl-histone H4, anti-phospho-Ser-10-histone H3 (Upstate), and β-actin (Sigma). One-twentieth of the precipitated chromatin was used for each PCR reaction. To ensure linearity, 28–38 cycles were performed, and one representative result is shown. Sequences of the promoter-specific primers and a detailed experimental protocol are available upon request.
Supplementary Material
Supplementary data 1
Supplementary data 2
Supplementary data 3
Acknowledgments
We thank Birgit Kohnke-Ertel and Konstanze Geiger for excellent technical support. We also thank Dr Nakayama for generously providing the skp2 expression plasmids. This work was funded by grants from the Deutsche Forschungsgemeinschaft (SFB456 to GS and RS).
References
- Albanese C, Wu K, D'Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze'ev A, Bromberg JF, Lamberti C, Verma U, Gaynor RB, Byers SW, Pestell RG (2003) IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol Biol Cell 14: 585–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anest V, Cogswell PC, Baldwin AS Jr (2004) IkappaB kinase alpha and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IkappaBalpha degradation. J Biol Chem 279: 31183–31189 [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature 423: 659–663 [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M (2004) Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428: 190–193 [DOI] [PubMed] [Google Scholar]
- Bloom J, Pagano M (2003) Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol 13: 41–47 [DOI] [PubMed] [Google Scholar]
- Bours V, Azarenko V, Dejardin E, Siebenlist U (1994) Human RelB (I-Rel) functions as a kappa B site-dependent transactivating member of the family of Rel-related proteins. Oncogene 9: 1699–1702 [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M (2001) IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 107: 763–775 [DOI] [PubMed] [Google Scholar]
- Carrano AC, Pagano M (2001) Role of the F-box protein Skp2 in adhesion-dependent cell cycle progression. J Cell Biol 153: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demicco EG, Kavanagh KT, Romieu-Mourez R, Wang X, Shin SR, Landesman-Bollag E, Seldin DC, Sonenshein GE (2005) RelB/p52 NF-kappaB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IkappaB-alpha expression and promote carcinogenesis of the mammary gland. Mol Cell Biol 25: 10136–10147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrzanski P, Ryseck RP, Bravo R (1994) Differential interactions of Rel–NF-kappa B complexes with I kappa B alpha determine pools of constitutive and inducible NF-kappa B activity. EMBO J 13: 4608–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296 [DOI] [PubMed] [Google Scholar]
- Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Krek W (2001) Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci USA 98: 5043–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr (1999) NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19: 5785–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M (1999) NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol 19: 2690–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg JE, Yeung F, Mayo MW (2004) SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol Cell 16: 245–255 [DOI] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC (2004) IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117: 225–237 [DOI] [PubMed] [Google Scholar]
- Imaki H, Nakayama K, Delehouzee S, Handa H, Kitagawa M, Kamura T, Nakayama KI (2003) Cell cycle-dependent regulation of the Skp2 promoter by GA-binding protein. Cancer Res 63: 4607–4613 [PubMed] [Google Scholar]
- Jacque E, Tchenio T, Piton G, Romeo PH, Baud V (2005) RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. Proc Natl Acad Sci USA 102: 14635–14640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP (2004) Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev 18: 2602–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latres E, Chiarle R, Schulman BA, Pavletich NP, Pellicer A, Inghirami G, Pagano M (2001) Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci USA 98: 2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Aggarwal BB, Shishodia S, Abbruzzese J, Kurzrock R (2004) Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer 101: 2351–2362 [DOI] [PubMed] [Google Scholar]
- Lin DI, Diehl JA (2004) Mechanism of cell-cycle control: ligating the ligase. Trends Biochem Sci 29: 453–455 [DOI] [PubMed] [Google Scholar]
- Liptay S, Weber CK, Ludwig L, Wagner M, Adler G, Schmid RM (2003) Mitogenic and antiapoptotic role of constitutive NF-kappaB/Rel activity in pancreatic cancer. Int J Cancer 105: 735–746 [DOI] [PubMed] [Google Scholar]
- Mamillapalli R, Gavrilova N, Mihaylova VT, Tsvetkov LM, Wu H, Zhang H, Sun H (2001) PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27(KIP1) through the ubiquitin E3 ligase SCF(SKP2). Curr Biol 11: 263–267 [DOI] [PubMed] [Google Scholar]
- Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M (2003) RelB forms transcriptionally inactive complexes with RelA/p65. J Biol Chem 278: 19852–19860 [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Hatakeyama S (2000) Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 19: 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI (2004) Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 6: 661–672 [DOI] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K (2005) Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol 16: 323–333 [DOI] [PubMed] [Google Scholar]
- Natoli G, Saccani S, Bosisio D, Marazzi I (2005) Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol 6: 439–445 [DOI] [PubMed] [Google Scholar]
- Park KJ, Krishnan V, O'Malley BW, Yamamoto Y, Gaynor RB (2005) Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell 18: 71–82 [DOI] [PubMed] [Google Scholar]
- Reed SI (2003) Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol 4: 855–864 [DOI] [PubMed] [Google Scholar]
- Rocha S, Martin AM, Meek DW, Perkins ND (2003) p53 represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-kappaB subunit with histone deacetylase 1. Mol Cell Biol 23: 4713–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE (1997) Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res 57: 1731–1734 [PubMed] [Google Scholar]
- Ruben SM, Klement JF, Coleman TA, Maher M, Chen CH, Rosen CA (1992) I-Rel: a novel rel-related protein that inhibits NF-kappa B transcriptional activity. Genes Dev 6: 745–760 [DOI] [PubMed] [Google Scholar]
- Ryseck RP, Bull P, Takamiya M, Bours V, Siebenlist U, Dobrzanski P, Bravo R (1992) RelB, a new Rel family transcription activator that can interact with p50–NF-kappa B. Mol Cell Biol 12: 674–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G (2003) Modulation of NF-kappaB activity by exchange of dimers. Mol Cell 11: 1563–1574 [DOI] [PubMed] [Google Scholar]
- Sarmento LM, Huang H, Limon A, Gordon W, Fernandes J, Tavares MJ, Miele L, Cardoso AA, Classon M, Carlesso N (2005) Notch1 modulates timing of G1–S progression by inducing SKP2 transcription and p27 Kip1 degradation. J Exp Med 202: 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G, Oswald F, Wahl C, Greten FR, Adler G, Schmid RM (2002) Cyclosporine inhibits growth through the activating transcription factor/cAMP-responsive element-binding protein binding site in the cyclin D1 promoter. J Biol Chem 277: 43599–43607 [DOI] [PubMed] [Google Scholar]
- Schneider G, Weber A, Zechner U, Oswald F, Friess HM, Schmid RM, Liptay S (2006) GADD45alpha is highly expressed in pancreatic ductal adenocarcinoma cells and required for tumor cell viability. Int J Cancer 118: 2405–2411 [DOI] [PubMed] [Google Scholar]
- Shim EH, Johnson L, Noh HL, Kim YJ, Sun H, Zeiss C, Zhang H (2003) Expression of the F-box protein SKP2 induces hyperplasia, dysplasia, and low-grade carcinoma in the mouse prostate. Cancer Res 63: 1583–1588 [PubMed] [Google Scholar]
- Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, Weih F (1996) B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med 184: 1537–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan NJ, Miyoshi H, Carmona EM, Bren GD, Paya CV (2002) RelB cellular regulation and transcriptional activity are regulated by p100. J Biol Chem 277: 1405–1418 [DOI] [PubMed] [Google Scholar]
- Suhasini M, Pilz RB (1999) Transcriptional elongation of c-myb is regulated by NF-kappaB (p50/RelB). Oncogene 18: 7360–7369 [DOI] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr (2004) Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428: 194–198 [DOI] [PubMed] [Google Scholar]
- Wirbelauer C, Sutterluty H, Blondel M, Gstaiger M, Peter M, Reymond F, Krek W (2000) The F-box protein Skp2 is a ubiquitylation target of a Cul1-based core ubiquitin ligase complex: evidence for a role of Cul1 in the suppression of Skp2 expression in quiescent fibroblasts. EMBO J 19: 5362–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Gaynor RB (2004) IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci 29: 72–79 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB (2003) Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature 423: 655–659 [DOI] [PubMed] [Google Scholar]
- Zhang H, Kobayashi R, Galaktionov K, Beach D (1995) p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell 82: 915–925 [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang C (2006) F-box protein Skp2: a novel transcriptional target of E2F. Oncogene 25: 2615–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data 1
Supplementary data 2
Supplementary data 3