

Abstract
We report here a synthetic-lethal screen in Caenorhabditis elegans that overcomes a number of obstacles associated with the analysis of functionally redundant genes. Using this approach, we have identified mutations that synthetically interact with lin-35/Rb, a SynMuv gene and the sole member of the Rb/pocket protein family in C. elegans. Unlike the original SynMuv screens, our approach is completely nonbiased and can theoretically be applied to any situation in which a mutation fails to produce a detectable phenotype. From this screen we have identified fzr-1, a gene that synthetically interacts with lin-35 to produce global defects in cell proliferation control. fzr-1 encodes the C. elegans homolog of Cdh1/Hct1/FZR, a gene product shown in other systems to regulate the APC cyclosome. We have also uncovered genetic interactions between fzr-1 and a subset of class B SynMuv genes, and between lin-35 and the putative SCF regulator lin-23. We propose that lin-35, fzr-1, and lin-23 function redundantly to control cell cycle progression through the regulation of cyclin levels.
Keywords: lin-35, retinoblastoma, fizzy related, fzr-1, C. elegans, hyperproliferation, synthetic screen
Genetic redundancy is a common feature of all eukaryotes, ensuring both a high degree of regulatory control and protection against mutational assault. It is also an impediment to biologists seeking to determine gene function through forward and reverse genetic approaches. Mutations in functionally redundant genes may show no obvious phenotype on their own but, when present in specific combinations, can lead to severe defects (i.e., a synthetic phenotype). Therefore, the ability to assign functions to this class of genes based solely on the availability and analysis of single mutants may be difficult or impossible.
Estimates from yeast suggest that ∼40% of all genes may fail to show even weak phenotypes when deleted (Smith et al. 1996; Winzeler et al. 1999), whereas a recent large-scale functional analysis of Caenorhabditis elegans genes using RNAi methods failed to detect phenotypes for >85% of the genes examined (Fraser et al. 2000). In addition, recent small-scale deletion studies as well as previous mutational analyses suggest that functional disruptions of most genes in C. elegans fail to produce obvious phenotypic effects (for review, see Hodgkin 2001). This apparent lack of an observable gene function may be the result of limitations at the level of detection as well as in the methodology used in the case of RNAi analysis. However, it is also likely a consequence of functional overlap conferred by either structurally related proteins or by molecularly distinct but functionally connected pathways. Evidence from yeast suggests that this latter explanation may, in fact, be the more common cause of genetic redundancy (Winzeler et al. 1999; for review, see Tautz 2000; Wagner 2000).
Given the difficulties in detecting synthetic interactions, relatively few examples of such nonhomologous genetic redundancies have been well characterized in C. elegans (Culotti et al. 1981; Johnson et al. 1981; Ferguson and Horvitz 1989; Davies et al. 1999). The best-known case of functional redundancy in C. elegans is the synthetic multivulval (SynMuv) genes (Ferguson et al. 1987; Ferguson and Horvitz 1989; for review, see Fay and Han 2000). SynMuv genes act in opposition to Ras/Map kinase pathway signaling to prevent the adoption of ectopic vulval cell fates during postembryonic development (Ferguson et al. 1987; Lu and Horvitz 1998). The relatively straightforward identification of SynMuv genes was made feasible by the ease with which Muv animals can be detected, the normally rare incidence of Muv animals in mutagenized populations, and the fact that double mutants are generally viable. SynMuv genes are commonly assigned membership into one of two classes, A or B. Pairwise combinations of mutations in both a class A gene and a class B gene lead to the expression of the Muv phenotype (Ferguson and Horvitz 1989).
The past several years have seen the molecular identification of many SynMuv genes through both forward and reverse genetic approaches. Although some encode peptides with unknown cellular or biochemical functions, a sizable fraction of class B genes have been linked (based on sequence homology) to transcriptional repression mechanisms. These include multiple members of the NuRD (nucleosome remodeling and histone deacetylase) complex (Lu and Horvitz 1998; Solari and Ahringer 2000; von Zelewsky et al. 2000), E2F and its binding partner Dp (Ceol and Horvitz 2001), and the solitary C. elegans Rb/pocket protein homolog LIN-35 (Lu and Horvitz 1998).
In dramatic contrast to Rb knockout mutations in flies and mice (Clarke et al. 1992; Jacks et al. 1992; Lee et al. 1992; Du and Dyson 1999; for review, see Lipinski and Jacks 1999), strong loss-of-function (LOF) mutations in lin-35 are not lethal and have relatively subtle effects on viability and development (Lu and Horvitz 1998; this paper). Thus, based on the SynMuv screens, the single function ascribed to lin-35 affected only a small number of postembryonic blast cells and, moreover, required the presence of a second-site mutation to be revealed. These observations and the finding that LIN-35 is expressed broadly throughout development (Lu and Horvitz 1998) led us to hypothesize that: (1) lin-35 must carry out additional functions during C. elegans development; and (2) the identification of such functions will necessitate the isolation of second-site mutations that interact synthetically with mutations in lin-35 to produce novel phenotypes.
Using a nonbiased genetic approach, we have isolated a set of mutations that interact synthetically with lin-35. We report here the molecular identification and phenotypic characterization of one of these mutants, fzr-1, that affects the function of the C. elegans homolog of Cdh1/Hct1/Fizzy Related (FZR; Schwab et al. 1997; Sigrist and Lehner 1997; Visintin et al. 1997). In combination, mutations in fzr-1 and lin-35 lead to a striking hyperproliferation phenotype, showing for the first time that LIN-35 functions globally to control cell proliferation in C. elegans. In addition, we have also identified a synthetic requirement for lin-35 and fzr-1 during embryonic development, and describe an additional synthetic interaction between lin-35 and the putative SCF complex (Skp1, Cullin, F-box) regulator, lin-23 (Kipreos et al. 2000).
Results
A targeted strategy to identify novel lin-35 synthetic mutations
As a starting point for identifying novel lin-35/Rb synthetic mutants, we engineered strain MH1461 with two key components. The strain is (1) homozygous for a strong LOF mutation at the endogenous lin-35 locus (n745) and (2) carries an extrachromosomal array (kuEx119) containing wild-type rescuing copies of lin-35 and a GFP reporter that is ubiquitously expressed (also see Materials and Methods). kuEx119 fails to be transmitted to ∼25% of progeny derived from kuEx119+ parents. Thus, kuEx119+ animals give rise to both kuEx119+ (GFP+/lin-35+) and kuEx119− (GFP−/lin35−) progeny (Figs. 1 and 2A,B).
Figure 1.

Outline of lin-35 synthetic screen. Following chemical mutagenesis, three consecutive generations of kuEx119+ animals are single-cloned and allowed to self-fertilize. Animals acquiring silent or otherwise viable mutations (m) that show synergy with lin-35 are identified by the absence of wild-type-appearing kuEx119− progeny in the final (F3) generation. Only animals carrying mutations are shown in subsequent generations. For further details, see text.
Figure 2.

Synergism between lin-35 and fzr-1. (A,C,E,G) DIC and corresponding (B,D,F,H) GFP fluorescence images. White arrows indicate positions of kuEx119− animals. (A,B) The starting strain for mutagenesis, MH1461 (lin-35; kuEx119). Note the identical appearance of kuEx119+/− adult animals. (C–F) kuEx119+/− progeny from strain MH1791 (lin-35; fzr-1; kuEx119), (C,D) ∼60 h and (E,F) ∼96 h posthatching. Note pronounced developmental-stage (C,D) and size (E,F) differences between kuEx119+/− animals. Black arrows indicate the location of nonviable embryos. (D) F1 progeny of MH1791 animals injected with lin-35 dsRNA (1.2 μg/μL). Large white arrowheads indicate the positions of kuEx119+; lin-35(RNAi) affected animals; small white arrowheads, GFP-expressing nuclei in untreated kuEx119+ adults. Note the similar appearance of kuEx119− animals to kuEx119+ animals affected by RNAi. Also note the absence of large GFP-expressing intestinal cells in the kuEx119+ RNAi-affected animals. Size bar, 100 μm.
MH1461 animals were mutagenized and an F2 clonal screen was carried out (also see Materials and Methods). Strains acquiring desirable mutations would be predicted to show clear-cut differences in the phenotypes of kuEx119+ and kuEx119− progeny in the F3 generation (Fig. 1). Through screening of ∼3500 haploid genomes, we isolated seven mutations defining seven distinct loci. Animals homozygous for mutations in lin-35 and the individual slr (synthetic with lin-35/Rb) genes show a range of phenotypes including reduced size, slow growth rates, larval lethality, and sterility (D.S. Fay and M. Han, unpubl.). As anticipated, the screen also uncovered a number of class A SynMuv mutations that were not further pursued. We describe here the phenotypic analysis and molecular identification of one lin-35 synthetic mutation, slr-1(ku298). Based on the molecular identity of the gene defined by this mutation (see below), we refer to this gene as fzr-1.
Synthetic interaction between lin-35 and fzr-1
In screening for lin-35 synthetic mutants, we isolated candidate strain MH1621. MH1621 animals that carry kuEx119 are healthy, fertile, and generally indistinguishable from wild-type animals. In contrast, kuEx119− animals derived from this strain are nonviable and show multiple defects (Fig. 2C–F; see below). Genetic analyses indicated that the mutant phenotype observed in kuEx119− progeny resulted from the combined effect of mutations in lin-35 and a second unlinked gene, fzr-1.
As a further test for specificity, we inactivated lin-35 using RNAi methods in strain MH1621. As expected, lin-35(RNAi) led to the highly penetrant expression of the kuEx119− phenotype in kuEx119+ animals (Fig. 2G,H; see below), as a result of eliminating LIN-35 activity derived from the array. Based on these criteria, we conclude that the phenotype observed in MH1621 kuEx119− progeny (hereafter referred to as lin-35; fzr-1 double mutants) is the direct result of a synthetic interaction between lin-35 and fzr-1.
fzr-1 single mutants isolated from strain MH1621 are viable and show no obvious anatomical or behavioral defects (see below). We did observe a low level of sterility in fzr-1 hermaphrodites (3%, n = 104) as well as an ∼50% reduction in brood size compared with wild type (142 ± 37, n = 8, vs. 264 ± 18, n = 50). In addition, fzr-1 hermaphrodites produce males at an elevated frequency compared with wild type (1.74%, n = 1205, vs. ∼0.2%; Sulston and Hodgkin 1988) and show an increase in the occurrence of embryonic lethality (11.9%, n = 1241, vs. <1%, n > 1000). An examination of lin-35 single mutants revealed similar low levels of sterility (4%, n = 100) and embryonic lethality (6%, n = 382), as well as an ∼65% reduction in brood size (98 ± 43, n = 10).
Whereas only 12% (n = 362) of kuEx119+ animals derived from MH1621 fail to complete embryogenesis, 42% (n = 98) of sibling lin-35; fzr-1 embryos fail to hatch, often arresting close to the end of elongation phase. For double mutants that successfully completed embryogenesis, the duration of embryonic development was similar to that observed for wild-type animals (∼12–16 h). In contrast, the length of postembryonic development was greatly extended in lin-35; fzr-1 animals, requiring 72 to >96 h at 20°C (n = 30) to complete larval stages versus 44 to 52 h (n = 200) for wild-type animals (Fig. 2C,D; data not shown). Adult lin-35; fzr-1 animals show an approximate threefold reduction in total body volume compared with wild-type animals (Fig. 2E,F). In addition, some adult lin-35; fzr-1 hermaphrodites produce small numbers of nonviable embryos, which remain within the uterus and show pleiotropic defects (Fig. 2E; data not shown; also see below).
lin-35; fzr-1 double mutants show hyperproliferation in multiple tissue types
An examination of lin-35; fzr-1 double-mutant adults revealed extensive tissue hyperproliferation affecting subsets of cells derived from all the major germ layers (Sulston and Horvitz 1977). The supernumerary divisions appear to take place exclusively during postembryonic development as newly hatched lin-35; fzr-1 L1 larvae show no evidence of excess cell divisions and are phenotypically indistinguishable from wild type (data not shown).
We measured the extent of hyperproliferation in three lineages derived from ectoderm in lin-35 and fzr-1 double mutants. Using a seam-cell-specific GFP marker (gift of J. Rothman, University of California, Santa Barbara), we observed up to a twofold increase in the number of hypodermal seam cells in lin-35; fzr-1 double mutants and in fzr-1 single mutants wherein lin-35 was inactivated by RNAi (Table 1). Interestingly, normal numbers of seam cells were observed in lin-35; fzr-1 double mutants derived from lin-35 heterozygous hermaphrodites, indicating that lin-35 maternal contributions are capable of suppressing hyperproliferation in at least some lineages (Table 1). Seam-cell proliferation was also normal in lin-35 and fzr-1 single mutants (Table 1).
Table 1.
Hyperproliferation of lin-35; fzr-1 double mutants
|
|
Average cell no.
|
Range
|
n
|
|---|---|---|---|
| Hypodermal seam-cell nucleia | |||
| Wild type | 15.9 ± 1.2 | 14–19 | 25 |
| lin-35 | 16.3 ± 0.9 | 15–18 | 8 |
| fzr-1 | 16.5 ± 1.1 | 15–19 | 25 |
| fzr-1; lin-35(RNAi) | 21.2 ± 3.5 | 16–30 | 30 |
| lin-35m+; fzr-1m+ | 17.1 ± 1.1 | 16–19 | 8 |
| lin-35m+; fzr-1 | 17.3 ± 1.5 | 16–21 | 17 |
| lin-35; fzr-1m+ | 23.2 ± 2.5 | 18–26 | 9 |
| Vulval cell nuclei | |||
| Wild type | 22.0 ± 0.0 | >100 | |
| lin-35 | 22.0 ± 0.0 | 20 | |
| fzr-1 | 22.0 ± 0.0 | 20 | |
| lin-35; fzr-1 | 30.6 ± 5.8 | 22–43 | 13 |
| unc-47::GFP-expressing GABA neuronsb | |||
| Wild type | 18.4 ± 0.8 | 17–19 | 12 |
| lin-35m+; fzr-1m+ | 18.9 ± 0.8 | 18–20 | 12 |
| lin-35m+; fzr-1 | 18.4 ± 0.7 | 17–19 | 7 |
| lin-35; fzr-1m+ | 18.1 ± 0.9 | 17–19 | 10 |
| fzr-1; lin-35(RNAi) | 18.5 ± 0.7 | 17–19 | 20 |
| Intestinal cell nucleic | |||
| Wild type | 26.3 ± 0.4 | 20–32 | 43 |
| fzr-1 | 35.8 ± 1.5 | 23–48 | 21 |
| lin-35(RNAi) | 29.0 ± 0.7 | 20–39 | 43 |
| fzr-1; lin-35(RNAi)d | 52.2 ± 7.1 | 44–67 | 20 |
Cell numbers were scored for wild-type and mutant animals either directly (vulval cells) or using cell-type-specific integrated GFP markers.
Hypodermal seam cells were scored using a seam-cell-specific GFP marker strain, JR632. m+ indicates the presence of maternal gene product.
unc-47::GFP-expressing GABA neurons were assayed using a GFP marker strain, oxln12. Only those cells located between the rectum and pharyngeal grinder were scored.
Intestinal cells were scored using an elt-2::GFP marker strain.
Owing to weak and variable GFP staining in fzr-1; lin-35(RNAi) animals, numbers obtained are likely to be underestimates of the total number of intestinal cells (also see text).
Hyperproliferation was also observed in vulval tissue (Table 1; Fig. 3B) as well as in cells that make up the somatic gonad (data not shown). An analysis of vulval development revealed complex and variable defects in both induction and cell division patterns. This was owing in part to the presence of extra vulval precursor cells (VPCs) that were present in L2/L3 animals and were competent to respond to the induction signal. Induction of these supernumerary VPCs appeared to account for most or all of the extra vulval cells observed. VPC induction in lin-35; fzr-1 mutants was dependent on lin-3 signal: 13/38 fzr-1; lin-3(e1417); lin-35(RNAi) animals failed to produce any vulval tissue, similar to results obtained for lin-3(e1417) mutants alone (10/21). Based on cell position assignments, the induced VPCs appeared to be derived exclusively from progenitor Pn.p cells that normally contribute to the vulva (P5.p, P6.p, and P7.p).
Figure 3.

Hyperproliferation of lin-35; fzr-1 double mutants. (A,C,G,I) lin-35; fzr-1; kuEx119 and (B,D,H,J) lin-35; fzr-1 animals. (A,B) L4-stage vulvae. The vulva in A contains 22 nuclei and is indistinguishable from wild type, whereas the vulva in B contains 32 nuclei and shows an abnormal morphology. (C,D) Morphology of the gonad arm in L4/young-adult animals. The gonad arm in C appears completely wild type whereas that in D contains a single bifurcation, the result of an extra division of the distal tip cell. (E,F) Distal tip cells (arrows) in fzr-1; lin-35(RNAi) animals expressing lag-2::GFP. The DTCs in E are in the process of dividing whereas F shows cells several hours after divisions have taken place. The images in E and F are of different animals. (G,H) DAPI staining of intestinal (arrowheads) and distal germ (arrows) cells. Contrast the large, brightly staining intestinal cells in G to the smaller, fainter intestinal cells in H. A relative indication of intestinal-cell DNA content can be inferred from comparing the intensity of intestinal-cell staining to that of the germ cells in each panel. (I,J) Proximal gonad and vulval region in adults. White arrowheads indicate the location of vulval eversions. Note the presence of multiple eversions in J. In addition, the animal in J shows a hyperproliferation defect of the proximal germ line (pg), whereas the animal in G contains wild-type embryos (em) in this region. The distal germ line (dg) appears normal in both I and J. Size bars, 10 μm.
Following induction, VPCs underwent at least two to three rounds of divisions that were markedly slower and asynchronous compared with wild type. Whereas induced VPCs normally divide every 2 h, we often observed division times of >4 h and failed to observe accelerated division cycles. In addition, the pattern of vulval cell fates based on division plane orientations and cell- fate-specific markers (lin-11::GFP) was highly variable (data not shown). These defects led to aberrant vulval morphologies; in some of these cases vulval cells failed to integrate into a single structure, producing multiple closely spaced invaginations reminiscent of a Muv phenotype (Fig. 3G,H).
In contrast to seam-cell and vulval lineages, ventral cord GABA neurons expressing unc-47::GFP (gift of E. Jorgensen, University of Utah, Salt Lake City) appeared to be completely unaffected in lin-35; fzr-1 double mutants (Table 1). This absence of a phenotype may be due to the lack of a requirement for lin-35 and fzr-1 in controlling proliferation in these cells, or may result from suppression by maternal products of fzr-1.
Cells derived from the mesoderm were also affected, including distal-tip cells (DTCs), which guide migration of gonad arms during postembryonic development. lin-35; fzr-1 double mutants showed a high frequency of extra DTCs, leading to a Shiva (Shv) phenotype, in which an abnormal bifurcation produces gonads with extra arms (Fig. 3C,D; Table 2). In general, only one bifurcation per gonad arm was observed, indicating that most DTCs were undergoing only a single extra division cycle. Consistent with this, the average number of DTCs detected using the DTC-expressing lag-2::GFP marker (Blelloch et al. 1999) was 1.67 ± 0.68 (range, 1–4; n = 60) per gonad arm, where only 5/60 animals contained >2 DTCs per arm. An analysis of DTC divisions in fzr-1; lag-2::GFP; lin-35(RNAi) animals indicated that most or all extra DTCs are generated through division of the DTC itself, and not from other cell types in the somatic gonad. This result was supported by the direct observation of lag-2::GFP-expressing DTCs in the process of division (Fig. 3E,F), and by experiments where DTCs were laser-ablated in L2-stage animals: 6/7 animals failed to produce additional DTCs based on gonad morphology and lag-2::GFP expression. This result contrasts with findings in cki-1(RNAi) animals where supernumerary DTCs were shown to originate from other cells within the somatic gonad (Hong et al. 1998). We cannot, however, rule out the possibility that a small percentage of DTCs in lin-35; fzr-1 animals may be derived from other sources.
Table 2.
fzr-1 genetic interactions with SynMuv genes
|
|
% Shv
|
n
|
|---|---|---|
| lin-35(n745) | 1 | 76 |
| fzr-1(ku298) | 0 | 40 |
| lin-35; fzr-1; kuEx119 | 2 | 97 |
| lin-35; fzr-1 | 58 | 45 |
| lin-35; fzr-1; kuEx119; lin-35(RNAi)a | 68 | 60 |
| lin-35; fzr-1; kuEx119; lin-36(RNAi)a | 69 | 36 |
| lin-35; fzr-1; kuEx119; efl-1(RNAi)a | 46 | 26 |
| fzr-1; lin-53(n833) | 0 | 25 |
| lin-35; fzr-1; kuEx119; lin-53(RNAi)b | 4 | 28 |
| lin-35; fzr-1; kuEx119; hda-1(RNAi)b | 0 | 25 |
| lin-35; fzr-1; kuEx119; chd-4(RNAi)b | 0 | 43 |
| fzr-1; lin-15a(n767) | 0 | 22 |
| lin-35; fzr-1; kuEx119; lin-15a(RNAi) | 7 | 81 |
| lin-35; kuEx119; fzr-1(RNAi) | 2 | 61 |
| lin-35; fzr-1(RNAi) | 37 | 49 |
Adult hermaphrodites of the indicated genotypes were placed on RNAi feeding plates, and adult F1 progeny were scored for the presence of the Shiva (Shv) phenotype. kuEx119 is the extrachromosomal array containing the lin-35+ gene.
Similar percentages of Shv animals were also observed following RNAi treatment of fzr-1 single mutants.
RNAi treatment led to a highly penetrant sterile phenotype among F1 progeny.
Endoderm was also strongly affected in lin-35; fzr-1 double mutants. Wild-type adults normally contain ∼26 gut cell nuclei that are polyploid (32N) as a consequence of four endoreduplication cycles (DNA replication without intervening mitosis) during larval development (Fig. 3G; Hedgecock and White 1985). In lin-35; fzr-1 double mutants, gut cell ploidy was highly variable, ranging from 2N to 16N (n = 20), with the majority of cells falling in the range of 4N to 8N (Fig. 3H; data not shown). In addition, we observed lin-35; fzr-1 DAPI-stained animals that contained up to four times the normal number of intestinal cell nuclei (Fig. 3H). Consistent with this, fzr-1; lin-35(RNAi) animals expressing the gut-cell-specific marker elt-2::GFP showed excess divisions (Table 1). The large number and reduced ploidy of mutant gut cells suggest that rather than executing endocycles during larval development, intestinal cells in the double mutants are undergoing cycles of DNA replication followed by mitosis. Interestingly, both lin-35 and fzr-1 single mutants showed a modest increase in the number of gut cell nuclei compared with wild type (Table 1).
Excess divisions were also observed in cells that make up the germ line. Severe effects were confined to proximal regions of the gonad, where rampant hyperproliferation led in some cases to a tumorous phenotype (Fig. 3I,J; Francis et al. 1995). The specific involvement of germ cells was evidenced by the lack of any detectable GFP expression in affected regions of the gonad in MH1621 animals in which lin-35 was inactivated with RNAi (data not shown). This failure of the tumorous cells to express GFP reflects the ability of germ-line cells to efficiently silence transgene expression, a characteristic not associated with the somatic gonad, in which we observed consistent GFP expression. Distal gonad regions were less affected and showed normal-appearing DAPI-stained mitotic, transition, and pachytene zones (data not shown). These results suggest that germ cells in lin-35; fzr-1 mutants abnormally reenter mitosis after failing to progress past meiotic prophase. These defects could reflect a direct requirement for lin-35 in germ cells, or may be caused by nonautonomous effects, such as signaling via the lag-2/glp-1 pathway from the somatic gonad.
fzr-1 shows synthetic phenotypes with a subset of class B SynMuv genes
As described in the introduction, a subset of class B SynMuv genes (including lin-35) have been linked to transcriptional regulation. However, it is currently unclear to what extent these functionally related genes are interchangeable with respect to their genetic interactions. To address this issue, we examined interactions between fzr-1 and six SynMuv genes. The class B genes efl-1, lin-53, hda-1, and chd-4 encode homologs of E2F (Ceol and Horvitz 2001), RbAp48, histone deacetylase (Lu and Horvitz 1998), and the NuRD complex component, Mi-2, respectively (Solari and Ahringer 2000; von Zelewsky et al. 2000). lin-15a and lin-36 (class A and B genes, respectively) encode novel proteins (Clark et al. 1994; Huang et al. 1994; Thomas and Horvitz 1999).
Similar to the effects observed for lin-35(RNAi) (Fig. 2G,H; Tables 1, 2), RNAi inactivation of either lin-36 or efl-1 in strain MH1621 led to the appearance of kuEx119+ animals that were phenotypically indistinguishable from lin-35; fzr-1 double mutants (Table 2; data not shown). In contrast, RNAi inactivation of lin-53, hda-1, chd-4, and lin-15a failed to induce hyperproliferation in kuEx119+ animals (Table 2; data not shown). In addition, we constructed double mutants between fzr-1 and lin-53 and lin-15a. In both cases, double-mutant animals were indistinguishable from wild type with respect to viability and cellular proliferation properties (Table 2; data not shown). We conclude that fzr-1 shows interactions with a subset of class B genes that may function more specifically to control cell cycle regulation.
fzr-1 encodes a C. elegans homolog of Cdh1/Hct1/FZR
Using several novel approaches to overcome a number of technical challenges (see Materials and Methods), we mapped ku298 to a small region on LGII between let-23 and unc-4. We attained rescue of the lin-35; fzr-1 double-mutant phenotype using microinjection with a single cosmid from the region ZK1307. Partial but clear rescue was subsequently obtained with a subclone of ZK1307 that contained only a single predicted ORF, ZK1307.6 (see Materials and Methods). A search of the database indicated that ZK1307.6 encodes the C. elegans homolog of Cdh1/Hct1/FZR.
To further support the claim that ku298 represents an allele of fzr-1, we sequenced fzr-1 genomic DNA from ku298 mutant animals. We detected a single missense mutation within the predicted coding region of fzr-1. The mutation (G → A) substitutes a highly conserved cysteine with tyrosine in the fourth WD-repeat domain (Fig. 4A). As an additional test, strain MH1461 was grown on fzr-1(RNAi) feeding plates. Although kuEx119+ animals appeared largely unaffected by this treatment, the majority of kuEx119− animals showed hyperproliferation defects similar to those of lin-35; fzr-1 double mutants (Fig. 5A; Table 2; data not shown).
Figure 4.


(A) Peptide alignment of C. elegans FZR-1, Drosophila melanogaster Fzr, and Homo sapiens CDH1. Black boxes signify identity; gray, similarity. The boundaries of the seven WD-repeat regions are indicated by arrows and corresponding numbers. The location of the ku298 lesion is indicated by an asterisk. (B) Unrooted phylogenetic tree diagram showing relationships between FZR (non-underlined) and FZY (underlined) homologs from C. elegans, D. melanogaster, Xenopus laevis, Mus musculus, H. sapiens, Saccharomyces cerevisiae, and Schizosaccharomyces pombe. Numbers indicate bootstrap values.
Figure 5.

fzr-1(RNAi) and fzr-1 expression. (A) Example of the Shv phenotype observed in lin-35 animals grown on fzr-1(RNAi) feeding plates. (B) Representative gonad from progeny of N2 animals injected with fzr-1 dsRNA (RNAi; 1.5 μg/μL). Note the small malformed gonad arm extension (arrow) protruding from the proximal gonad region (pg). (C,D) Terminal embryos derived from lin-35 animals injected with fzr-1 dsRNA. Arrows indicate the locations of vacuoles. Note the variability in embryonic (C,D) arrest point and (D) size. (E,F) Corresponding DIC and GFP fluorescence images of an fzr-1::GFP reporter construct expression in wild-type embryos. Size bars, 10 μm.
Using a cDNA clone (gift of Y. Kohara, National Institute of Genetics, Mishima, Japan), we confirmed the predicted gene structure of fzr-1 and identified an SL1 transplice leader sequence (Krause and Hirsh 1987) at the 5′ end of the clone, indicating that it is full length. The highly conserved C-terminal domain of FZR-1 is 68% identical to both Drosophila FZR and human CDH1 (Fig. 4A). The sizable N-terminal domain of FZR-1 contains scattered regions of homology to the fly and human gene products as well as novel sequences (Fig. 4A). A phylogenetic analysis indicates that FZR-1 is more similar to members of the Cdh1/Hct1/FZR family than to the closely related but functionally distinct gene family composed of Cdc20/FZY (Fig. 4B).
FZR-1 function is required for fertility
The nature of the genetic lesion suggested that ku298 might not represent a complete LOF allele of fzr-1. Because dsRNA injection techniques may produce RNAi phenotypes that more closely mimic that of the molecular null than do those produced by feeding methods (M. Han, unpubl.), we inactivated FZR-1 function in wild-type animals using RNAi injection methods. Injection led to a severe reduction in the fertility of the derived F1 progeny: 47% of F1 animals were completely sterile, whereas an additional 39% had brood sizes of <10 (n = 210). An analysis of affected animals revealed pleiotropic defects within the germ-line and somatic gonad. These included a failure of gonad arms to elongate, missing or abnormal oocytes and sperm, and morphogenetic defects of the uterus and vulva (Fig. 5B; data not shown). From these results, we conclude that FZR-1 function is required for viability and that ku298 encodes a hypomorphic allele of fzr-1.
We also examined the effect of inactivating fzr-1 using RNAi injection methods in a lin-35 mutant background. Interestingly, this led to embryonic lethality in 88% of F1 progeny, with the remaining 12% arresting during early larval stages (n = 269). Nonviable embryos showed a variety of phenotypes ranging from premorphogenetic to late-stage arrest (Fig. 5C,D). In the majority of cases, arrested embryos showed clear signs of tissue-specific differentiation but failed to complete morphogenesis and contained vacuoles and necrotic regions (Fig. 5C,D). In addition, a significant percentage of embryos (∼30%) were misshapen or of abnormal size (Fig. 5D). We conclude that, in addition to controlling cell proliferation during postembryonic development, lin-35 and fzr-1 play essential, albeit redundant, roles during embryonic development.
Consistent with these findings, an fzr-1::GFP transcriptional reporter showed robust expression in most or all cells during embryonic development in wild-type animals (Fig. 5E,F). Expression was also observed at lower levels in many cell types during larval development, including those showing proliferation defects in lin-35; fzr-1 double mutants (data not shown).
lin-35 shows a synthetic interaction with the putative SCF regulator lin-23
lin-23 encodes a WD-repeat protein with homology to regulators of the SCF proteosome complex (for review, see Deshaies 1999; Kipreos et al. 2000). Loss of LIN-23 function leads to a widespread hyperproliferation phenotype in mutant animals, presumably resulting from a failure to degrade G1 cyclins. We had previously identified an allele of lin-23 in an earlier screen for lin-35 synthetic mutants (D.S.F. and M.H., unpubl.). lin-23(ku294) contains a mutation in the conserved 5′ splice site (GT → AT) between exons 4 and 5 of lin-23, resulting in a peptide that is predicted to terminate following amino acid 245 of the 665-amino-acid peptide. Based on the size of the truncation and the predicted instability of the resulting mRNA, ku294 is likely to represent a strong or complete LOF allele of lin-23.
In contrast to lin-23 and lin-35 single mutants, lin-35; lin-23(ku294) double mutants generally fail to progress past the L3 larval stage and are severely growth retarded relative to the single mutants (Fig. 6A; data not shown). Because of the extremely poor viability of lin-35; lin-23 double mutants, and the fact that lin-23 single mutants themselves show a strong hyperproliferation phenotype on their own, it was not possible for us to discern whether the absence of LIN-35 function exacerbated the lin-23 hyperproliferation defect in the double mutants.
Figure 6.
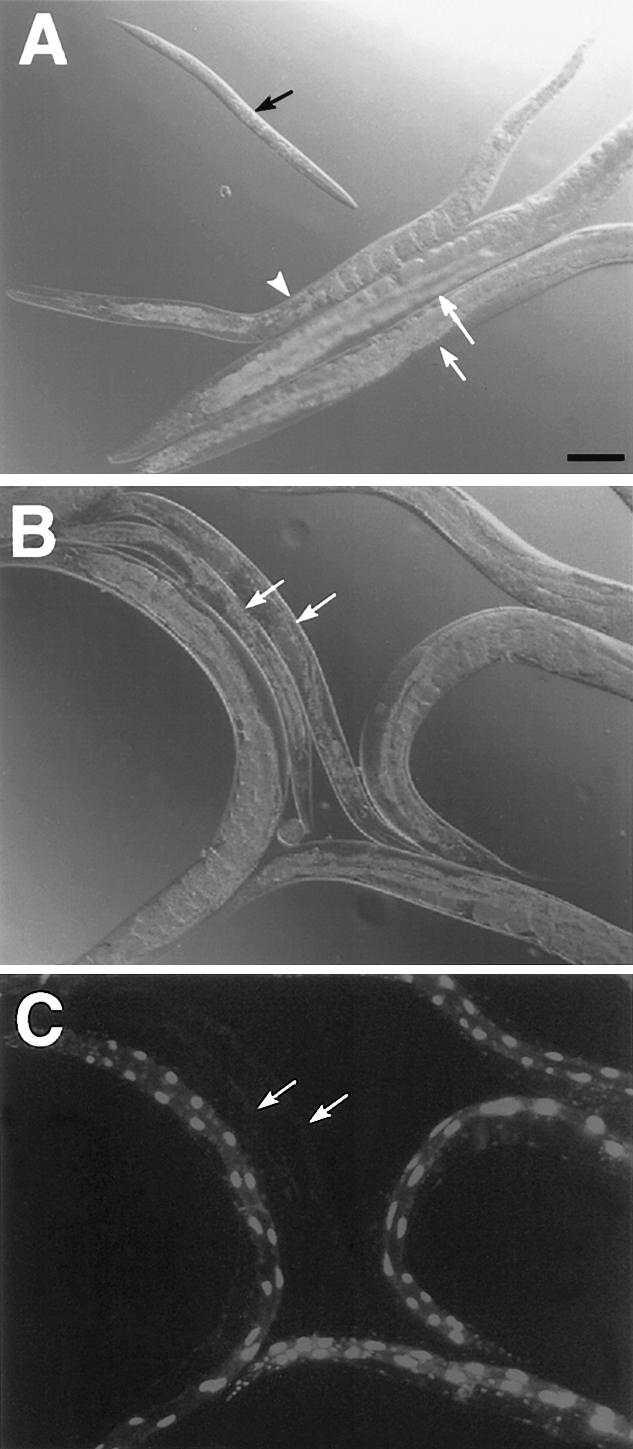
lin-35; lin-23 synthetic interactions. (A) lin-35 single mutant (white arrows), lin-23(ku294) single mutant (white arrowhead), and lin-35; lin-23 double mutants (black arrow). The lin-35; lin-23 animal shown is >96 h posthatching and shows the typical midlarval-stage terminal arrest phenotype. (B,C) Corresponding DIC and GFP fluorescence images of representative adult MH1461 animals propagated on lin-23(RNAi) feeding plates. White arrows indicate the positions of Ex119− animals. Note the preferential effect of lin-23(RNAi) on the kuEx119− animals. Size bar, 100 μm.
As an alternative approach, we reduced lin-23 function in MH1461 animals using RNAi feeding methods and looked for differences in the sensitivity of lin-35; kuEx119+ and lin-35; kuEx119− sibling progeny. Interestingly, whereas 82% (n = 177) of kuEx119− animals exposed to lin-23(RNAi) showed the characteristic lin-23 hyperproliferation phenotype, only 43% (n = 236) of sibling kuEx119+ animals were similarly affected (Fig. 6B,C). Thus the presence of functional LIN-35 significantly decreased sensitivity to diminished LIN-23 activity. This effect was not due to a generic hypersensitivity of lin-35 mutant animals to RNAi as both kuEx119+ and kuEx119− animals were equally sensitive to unc-22(RNAi) feeding (data not shown).
LIN-35 and FZR-1 likely coregulate G1 cyclin levels
Based on work from other systems (see Discussion), loss of LIN-35 function would be predicted to disrupt the activity of Rb/E2F transcriptional inhibitory complexes, leading to elevated levels of cyclin E and cyclin A mRNA. In turn, loss of either FZR-1 or LIN-23 would result in decreased APC or SCF functions, leading to an increase in the stability of cyclin A and cyclin E proteins, respectively. A simple model would then predict that in the lin-35; fzr-1 double mutant, the combination of both increased cyclin transcription and stability leads to cyclin protein levels that are sufficient to override the normal G1 arrest.
To test this model, we engineered strains containing temperature-inducible cyclin A or cyclin E constructs expressed from extrachromosomal arrays. As shown in Table 3, in an otherwise wild-type background, either cyclin A or cyclin E overexpression can lead to the production of extra distal tip cells in a small percentage of animals. Notably, this effect is strongly enhanced in an fzr-1 mutant background. The synthetic interaction observed between cyclin overexpression and fzr-1 mutants is consistent with the model that LIN-35 and FZR-1 cooperate to control the levels of G1 cyclins through transcriptional repression and protein destabilization, respectively.
Table 3.
Heat shock of cyclin A and cyclin E
|
|
% Shv
|
n
|
|---|---|---|
| Cyclin A overexpression | ||
| fzr-1; (fdEx1) | 16 | 154 |
| fzr-1 | 0 | 91 |
| N2; (fdEx1) | 5 | 78 |
| N2 | 0 | 70 |
| fzr-1; (fdEx2) | 32 | 122 |
| fzr-1 | 1 | 69 |
| N2; fdEx2 | 5 | 86 |
| N2 | 0 | 63 |
| Cyclin E overexpression | ||
| fzr-1; (fdEx6) | 18 | 122 |
| fzr-1 | 0 | 115 |
| N2; (fdEx6) | 3 | 64 |
| N2 | 0 | 42 |
| fzr-1; (fdEx7) | 5 | 124 |
| fzr-1 | 1 | 127 |
| N2; (fdEx7) | 1 | 62 |
| N2 | 0 | 50 |
Strains harboring cyclin A (fdEx1, fdEx2) or cyclin E (fdEx6, fdEx7) heat-shock expression constructs contained on extrachromosomal arrays were generated as described in Materials and Methods. Mixed-stage L1/L2 larval animals were heat-shocked at 33°C for 4 h and allowed to recover at 20°C for ∼52 h. Sibling Ex+/Ex− young adult animals were then analyzed for the Shiva phenotype. Percentages indicate the averages obtained from two separate experiments.
Discussion
lin-35 and the control of cell proliferation
We report here the cloning and characterization of fzr-1, a gene that cooperates with lin-35 to control cell proliferation. A relatively small number of genes have been described that cause widespread hyperproliferation in C. elegans. These include the putative SCF components cul-1 and lin-23 (Kipreos et al. 1996, 2000), the CIP/KIP family member cki-1 (Hong et al. 1998), and the CBP/p300 homolog cbp-1 (Shi and Mello 1998). In the cases examined thus far, a hyperproliferation phenotype is observed following inactivation of a single gene product. We have shown that loss of cell proliferation control can also result from a synthetic genetic interaction. Although single mutants of lin-35 and fzr-1 showed only subtle or low-penetrance phenotypes, lin-35; fzr-1 double mutants showed extensive tissue hyperproliferation affecting a wide range of cell types (Figs. 3–5; Tables 1, 2). Thus, uncontrolled proliferation in C. elegans can in essence follow the same genetic pattern as multistep carcinogenesis in mammals. Namely, proliferation control is abolished through the sequential loss of genes that function to restrain cell cycle progression.
Given our findings, and the large body of evidence implicating Rb in human cancers (for review, see Nevins 2001), it seems reasonable to suggest that our technical approach may facilitate the study of multistep carcinogenesis using C. elegans. Along these lines, it will be interesting to determine whether the human homolog of fzr-1, hCDH1, can function as a tumor-suppressor gene, and if so, whether it does so in cooperation with Rb.
Inactivation of fzr-1 function using RNAi injection led to sterility and aberrancies in germ cell proliferation (Fig. 5A,B). Although we have not determined the specific cause of this phenotype, previous studies would implicate defects in either the execution of G1 arrest (Irniger and Nasymth 1997; Sigrist and Lehner 1997; Zachariae et al. 1998) or in late-stage mitotic events such as cytokinesis (Schwab et al. 1997; Zur and Brandeis 2001). We also observed embryonic lethality when fzr-1 was inactivated using RNAi injection in a lin-35 mutant background (Fig. 5C,D). The cause for this lethality is presently unknown. These embryos do not show obvious hallmarks associated with either excess cellular proliferation or grossly elevated levels of apoptosis (data not shown). Although additional work will be necessary to determine the nature of this embryonic requirement, a role during embryonic development is consistent with the expression patterns observed for both fzr-1 (Fig. 5E,F) and lin-35 (Lu and Horvitz 1998). The lack of an apparent hyperproliferation phenotype in embryos likely reflects significant differences in the means by which embryonic and postembryonic cell cycles are regulated. For example, cyclin D, an upstream regulator of Rb, has been shown to be required exclusively for the execution of postembryonic division cycles in C. elegans (Park and Krause 1999; Boxem and van den Heuvel 2001).
Differential properties of SynMuv genes
Work carried out over the past several years has produced an explosion in the number of identified SynMuv genes (see Fay and Han 2000). Although certain functional classifications, such as transcriptional repressors, may accurately describe some of the SynMuv genes, others clearly defy straightforward categorization. This fact alone suggests that SynMuv genes most likely do not all act through the same mechanisms or pathways.
We have found that both lin-36 and efl-1(RNAi) can phenocopy the effect of lin-35 LOF in an fzr-1 mutant background (Table 2). However other class B genes, including lin-53, hda-1, and chd-4, did not show genetic interactions with fzr-1, nor did the class A gene lin-15a (Table 2). These experiments are complicated by the fact that lin-53, hda-1, and chd-4 encode for essential genes, and RNAi leads to a highly penetrant sterile or lethal phenotype within several generations. Nevertheless, we saw no evidence for hyperproliferation in either the affected or unaffected classes of RNAi-treated animals. This suggests that neither a weak nor a severe reduction in the function of these genes is capable of producing a synthetic hyperproliferation phenotype with fzr-1. In addition, we detected no evidence for an interaction in lin-53(n833); fzr-1 double-mutant animals. Although n833 results in only a partial loss of LIN-53 function (Lu and Horvitz 1998), this allele does lead to a highly penetrant Muv phenotype in conjunction with class A SynMuv mutations (Ferguson and Horvitz 1989).
These data indicate a functional distinction between those class B genes that may play relatively direct roles in the regulation of the cell-cycle machinery (lin-35, lin-36, and efl-1) and others that may function more generally in the regulation of chromatin modification and transcriptional control (lin-53, hda-1, and chd-4). Moreover, our data correlate well with recent studies on vulval-cell fate specification, in which clear functional differences were observed between similar subsets of class B genes (Chen and Han 2001).
lin-35, fzr-1, lin-23, and cyclin regulation
Rb and its family members p107 and p130 have been shown in multiple systems to modulate transcription through direct interactions with a variety of transcriptional regulators (for review, see Kaelin 1999; Zheng and Lee 2001). The majority of work indicates that Rb serves primarily as a transcriptional repressor, acting through a number of mechanisms including the recruitment of chromatin-modifying enzymes (Dunaief et al. 1994; Brehm et al. 1998; Luo et al. 1998; Magnaghi-Jaulin et al. 1998; Zhang et al. 2000; Nielsen et al. 2001) and the steric interference of transactivation domains (Flemington et al. 1993; Helin et al. 1993; Weintraub et al. 1995). Acting as transcriptional corepressors with E2F (for review, see Dyson 1998; Harbour and Dean 2000; Classon and Dyson 2001), Rb and its family members regulate the expression of many key genes required for entry and progression through S-phase (Dou et al. 1994) including cyclin E (DeGregori et al. 1995; Duronio and O'Farrell 1995; Ohtani et al. 1995) and cyclin A (Degregori et al. 1995; Schulze et al. 1995). Consistent with these reports, we have observed a significant increase in the levels of ribonucleotide reductase mRNA, an E2F-regulated gene, in lin-35 mutant animals (D.S. Fay and M. Han, unpubl.).
In contrast, Cdh1/Hct1/FZR and its close relative Cdc20/FZY function as regulatory subunits for the anaphase-promoting complex (APC) cyclosome (Dawson et al. 1995; Sigrist et al. 1995; Schwab et al. 1997; Visintin et al. 1997; for review, see Morgan 1999; Zachariae and Nasmyth 1999). This multisubunit peptide complex is thought to function as a ubiquitin ligase, and targets the degradation of several key factors during mitosis, including yeast Pds1 (Cohen-Fix et al. 1996), human securin/PTTG (Zur and Brandeis 2001), and cyclins A and B in higher eukaryotes (Dawson et al. 1995; Irniger et al. 1995; King et al. 1995; Sigrist et al. 1995; Sudakin et al. 1995). APC function requires activation first by Cdc20/FZY during metaphase/anaphase, and then by Cdh1/Hct/FZR during late mitosis and G1. It is believed that Cdh1 and Cdc20 differentially target distinct but overlapping subsets of proteins for degradation by the APC cyclosome (see above references; for review, see Morgan 1999; Zachariae and Nasmyth 1999).
In Drosophila, loss of fzr function leads to reentry into the cell cycle following embryonic cycle 16, thereby bypassing the normal G1 arrest (Sigrist and Lehner 1997). This ectopic division cycle is correlated with excess levels of cyclin A (Sigrist and Lehner 1997), which when overexpressed during G1 can lead to ectopic entry into S-phase (Sprenger et al. 1997). Interestingly, mutations in the Drosophila Rb homolog rbf (Du and Dyson 1999), as well as in the CDK inhibitor decapo (de Nooij et al. 1996), show cell cycle defects similar to those of fzr mutants, suggesting complementary roles in G1/S-phase regulation. However, we note that conclusions regarding fzr functions were inferred from the analysis of a large deletion that removed several genes in addition to fzr (Sigrist and Lehner 1997). Therefore, fzr-1(ku298) is the first reported mutation in metazoans that specifically reduces CDH1/HCT1/fzr activity.
Our analysis of DTC hyperproduction in strains that overexpress either cyclin A or cyclin E mRNA (Table 3) supports the model that lin-35 and fzr-1 are likely to coregulate cyclin levels during G1. In addition, our finding that the E2F homolog efl-1 synergizes with fzr-1 (Table 2) adds further credence to this model. The ability of both cyclin A and cyclin E to induce extra DTCs in fzr-1 mutants could indicate that these cyclins may be functionally interchangeable and that sufficient levels of either cyclin A or cyclin E, or possibly both in combination, can work to override G1 arrest.
Multiple mutations show synthetic lethality with lin-35
By screening ∼3500 haploid genomes, we uncovered seven Slr mutations that show synthetic lethality or inviability with mutations in lin-35. The phenotypes observed in the double mutants range from embryonic and early larval arrest to adult sterility and size defects (D.S.Fay and M. Han, unpubl.). About half of the Slr mutations that we have identified show only little or no phenotype as single mutants, whereas others show moderate effects on brood size and/or growth rates. In addition, we note that the penetrance and severity of the double-mutant phenotypes appear in several cases to be substantially reduced by the presence of low levels of maternal lin-35 expressed from the array, consistent with previous reports that maternal lin-35 can also suppress expression of the Muv phenotype (Ferguson and Horvitz 1989).
Other than fzr-1 and lin-23, we have not yet identified any Slr mutations that produce an obvious synthetic hyperproliferation phenotype with lin-35. However, as inferred from our comparison of lin-35(n745); fzr-1(ku298) and lin-35(n745); fzr-1(RNAi) phenotypes, this may depend to a significant extent on the specific nature of the allele isolated. Therefore, whether or not additional Slr genes will turn out to function in cell cycle control mechanisms or will cooperate with lin-35 in activities not directly related to the cell cycle remains to be seen.
A screen of general utility
We believe the means used to uncover the genetic interaction between lin-35 and fzr-1 will be of general use for those wishing to assign functions to genes lacking known biological roles or to identify novel functions for genes with previously characterized activities. In addition, this genetic approach serves to identify functional copartners through the isolation and cloning of the affected second-site mutations. Importantly, this method in no way depends on prior knowledge of the synthetic double-mutant phenotype, thereby permitting a nonbiased search for genetic modifiers of any gene of interest.
Given the inevitable saturation of the genome for mutations that cause easily detectable phenotypes, the ability to identify synthetic mutations will become increasingly important. Large-scale analyses carried out in yeast and C. elegans suggest that a large percentage of genes in higher organisms may fail to show easily discernable phenotypes when mutated (Smith et al. 1996; Winzeler et al. 1999; Fraser et al. 2000). It is likely that the vast majority of these no-phenotype genes may nevertheless confer a weak selective advantage to the organism, thus accounting for their presence in the genome (Nowak et al. 1997; for review, see Tautz 2000). At the same time, it can be argued that many of these genes fail to show mutational effects owing to genetic redundancy. Importantly, these two explanations are in no way mutually exclusive. By devising methods to experimentally address this latter issue, we may hope to assign biological roles to many genes that would normally not be amenable to straightforward functional analyses.
Materials and methods
Culture methods
Maintenance, culturing, and genetic manipulations of C. elegans strains were carried out according to standard procedures (Sulston and Hodgkin 1988) and conducted at 20°C.
Strains used/generated
The strains used or generated were as follows: MT1624 [lin-35(n745)I; lin-8(n111)II], MH1461 [lin-35(n745)I; kuEx119], MH1621 [lin-35(n745)I; fzr-1(ku298)II; kuEx119], MH1829 [fzr-1(ku298), unc-4(e120)II], MH1832 [dpy-10(e128), fzr-1(ku298), unc-4(e120)II], CB4856, MH1791 [fzr-1(ku298)II], MH1640 [lin-35(n745)I; dpy-10(e128), unc-4(e120)II], MH1630 [lin-35(n745)I; bli-2(e768), rol-6(e187)II], MH1758 [lin-35(n745)I; let-23(sy15), unc-4(e120)/dpy-10(e128), unc-4(e120)II], JK2533 [qC1(lag-2::GFP qIs26)/eT1 (Blelloch et al. 1999)], GR1314 [mgIs21 (lin-11::GFP (Hobert et al. 1998)], JR632 [seam cell::GFP (Terns et al. 1997)], oxIn12 [unc-47::GFP (McIntire et al. 1997), and JM63 [elt-2::GFP (Fukushige et al. 1998)].
DNA constructs
RNAi
Target sequences for insertion into pPD129.36 (gift of A. Fire) were generated by PCR amplification from appropriate cDNA or genomic clones. cDNA clones of lin-35, efl-1, lin-36, hda-1, and lin-53 were obtained from Y. Kohara, and a cDNA clone of lin-15a was obtained from P. Sternberg (California Institute of Technology, Pasadena). Inserts into pPD129.36 were of the following sizes: lin-35, 2.7-kb cDNA spanning exons 3–10. fzr-1, 750-bp genomic spanning exons 1–2. lin-23, 1.1-kb genomic spanning exons 2–5. lin-15a, 1.1-kb cDNA spanning exons 2–6. chd-4, 1.4-kb genomic spanning exons 1–2. efl-1, 370-bp cDNA spanning exons 1–3. lin-36, 1.0-kb cDNA spanning exons 3–5. hda-1, 1.4-kb cDNA spanning exons 1–4. lin-53, 1.3-kb insert spanning exons 1–3.
fzr-1 subclones
pDF63 contains a 7960-bp XbaI/SalI fragment from cosmid ZK1307 subcloned into pBluescript. pDF65 contains a 3250-bp PCR-amplified fragment from the 5′ UTR of fzr-1 cloned into plasmid pPD95.69 (gift of A. Fire). The construct creates an in-frame fusion of GFP containing an NLS to the first 6 amino acids of fzr-1.
Cyclin heat-shock constructs
Full-length cyclin A cDNA from clone yk610 (from Y. Kohara) was amplified and inserted into pPD49.78 and pPD49.83 heat-shock vectors (from A. Fire). Cyclin E-heat shock vectors were a gift of M. Krause (NIH, Bethesda, MD).
Screen methodology
lin-35(n745); lin-8(n111) (MT1624) animals were injected with a mixture of cosmid C32F10 (100 μg/μL; Gu et al. 1998) and sur-5::GFP (100 μg/μL) to generate multiple independent strains carrying stable extrachromosomal arrays. Array kuEx119 suppressed expression of the Muv phenotype in >90% of lin-35; lin-8 double mutants and showed an ∼75% transmission frequency. After crossing out the lin-8 mutation to generate strain MH1461, animals were mutagenized with ethylmethanesulfonate (EMS) using standard procedures (Sulston and Hodgkin 1988). An F2 clonal screen was carried out in which four F2 animals were cloned per F1 (for additional details, see Results), resulting in an expected coverage of ∼1.35 haploid genomes per F1 animal. Based on this estimate, we have screened ∼3500 haploid genomes, or ∼2500 F1 (10,000 F2) clones, to obtain seven unlinked mutations.
Mapping
Positional mapping of fzr-1 presented a number of technical challenges: (1) both fzr-1(ku298) and lin-35(n745) single mutants lack discernable plate phenotypes; (2) double-mutant animals are nonviable and difficult to distinguish under a dissecting scope; and (3) the severity of the double-mutant phenotype can be at least partially suppressed by maternal products. These difficulties were circumvented by the procedures described below.
Two- and three-point mapping
Triple-mutant mapping strains carrying lin-35(n745) and two linked visible marker mutations (e.g., dpy and unc) were generated for each chromosome. Homozygous lin-35 males were mated to mapping strains, and male cross progeny were in turn mated to strain MH1621 (fzr-1; lin-35; kuEx119). kuEx119+ cross progeny that segregated the marker mutations were then identified and used for either two- or three-point mapping.
For two-point mapping, ∼100 kuEx119+ wild-type sibling animals were cloned to single plates and, based on the segregated progeny, were scored as either homozygous or nonhomozygous for ku298 and plus or minus for the marker mutations. ku298 showed strong linkage to markers on LGII: 31/31 ku298 homozygous animals failed to segregate dpy-10, unc-4 markers, and 25/26 ku298 homozygous animals failed to segregate bli-2, rol-6 markers.
For three-point mapping, kuEx119+ sibling recombinants (e.g., Dpy–non-Unc) were identified and allowed to self-fertilize. Then 10–15 kuEx119+ self-progeny of the recombinant phenotype were cloned, allowed to self-fertilize, and then scored both for the presence of homozygous ku298 and of the recombinant chromosomes. We obtained the following data for three-point mapping of ku298: 3/19 unc-4–non-dpy-10 recombinants acquired ku298; 28/29 bli-2–non-rol-6 recombinants acquired ku298; 0/5 rol-6–non-bli-2 recombinants acquired ku298; 9/38 unc-4–non-let-23 recombinants acquired ku298.
SNP mapping
We constructed the triple mutant strain MH1832 (dpy-10, ku298, unc-4) and verified the strain's ability to phenocopy the lin-35; ku298 double-mutant phenotype using lin-35(RNAi) feeding methods (Fraser et al. 2000). MH1832 hermaphrodites were then mated to CB4856 homozygous males, and hermaphrodite cross progeny were cloned and allowed to self-fertilize. Recombinant animals (e.g., Unc–non-Dpy) were identified, and homozygous recombinant animals were isolated in the next generation and tested for the presence of ku298 using lin-35(RNAi). Predicted SNPs in the region were identified using the C. elegans SNP database and were further verified by experimentation.
We obtained the following mapping data for SNP vr77f08.s1@477 on cosmid T15H9: For Unc–non-Dpy recombinants that retained ku298, 54/54 were N2 for the SNP; 7/7 Unc–non-Dpy recombinants that lost ku298 were CB4856 for the SNP; 5/5 Dpy–non-Unc recombinants that retained ku298 were N2 for the SNP. For SNP vr89b04.s1@165 on cosmid T10B9 the following data were obtained: 2/7 Unc–non-Dpy recombinants that lost ku298 were CB4856 for the SNP; 3/5 Dpy–non-Unc recombinants that retained ku298 were CB4856 for the SNP.
Other methods
Cosmid rescue
Rescue was obtained by coinjection of either ZK1307 or pDF63 with pRF4 into MH1621 using standard procedures (Mello and Fire 1995). Rescued animals were fertile, kuEx119−, and showed a Rol phenotype.
RNAi
RNAi was carried out according to standard methods (Fire et al. 1998; Fraser et al. 2000). DAPI staining was according to Sulston and Hodgkin (1988).
Phylogenetic analysis
Phylogenetic analysis was carried out with PAUP (Swafford 2000) using neighbor joining and maximum parsimony analysis with 500 bootstrap replicates. For each bootstrap, 10 heuristic searches of random addition sequence replicates were carried out.
Cyclin heat-shock strains
Strains containing temperature-inducible cyclin E or cyclin A expression vectors were generated by injecting corresponding pPD49.78- and pPD49.83-derived constructs (50 μg/μL each) along with sur-5::GFP (100 μg/μL) into unc-4, fzr-1 animals to obtain multiple stable extrachromosomal lines (cyclin A, fdEx1 and fdEx2; cyclin E, fdEx6 and fdEx7). Crossing to N2 generated Ex+ lines in the wild-type background.
Acknowledgments
We thank the following people: Andy Fire for vectors; Yuji Kohara, Mike Krause, Paul Sternberg, and Karen Bennett for cDNAs and other clones; Joel Rothman, Eric Jorgensen, David Greenstein, Judith Kimble, and Jim McGhee for strains; Mike Krause, Ed Kipreos, Tin Tin Su, and Ann Rose for helpful discussions; Dan Starr and Amy Fluet for comments on the manuscript; and Scott Kelly for help with the phylogentic comparison. Some of the strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Center for Research Resources of the NIH. D.S.F. was supported by an NRSA fellowship (F32GM20029-01). M.H. is an HHMI investigator. This work was supported by NIH grant GM37869.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL davidfay@uwyo.edu; FAX (307) 766-5098.
E-MAIL mhan@colorado.edu; FAX (303) 735-0175.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.952302.
References
- Blelloch R, Anna-Arriola SS, Gao D, Li Y, Hodgkin J, Kimble J. The gon-1 gene is required for gonadal morphogenesis in Caenorhabditis elegans. Dev Biol. 1999;216:382–393. doi: 10.1006/dbio.1999.9491. [DOI] [PubMed] [Google Scholar]
- Boxem M, van den Heuvel S. lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development. 2001;128:4349–4359. doi: 10.1242/dev.128.21.4349. [DOI] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Ceol CJ, Horvitz HR. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol Cell. 2001;7:461–473. doi: 10.1016/s1097-2765(01)00194-0. [DOI] [PubMed] [Google Scholar]
- Chen Z, Han M. C. elegans Rb, NuRD and Ras regulate lin-39-mediated cell fusion during vulval fates specification. Curr Biol. 2001;11:1874–1879. doi: 10.1016/s0960-9822(01)00596-6. [DOI] [PubMed] [Google Scholar]
- Clark SG, Lu X, Horvitz HR. The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics. 1994;137:987–997. doi: 10.1093/genetics/137.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328–330. doi: 10.1038/359328a0. [DOI] [PubMed] [Google Scholar]
- Classon M, Dyson N. p107 and p130: Versatile proteins with interesting pockets. Exp Cell Res. 2001;264:135–147. doi: 10.1006/excr.2000.5135. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes & Dev. 1996;10:3081–3093. doi: 10.1101/gad.10.24.3081. [DOI] [PubMed] [Google Scholar]
- Culotti JG, Von Ehrenstein G, Culotti MR, Russell RL. A second class of acetylcholinesterase-deficient mutants of the nematode Caenorhabditis elegans. Genetics. 1981;97:281–305. doi: 10.1093/genetics/97.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AG, Spike CA, Shaw JE, Herman RK. Functional overlap between the mec-8 gene and five sym genes in Caenorhabditis elegans. Genetics. 1999;153:117–134. doi: 10.1093/genetics/153.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson IA, Roth S, Artavanis-Tsakonas S. The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces cerevisiae. J Cell Biol. 1995;129:725–737. doi: 10.1083/jcb.129.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol. 1995;15:4215–4224. doi: 10.1128/mcb.15.8.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Nooij JC, Letendre MA, Hariharan IK. A cyclin-dependent kinase inhibitor, Dacapo, is necessary for timely exit from the cell cycle during Drosophila embryogenesis. Cell. 1996;87:1237–1247. doi: 10.1016/s0092-8674(00)81819-x. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;115:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- Dou QP, Zhao S, Levin AH, Wang J, Helin K, Pardee AB. G1/S-regulated E2F-containing protein complexes bind to the mouse thymidine kinase gene promoter. J Biol Chem. 1994;269:1306–1313. [PubMed] [Google Scholar]
- Du W, Dyson N. The role of RBF in the introduction of G1 regulation during Drosophila embryogenesis. EMBO J. 1999;18:916–925. doi: 10.1093/emboj/18.4.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, O'Farrell PH. Developmental control of the G1 to S transition in Drosophila: Cyclin E is a limiting downstream target of E2F. Genes & Dev. 1995;9:1456–1468. doi: 10.1101/gad.9.12.1456. [DOI] [PubMed] [Google Scholar]
- Dyson N. The regulation of E2F by pRB-family proteins. Genes & Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- Fay DS, Han M. The synthetic multivulval genes of C. elegans: Functional redundancy, Ras-antagonism, and cell fate determination. Genesis. 2000;26:279–284. doi: 10.1002/(sici)1526-968x(200004)26:4<279::aid-gene100>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Ferguson EL, Horvitz HR. The multivulva phenotype of certain Caenorhabditis elegans mutants results from defects in two functionally redundant pathways. Genetics. 1989;123:109–121. doi: 10.1093/genetics/123.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson EL, Sternberg PW, Horvitz HR. A genetic pathway for the specification of the vulval cell lineages of Caenorhabditis elegans. Nature. 1987;326:259–267. doi: 10.1038/326259a0. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Flemington EK, Speck SH, Kaelin WG., Jr E2F-1-mediated transactivation is inhibited by complex formation with the retinoblastoma susceptibility gene product. Proc Natl Acad Sci. 1993;90:6914–6918. doi: 10.1073/pnas.90.15.6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, Barton MK, Kimble J, Schedl T. gld-1, a tumor suppressor gene required for oocyte development in Caenorhabditis elegans. Genetics. 1995;139:579–606. doi: 10.1093/genetics/139.2.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- Fukushige T, Hawkins MG, McGhee JD. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol. 1998;198:286–302. [PubMed] [Google Scholar]
- Gu T, Orita S, Han M. Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol Cell Biol. 1998;18:4556–4564. doi: 10.1128/mcb.18.8.4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Dean DC. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes & Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, White JG. Polyploid tissues in the nematode Caenorhabditis elegans. Dev Biol. 1985;107:128–133. doi: 10.1016/0012-1606(85)90381-1. [DOI] [PubMed] [Google Scholar]
- Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O, D'Alberti T, Liu Y, Ruvkun G. Control of neural development and function in a thermoregulatory network by the LIM homeobox gene lin-11. J Neurosci. 1998;18:2084–2096. doi: 10.1523/JNEUROSCI.18-06-02084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J. What does a worm need with 20,000 genes? Genome Biol. 2001;2:2008.1–2008.4. doi: 10.1186/gb-2001-2-11-comment2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Roy R, Ambros V. Developmental regulation of a cyclin-dependent kinase inhibitor controls postembryonic cell cycle progression in Caenorhabditis elegans. Development. 1998;125:3585–3597. doi: 10.1242/dev.125.18.3585. [DOI] [PubMed] [Google Scholar]
- Huang LS, Tzou P, Sternberg PW. The lin-15 locus encodes two negative regulators of Caenorhabditis elegans vulval development. Mol Biol Cell. 1994;5:395–411. doi: 10.1091/mbc.5.4.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irniger S, Nasmyth K. The anaphase-promoting complex is required in G1 arrested yeast cells to inhibit B-type cyclin accumulation and to prevent uncontrolled entry into S-phase. J Cell Sci. 1997;110:1523–1531. doi: 10.1242/jcs.110.13.1523. [DOI] [PubMed] [Google Scholar]
- Irniger S, Piatti S, Michaelis C, Nasmyth K. Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell. 1995;81:269–278. doi: 10.1016/0092-8674(95)90337-2. [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- Johnson CD, Duckett JG, Culotti JG, Herman RK, Meneely PM, Russell RL. An acetylcholinesterase-deficient mutant of the nematode Caenorhabditis elegans. Genetics. 1981;97:261–279. doi: 10.1093/genetics/97.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr Functions of the retinoblastoma protein. Bioessays. 1999;21:950–958. doi: 10.1002/(SICI)1521-1878(199911)21:11<950::AID-BIES7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell. 1995;81:279–288. doi: 10.1016/0092-8674(95)90338-0. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Lander LE, Wing JP, He WW, Hedgecock EM. cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell. 1996;85:829–839. doi: 10.1016/s0092-8674(00)81267-2. [DOI] [PubMed] [Google Scholar]
- Kipreos ET, Gohel SP, Hedgecock EM. The C. elegans F-box/WD-repeat protein LIN-23 functions to limit cell division during development. Development. 2000;127:5071–5082. doi: 10.1242/dev.127.23.5071. [DOI] [PubMed] [Google Scholar]
- Krause M, Hirsh D. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell. 1987;49:753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999;18:7873–7882. doi: 10.1038/sj.onc.1203244. [DOI] [PubMed] [Google Scholar]
- Lu X, Horvitz HR. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell. 1998;95:981–991. doi: 10.1016/s0092-8674(00)81722-5. [DOI] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Morgan DO. Regulation of the APC and the exit from mitosis. Nat Cell Biol. 1999;1:E47–E53. doi: 10.1038/10039. [DOI] [PubMed] [Google Scholar]
- Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci. 1995;92:12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Krause MW. Regulation of postembryonic G(1) cell cycle progression in Caenorhabditis elegans by a cyclin D/CDK-like complex. Development. 1999;126:4849–4860. doi: 10.1242/dev.126.21.4849. [DOI] [PubMed] [Google Scholar]
- Schulze A, Zerfass K, Spitkovsky D, Middendorp S, Berges J, Helin K, Jansen-Durr P, Henglein B. Cell cycle regulation of the cyclin A gene promoter is mediated by a variant E2F site. Proc Natl Acad Sci. 1995;92:11264–11268. doi: 10.1073/pnas.92.24.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, Seufert W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell. 1997;90:683–693. doi: 10.1016/s0092-8674(00)80529-2. [DOI] [PubMed] [Google Scholar]
- Shi Y, Mello C. A CBP/p300 homolog specifies multiple differentiation pathways in Caenorhabditis elegans. Genes & Dev. 1998;12:943–955. doi: 10.1101/gad.12.7.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist SJ, Lehner CF. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell. 1997;90:671–681. doi: 10.1016/s0092-8674(00)80528-0. [DOI] [PubMed] [Google Scholar]
- Sigrist S, Jacobs H, Stratmann R, Lehner CF. Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J. 1995;14:4827–4838. doi: 10.1002/j.1460-2075.1995.tb00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith V, Chou KN, Lashkari D, Botstein D, Brown PO. Functional analysis of the genes of yeast chromosome V by genetic footprinting. Science. 1996;274:2069–2074. doi: 10.1126/science.274.5295.2069. [DOI] [PubMed] [Google Scholar]
- Solari F, Ahringer J. NURD-complex genes antagonise Ras-induced vulval development in Caenorhabditis elegans. Curr Biol. 2000;10:223–226. doi: 10.1016/s0960-9822(00)00343-2. [DOI] [PubMed] [Google Scholar]
- Sprenger F, Yakubovich N, O'Farrell PH. S-phase function of Drosophila cyclin A and its downregulation in G1 phase. Curr Biol. 1997;7:488–499. doi: 10.1016/s0960-9822(06)00220-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell. 1995;6:185–197. doi: 10.1091/mbc.6.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston, J.E. and Hodgkin, J. 1988. Methods. In The nematode Caenorhabditis elegans (eds. W.B. Wood and the Community of C. elegans Researchers), pp. 587–606. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Swafford DL. PAUP phylogenetic analysis using parsimony (& other methods) 2000. version 4. Sinauer Associates, Sunderland, MA. [Google Scholar]
- Tautz D. A genetic uncertainty problem. Trends Genet. 2000;16:475–477. doi: 10.1016/s0168-9525(00)02118-1. [DOI] [PubMed] [Google Scholar]
- Terns RM, Kroll-Conner P, Zhu J, Chung S, Rothman JH. A deficiency screen for zygotic loci required for establishment and patterning of the epidermis in Caenorhabditis elegans. Genetics. 1997;146:185–206. doi: 10.1093/genetics/146.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH, Horvitz HR. The C. elegans gene lin-36 acts cell autonomously in the lin-35 Rb pathway. Development. 1999;126:3449–3459. doi: 10.1242/dev.126.15.3449. [DOI] [PubMed] [Google Scholar]
- Visintin R, Prinz S, Amon A. CDC20 and CDH1: A family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- von Zelewsky T, Palladino F, Brunschwig K, Tobler H, Hajnal A, Muller F. The C. elegans Mi-2 chromatin-remodelling proteins function in vulval cell fate determination. Development. 2000;127:5277–5284. doi: 10.1242/dev.127.24.5277. [DOI] [PubMed] [Google Scholar]
- Wagner A. Robustness against mutations in genetic networks of yeast. Nat Genet. 2000;24:355–361. doi: 10.1038/74174. [DOI] [PubMed] [Google Scholar]
- Weintraub SJ, Chow KN, Luo RX, Zhang SH, He S, Dean DC. Mechanism of active transcriptional repression by the retinoblastoma protein. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Nasmyth K. Whose end is destruction: Cell division and the anaphase-promoting complex. Genes & Dev. 1999;13:2039–2058. doi: 10.1101/gad.13.16.2039. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- Zheng L, Lee WH. The retinoblastoma gene: A prototypic and multifunctional tumor suppressor. Exp Cell Res. 2001;264:2–18. doi: 10.1006/excr.2000.5129. [DOI] [PubMed] [Google Scholar]
- Zur A, Brandeis M. Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis. EMBO J. 2001;20:792–801. doi: 10.1093/emboj/20.4.792. [DOI] [PMC free article] [PubMed] [Google Scholar]