

Abstract
Protein–protein interactions play pivotal roles in various aspects of the structural and functional organization of the cell, and their complete description is indispensable to thorough understanding of the cell. As an approach toward this goal, here we report a comprehensive system to examine two-hybrid interactions in all of the possible combinations between proteins of Saccharomyces cerevisiae. We cloned all of the yeast ORFs individually as a DNA-binding domain fusion (“bait”) in a MATa strain and as an activation domain fusion (“prey”) in a MATα strain, and subsequently divided them into pools, each containing 96 clones. These bait and prey clone pools were systematically mated with each other, and the transformants were subjected to strict selection for the activation of three reporter genes followed by sequence tagging. Our initial examination of ≈4 × 106 different combinations, constituting ≈10% of the total to be tested, has revealed 183 independent two-hybrid interactions, more than half of which are entirely novel. Notably, the obtained binary data allow us to extract more complex interaction networks, including the one that may explain a currently unsolved mechanism for the connection between distinct steps of vesicular transport. The approach described here thus will provide many leads for integration of various cellular functions and serve as a major driving force in the completion of the protein–protein interaction map.
It is well recognized that protein–protein interactions play key roles in structural and functional organization of the cell. Uncovering of protein interaction schemes often sheds light on molecular mechanisms underlying biological processes. Hence increasing attention is being paid to protein–protein interactions in both structural and functional studies. Although such studies are intensively conducted on the proteins of interest to a wide audience of researchers, genomes harbor a number of novel proteins currently lacking any hint as to their specific functions. For such novel proteins, interactions with known proteins serve as invaluable clues to their functions or biological roles. These interactions also suggest directions one may take for more detailed phenotypic examination of mutants for the novel genes. Organisms with completely sequenced genomes are quite suitable for such studies, because all of the proteins can be predicted and used for the comprehensive examination of protein–protein interactions. Such genome-wide interaction mapping would be a novel type of genomic data and strongly accelerate the comprehensive understanding of the cell as a molecular machinery. Therefore, the study of protein–protein interactions is one of the most important issues in functional genomics.
It also should be emphasized that recent studies of protein–protein interactions, in particular, those involved in signal transduction, uncovered a number of protein-binding domains or motifs, which are evolutionarily conserved and used in various signaling pathways (1). Thus, the identification of novel protein interaction modules contributes not only to the studies of the particular pathway but also to much wider fields of biomedical research. A large set of protein–protein interaction data would lay a foundation for the search of such modules by both experimental and computational means.
Despite the need for comprehensive studies on protein–protein interactions, no efforts for a genome-wide scale screening have ever been reported, although pioneering works using the yeast two-hybrid system (2) were reported on Drosophila cell cycle regulators (3), proteins of T7 phage (4), and yeast proteins involved in mRNA splicing (5).
We have launched a systematic identification of mutually interacting protein pairs in the budding yeast Saccharomyces cerevisiae. We use the budding yeast, because its genome is completely sequenced, and all of the ORFs have been predicted. In addition, a huge amount of knowledge in molecular cell biology, genetics, and biochemistry is accumulated for this simple eukaryotic organism to help evaluate the biological relevance of the identified interactions. It also should be noted that the ease of genome modification in this yeast enables one to create mutants defective for each interaction to examine its biological role.
To examine all of the possible protein–protein interactions, we established a comprehensive two-hybrid screening system, in which all yeast ORFs are cloned as both bait and prey, pooled in sets of 96 clones, and used for screening by systematic mating. After strict selection to minimize the background signals, the interacting proteins are decoded by sequence tagging of the plasmid inserts. Characterization of the positive clones obtained in the initial phase of the experiments, where ≈4 × 106 different combinations (≈10% of all possible ones) were tested, clearly demonstrated that the approach is feasible and useful for rapid collection of candidates for novel interactions. This system could serve as a prototype for the deciphering of entire protein–protein interaction networks within the cell.
Materials and Methods
PCR Amplification of Yeast ORFs.
PCR primers for each ORF (6) were purchased from Research Genetics (Huntsville, AL) and used for initial amplification from yeast genomic DNA. Amplified fragments were subjected to secondary PCR with common primers to add rare-cutter sites to both ends of each ORF, digested with the enzymes, and used for the cloning into the two-hybrid vectors described below. To minimize potential misincorporation during PCR, the high-fidelity enzyme, Pfu DNA polymerase (Stratagene), was used.
Construction of Two-Hybrid Vectors.
For a GAL4 activation domain-fusion vector, we constructed pGAD-RC by inserting a multiple cloning site (shown below) between the BamHI site and the PstI site of pGAD424g (7).
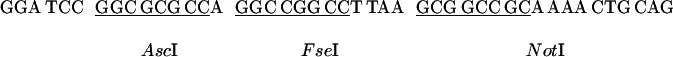 |
Similarly, a GAL4 DNA-binding domain fusion vector pGBK-RC was constructed by insertion of the multiple cloning site between the BamHI and PstI sites of pGBK, which carries the Kanr gene for efficient separation from pGAD marked with Ampr (7). These vectors allow in-frame cloning of all of the amplified ORF fragments digested with AscI or FseI and NotI or Sse8387I.
Two-Hybrid Strains.
Two strains were used in this system. One was PJ69–2A (MATa,trp1–901,leu2–3,112,ura3–52,his3Δ200, gal4Δ,gal80Δ,GAL2∷ADE2,GAL1∷HIS3), which was purchased from CLONTECH. The other strain was MaV204K (MATα,trp1–901,leu2–3,112,his3Δ200,ade2–101Δ∷kanMX,gal4Δ, gal80Δ,SPAL10∷URA3,UASGAL1∷HIS3,GAL1∷lacZ), which was constructed from MaV203 (8) by deleting ADE2 using the kanMX cassette (9). Although MaV203 is genetically ade2, it shows Ade+ phenotype for unknown reasons. Thus, we deleted for ADE2 and confirmed Ade− phenotype of the deletant, MaV204K.
Transformation and Construction of Screening Pools.
The two-hybrid strains were cultured overnight in YPAD (1% yeast extract, 2% peptone, 0.004% adenine sulfate, 2% glucose) media (10), and competent cells were prepared as described (11), except for the use of 10% DMSO. The suspended competent cells (24 μl) were added to plasmid DNA (1 μl) dispensed in each well of a 96-well PCR plate, and thoroughly mixed by pipetting. After incubation at 42°C for 20 min, an aliquot of each PJ69–2A transformant, bearing a defined pGBK plasmid marked with TRP1, was spotted onto agar plates of the synthetic complete (SC) media (10) lacking Trp (SC-Trp) and SC-Trp, -Ade, -His. Similarly prepared MaV204K clones bearing pGAD plasmids with LEU2 marker were spotted onto SC-Leu plates and SC-Leu, -His plates supplemented with 10 mM 3-aminotriazole. These plates were incubated for ≈14 days to screen out clones that autonomously activate the reporter genes. For the construction of the screening pool, an aliquot of each transformation was directly inoculated into a well of flat-bottom 96-well plates filled with 150 μl of SC-Trp or SC-Leu for pGBK-bearing PJ69-2A or pGAD-bearing MaV204K clones, respectively. After incubation at 30°C for several days, colonies formed on the well bottoms were resuspended by pipetting. Aliquots (≈50 μl) of each suspension were mixed with equal amounts of 30% glycerol and stored at −80°C. Another aliquot of each suspension was collected into a single flask containing YPAD media, which was shaken for ≈4 hr at 30°C. The yeast cells were collected by centrifugation, resuspended in 10 ml of YPAD containing 15% glycerol, and divided into 96 tubes, each containing 100 μl of pooled yeast. These screening pools were stored at −80°C until use.
Screening by Mating.
A bait pool and a prey pool were thawed, mixed, and collected onto a Millipore HA filter by filtration. The filter was placed on a prewetted YPAD (1% yeast extract, 2% peptone, 0.004% adenine sulfate, 2% glucose) agar plate. After incubation at 30°C for 5 hr, the filter was washed extensively in sterile water by vortexing. The yeast cells were collected by centrifugation, resuspended in an appropriate volume of water, and spread onto SC-Trp, -Leu, -His, -Ura, -Ade plates supplemented with 10 mM 3-aminotriazole and 200 μg/ml Geneticin (G418). An aliquot of the cells was spread onto an SC-Trp, -Leu to score the number of diploid cells. After incubation at 30°C for 1≈2 weeks, positive colonies were restreaked onto a new SC-Trp, -Leu, -His, -Ura, -Ade plate supplemented with 10 mM 3-aminotriazole and 5-bromo-4-chloro-3-indolyl-α-d-galactopyranoside (CLONTECH).
DNA Sequencing.
Yeast cells were suspended in water containing Zymolyase (Seikagaku Kogyo, Tokyo) and incubated at 37°C for 30 min followed by heating at 94°C for 10 min. An aliquot of the suspension was used for PCR amplification of the plasmid insert. The PCR product was treated with exonuclease I and shrimp alkaline phosphatase (Amersham Pharmacia) and subjected to cycle sequencing. Obtained sequences were sent to the blast server at the Saccharomyces Genome Database (12) to be analyzed.
Results
Design of the System for Comprehensive Two-Hybrid Screening.
The outline of our screening system is shown in Fig. 1. To establish this system, we developed a modified two-hybrid strain and vectors, GAL4 DNA-binding domain fusion or “bait” clone pools and GAL4 activation domain fusion or “prey” clone pools.
Figure 1.

Outline of the strategy for comprehensive two-hybrid screening for mutually interacting proteins. The genotypes of the two strains used in the system are also shown. See text for details.
In this project, we used two two-hybrid strains, PJ69–2A and MaV204K. The PJ69–2A strain was used for DNA-binding domain fusions (baits). This strain harbors an ADE2 reporter gene driven by the GAL2 promoter, which was shown to give minimum false positives (13), in addition to the conventional HIS3 reporter. The MaV204K strain for activation domain fusions (preys) was constructed by deleting ADE2 from MaV203 (8) using the kanMX cassette, which confers G418 resistance. This strain harbors a unique URA3 reporter, in addition to the conventional HIS3 and lacZ reporters, for the so-called reverse two-hybrid selection (8). Importantly, PJ69–2A and MaV204K have opposite mating types so that they can be mated with each other to perform two-hybrid assays in diploid cells. Because MaV204K harbors the kanMX cassette, we can add G418 to the selection plates for the diploids to substantially suppress the growth of contaminating fungi, which inevitably happens when handling a large number of plates.
To clone all of the ORFs in the budding yeast genome, we amplified each of them by a two-step procedure, in which PCR first is performed with ORF-specific primers and then with common primers to attach eight-base cutter sites to both ends of the amplified fragment. To minimize potential misincorporation during PCR, we used a high-fidelity enzyme, Pfu DNA polymerase, throughout the process. Amplified products were digested with appropriate eight-base cutters and cloned into a GAL4 DNA-binding domain fusion vector, pGBK-RC, and a GAL4 activation domain fusion vector, pGAD-RC, whose multiple cloning sites also contain the rare-cutter sites.
The plasmids isolated from Escherichia coli transformants were introduced into the two yeast strains by using a simple transformation procedure, which we developed for the 96-well plate format to facilitate the process. After the removal of transformants, which autonomously activated the reporter genes in the absence of interacting partners, clones in each plate were pooled and divided into aliquots to be used for multiple rounds of screening. So far, we have prepared 60 pools for both bait and prey clones, thereby covering ≈92% of the ORFs. The remaining ORFs, which were refractory to cloning, are being added to the collection.
For each screening, one bait pool and one prey pool were thawed, mixed, and collected onto a filter membrane by aspiration. After incubation to allow the yeasts to mate, the cells were collected and spread onto a plate selecting for the activation of ADE2, HIS3, and URA3 reporter genes. As we confirmed that more than 105 diploids were generated, each screening well covered 96 × 96 combinations. Transformants were restreaked onto a second selection plate supplemented with 5-bromo-4-chloro-3-indolyl-α-d-galatopyranoside to examine the activation of the endogenous MEL1 gene, which is a target gene for Gal4 transcription factor and hence functions as another reporter gene. Use of multiple reporter genes driven by different Gal4-responsive promoters was reported as critical in the elimination of false positives by fortuitous activation of a particular promoter (13).
The number of positive colonies was variable from one plate to the next. Although some plates gave no colonies at all, hundreds of survivors are found in others. In the latter case, it often happened that the same pGBK insert and variable pGAD inserts were recovered from the colonies, thereby indicating that some pGBK constructs in the pool can activate the reporter genes regardless of the partners. For such cases, we sequenced the pGBK insert to identify the problematic clone, which subsequently was removed from the bait pool. We then re-examined the “cleaned” pools by longer incubation to prove that they no longer harbor any problematic clones. Such careful elimination was crucial to avoid unnecessary sequencing, as was reported for the analysis of T7 phage proteins (4).
After confirmation of activation of at least three reporter genes, the yeast cells were subjected to colony PCR to amplify the cohabiting plasmid inserts. The obtained fragments were directly sequenced and subjected to blast analysis at the Saccharomyces Genome Database (12).
Feasibility Study of the Strategy.
As a pilot phase of this project, we analyzed 866 colonies obtained from 430 mating reactions, which should include ≈4 × 106 different combinations constituting ≈10% of the total permutations to be examined (Table 1). Of 866 colonies, we obtained 750 sets of sequence data. After the removal of repeatedly detected interactions, we operationally omitted proteins that interacted more than three unrelated partners without any supporting evidence for their biological relevance. As a result of this processing, 183 independent interactions were remained (Table 1 and Table 2, which is published as supplemental material on the PNAS web site, www.pnas.org), of which 16 were detected in both orientations. For instance, two-hybrid interactions were detected between pGAD-VMA7 and pGBK-VMA22 as well as pGAD-VMA22 and pGBK-VMA7. These two-hybrid interactions are synonymous, and hence we finally obtained 175 independent interactions. At the time of preparation of this manuscript, 12 of these interactions were classified as “known” interactions according to the descriptions in the Yeast Proteome Database (14) and the remaining 163 interactions were classified as “unknown” ones. These unknown interactions contain 32 homotypic interactions, which indicate that these proteins can dimerize or oligomerize.
Table 1.
Two-hybrid screening summary
| Mating reactions | 430 |
| Combinations to be examined | ∼4 × 106 |
| Positive colonies | 866 |
| Sequence tag pairs obtained | 750 |
| Independent two-hybrid interactions | 183 |
| Bidirectionally detected interactions | 16 |
| Total independent interactions | 175 |
| Known interactions | 12 |
| Previously unreported interactions | 163 |
| Highly likely | 26 |
| Homotypic | 32 |
| Novel | 105 |
It should be noted that the “unknown” category includes 26 interactions that seem to be biologically relevant or are “highly likely” ones. For example, Srp14 and Srp21 are both included in the signal recognition particle (SRP), although no direct evidence for the interaction between these two subunits has been substantiated. In addition to SRP, we detected two-hybrid interactions between the components of various protein complexes, including spindle pole body, ribosome, vacuolar H+-ATPase complex, EGD complex (nascent polypeptide-associated complex), TRAPP (transport particle protein) complex, spliceosome, exosome, TAF (II) complex, Arp2/3 actin-organizing complex, and small nuclear ribonucleoproteins. These data would contribute to the molecular dissection of such large protein complexes. In addition to these complexes, two-hybrid interactions were detected between proteins involved in the same specific biological process, such as autophagy and sporulation, suggesting the biological relevance of these interactions. This “highly likely” category also involves interactions between the subunits of enzymes, including RNA polymerase I and C-terminal domain kinase, as well as those between enzymes and substrates, such as ubiquitin-conjugating enzyme Ubc12 and ubiquitin-like protein Rub1.
The rest of the interactions are classified as “novel” ones. No strong evidence is currently available for their biological relevance. However, some of them consist of proteins that are both involved in the same cellular process in a broad sense of the phrase. For instance, both Yip1 and Tlg1 are involved in vesicular transport. These may reflect currently unreported but significant interactions.
The interactions between known proteins and those of unknown function would provide an invaluable hint in the search of functions for the latter. In fact, we could propose the function of Ybr254c. Because we found that Bet3 interacts with Ybr254c as well as Trs31, a protein of the TRAPP complex, we assumed that Ybr254c also may be involved in TRAPP complex function. Indeed, Ybr254c recently was shown identical with the 20K component of the TRAPP complex (15). Note that 50 of the 175 interactions detected are those between a known protein and a protein of unknown function. Some of these interaction also may play a role in the functional analysis of the proteins of unknown functions.
Complex Interaction Schemes Extracted from the Binary Interaction Data.
Careful analysis of the binary interactions obtained above revealed more complex protein interaction schemes or networks. So far, we have detected 16 networks composed of more than four proteins. An example of such two-hybrid interactions is found around Spc19 and Spc34, both of which are components of the spindle pole body (16) (Fig. 2A). Intriguingly, temperature-sensitive mutations and overexpression of DUO1 were reported to cause an arrest with abnormal microtubule organization, and its product Duo1 interacts with Dam1, another protein required for maintenance of spindle integrity during mitosis (17). These pieces of circumstantial evidence support that the interaction between Spc34 and Duo1 is biologically relevant. Note that Ydr016c, a protein of unknown function, shows two-hybrid interactions with two proteins related to the spindle pole body, namely Spc34 and Duo1. It thus seems quite reasonable to examine a role for Ydr016c in microtubule function.
Figure 2.
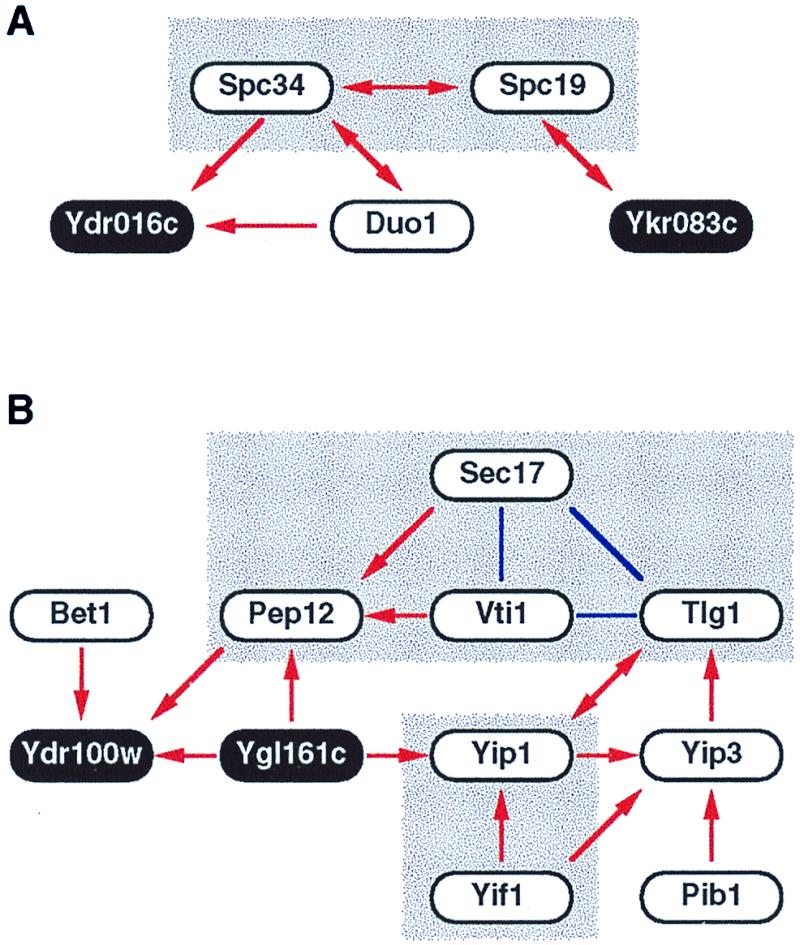
Complex two-hybrid interaction networks. Two-hybrid interaction networks for proteins related to spindle pole body (A) and vesicular transport (B) are shown. Red arrows indicate two-hybrid interactions, beginning from the bait and ending at the prey. Double-headed arrows mean that the interactions were detected bidirectionally. Note that arrows indicate the direction of two-hybrid interactions but not any biological orientation. Solid blue lines indicate known interactions recorded in the Yeast Proteome Database (14) but not yet detected by our two-hybrid screening. Shaded boxes are complexes or networks that were described in previous studies. Proteins of unknown function are indicated as black circles with white letters.
A more impressive two-hybrid interaction network is shown in Fig. 2B. Notably, all the known proteins in this network are implicated in the membrane fusion process of vesicular transport. This process requires GTPase Ypt, integral membrane soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptors (SNAREs), and Sec18/NSF with its accessory protein Sec17/SNAP. The GTPase Ypt1 currently is thought to tether endoplasmic reticulum-derived vesicles to Golgi (18), and Yip1 is a Golgi membrane protein implicated in the recruiting of Ypt1 to this organelle (19). [Yip3 is another Ypt-binding protein, whereas Yif1 is a Yip-interacting factor. Pib1 harbors a RING FYVE domain that interacts with phosphatidylinositol 3-phosphate and also plays a role in vesicular transport (20).] After the “tethering” by the GTPase Ypt, an unknown mechanism induces “docking” between vesicle-bound (v-) and target membrane (t-) SNAREs, the former of which includes Bet1 and Vti1 and the latter Pep12 and Tlg1. The docking between v- and t-SNAREs subsequently is disassembled by the action of Sec18 and Sec17 (21). Therefore, all of the known proteins participating in this two-hybrid interaction network are functionally related to one another in the membrane fusion process. Moreover, the network may serve as a tempting molecular model for functional connection between the two distinct steps of vesicular transport; Yip1, with its associated proteins, interacts with both Ypt and SNAREs, thereby transmitting some signal from the former to the latter to link the tethering step to the docking step. This hypothesis is a testable one, which we think is worth further pursuit. It is also conceivable that the two proteins of unknown function, Ygl161c and Ydr100w, play some roles in signaling from Ypt to SNAREs. It is reasonable to examine the phenotypes of deletants for these novel genes by putting particular emphasis on the defects in vesicular transport.
Discussion
We established a system for the examination of all of the possible two-hybrid interactions between the budding yeast proteins (Fig. 1). Pioneering works in much smaller scales have been reported on Drosophila cell cycle regulators (3), proteins of T7 phage (4), and yeast proteins involved in mRNA splicing (5). Although others also cloned all of the yeast ORFs as activation domain fusions to be used for two-hybrid screening (6), our approach is more comprehensive in that every ORF is cloned not only as prey but also as bait, thereby providing a unique resource for genome-wide hunting of protein–protein interactions. The bait pools can be used for the screening of full-length prey collection of ours and others (6) as well as conventional two-hybrid libraries containing variously truncated ORFs. The prey collection is also useful for the detection of interactions occurring at the N-terminal end portions, which are the regions inevitably underrepresented in conventional cDNA and genomic two-hybrid libraries.
Our initial experiment with these resources is the comprehensive examination of interactions between bait and prey both in full-length forms. The pilot phase described in this report appears promising; it already has revealed a number of (candidates for) interesting interactions (supplemental Table 2), and, in some cases, complex interaction networks, which may be of novel biological significance (Fig. 2).
Of course, the two-hybrid system has many pitfalls. First, any two-hybrid system inevitably suffers from false positives. We thus use four reporter genes, namely ADE2, HIS3, URA3, and MEL1, driven by different Gal4-responsive promoters to minimize false positives because of fortuitous activation of a particular promoter. Nevertheless, the data have to be carefully evaluated. For this, integration of these two-hybrid data with those of genetic interactions, subcellular localizations, and expression profiles would be quite useful, at least, in the elimination of apparently meaningless ones.
On the other hand, it should be noted that many interactions escape the screening (i.e., false negatives). Because the ORFs used are PCR products, it is inevitable that some populations bear mutations that may well abrogate the interactions with other proteins because of misincorporation during PCR. To minimize the frequency of such false negatives, we used a high-fidelity DNA polymerase for amplification and are examining all of the interactions in both directions (i.e., pGAD-X vs. pGBK-Y, pGAD-Y vs. pGBK-X). Another problem causing false negatives inherent to two-hybrid assay is the masking of interactions. It often happens that two-hybrid interaction is hardly detectable between two full-length proteins but becomes significantly stronger when bait and/or prey are appropriately trimmed. We are thus planning to use the bait pools for the screening of conventional genomic libraries to unmask such interactions in the second phase of our project. Also, some proteins would not interact with their partners unless they are first activated. This is well exemplified in the case of small GTPases, which show much stronger interaction with effector proteins in their activated or GTP-bound forms. In addition, because two-hybrid interactions are assumed to occur in the nucleoplasm, membrane proteins would be misfolded and show no meaningful interactions, although some membrane proteins did show biologically relevant two-hybrid interactions. Introduction of another two-hybrid system for the interaction between membrane proteins (22) may be necessary to overcome this problem.
Finally, one should bear in mind that two-hybrid interactions do not always indicate direct binding of bait and prey. A certain fraction of two-hybrid interactions detected between two yeast proteins are mediated by third-party endogenous yeast proteins that link the bait to the prey.
Nevertheless, this study clearly demonstrated that we can learn much from comprehensive two-hybrid screening. The 187 two-hybrid interactions, which were strictly selected for the activation of three independent reporter genes, were obtained from 430 mating reactions, presumably covering ≈4 × 106 different combinations. Because 4,225 mating reactions (65 pools × 65 pools) are required for the examination of all the possible combinations, we can roughly assume that ≈1,800 two-hybrid interactions will be finally obtained and that most of them are entirely novel. If one notes that only ≈600 two-hybrid interactions have so far been reported between the yeast proteins, the power of this comprehensive screening is obvious. To fully exploit such large two-hybrid data, they should be carefully curated as described above and finally integrated with the interaction data obtained by other means, including coprecipitation, in vitro binding assays, and more sophisticated methodologies (23). A comprehensive approach like ours will provide a core for such integration, thereby playing a major role in the completion of the protein–protein interaction map, which is indispensable to thorough understanding of the cell at the molecular level.
Can we apply a similar comprehensive approach to organisms with much larger genomes? Although the human genome contains some 100,000 genes, the number of those expressed in each cell or tissue is much smaller. If we assume we have to handle 30,000 genes, the number of combinations to be examined is 25 times larger than those in the yeast system. This will be an attainable goal with the aid of robotics and high throughput sequencers. Obstacles for the preparation of intact ORFs remarkably are being solved as in recent progress in full-length and long-insert cDNA cloning technologies (24–27). We thus believe that the system described here may well serve as a prototype for the comprehensive studies of protein–protein interactions in cells with much larger genomes such as those of our own.
Supplementary Material
Acknowledgments
We thank Haretsugu Hishigaki (Univ. of Tokyo) and Todd Taylor (RIKEN) for the analysis of two-hybrid interactions deposited in the databases and useful comments on the manuscript, respectively. This work was supported by research grants from the Ministry of Education, Science, Sports and Culture (MESSC) and Science and Technology Agency (STA) of Japan.
Abbreviations
- SC
synthetic complete
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
References
- 1.Pawson A J, editor. Protein Modules in Signal Transduction. Berlin: Springer; 1998. [Google Scholar]
- 2.Chien C T, Bartel P L, Sternglanz R, Fields S. Proc Natl Acad Sci USA. 1991;88:9578–9582. doi: 10.1073/pnas.88.21.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finley R L, Jr, Brent R. Proc Natl Acad Sci USA. 1994;91:12980–12984. doi: 10.1073/pnas.91.26.12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel P L, Roecklein J A, SenGupta D, Fields S. Nat Genet. 1996;12:72–77. doi: 10.1038/ng0196-72. [DOI] [PubMed] [Google Scholar]
- 5.Fromont-Racine M, Rain J C, Legrain P. Nat Genet. 1997;16:277–282. doi: 10.1038/ng0797-277. [DOI] [PubMed] [Google Scholar]
- 6.Hudson J R, Dawson E P, Rushing K L, Jackson C H, Lockshon D, Conover D, Lanciault C, Harris J R, Simmons S J, Rothstein R, Fields S. Genome Res. 1997;7:1169–1173. doi: 10.1101/gr.7.12.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito T, Nakamura R, Sumimoto H, Takeshige K, Sakaki Y. FEBS Lett. 1996;385:229–232. doi: 10.1016/0014-5793(96)00387-0. [DOI] [PubMed] [Google Scholar]
- 8.Vidal M, Brachmann R K, Fattaey A, Harlow E, Boeke J D. Proc Natl Acad Sci USA. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann J H. Nucleic Acids Res. 1996;24:2519–2524. doi: 10.1093/nar/24.13.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaiser C, Michaekis S, Mitchell A. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1994. [Google Scholar]
- 11.Gietz R D, Schiestl R H. Methods Mol Cell Biol. 1995;5:255–269. [Google Scholar]
- 12.Chervitz S A, Hester E T, Ball C A, Dolinski K, Dwight S S, Harris M A, Juvik G, Malekian A, Roberts S, Roe T, et al. Nucleic Acids Res. 1999;27:74–78. doi: 10.1093/nar/27.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James P, Halladay J, Craig E A. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodges P E, McKee A H Z, Davis B P, Payne W E, Garrels J I. Nucleic Acids Res. 1999;27:69–73. doi: 10.1093/nar/27.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sacher M, Jiang Y, Barrowman J, Scarpa A, Burston J, Zhang L, Schieltz D, Yates J R, Abeliovich H, Ferro-Novick S. EMBO J. 1998;17:2494–2503. doi: 10.1093/emboj/17.9.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wigge P A, Jensen O N, Holmes S, Soues S, Mann M, Kilmartin J V. J Cell Biol. 1998;141:967–977. doi: 10.1083/jcb.141.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hofmann C, Cheeseman I M, Goode B L, McDonald K L, Barnes G, Drubin D G. J Cell Biol. 1998;143:1029–1040. doi: 10.1083/jcb.143.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao X, Ballew N, Barlow C. EMBO J. 1998;17:2156–2165. doi: 10.1093/emboj/17.8.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang X P, Matern H T, Gallwitz D. EMBO J. 1998;17:4954–4963. doi: 10.1093/emboj/17.17.4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burd C G, Emr S D. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- 21.Ungermann C, Nichols B J, Pelham H R B, Wickner W. J Cell Biol. 1998;140:61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stagljar I, Korostensky C, Johnsson N, te Heesen S. Proc Natl Acad Sci USA. 1998;95:5187–5192. doi: 10.1073/pnas.95.9.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendelsohn A R, Brent R. Science. 1999;284:1948–1950. doi: 10.1126/science.284.5422.1948. [DOI] [PubMed] [Google Scholar]
- 24.Kato S, Shingo S, Oh S-W, Kim N-S, Umezawa Y, Abe N, Yokoyama-Kobayashi M, Aoki T. Gene. 1994;150:243–250. doi: 10.1016/0378-1119(94)90433-2. [DOI] [PubMed] [Google Scholar]
- 25.Carninci P, Kvam C, Kitamura A, Ohsumi T, Okazaki Y, Itoh M, Kamiya M, Shibata K, Sasaki N, Izawa M, et al. Genomics. 1996;37:327–336. doi: 10.1006/geno.1996.0567. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki Y, Yoshitomo K, Maruyama K, Sugano S. Gene. 1997;200:149–156. doi: 10.1016/s0378-1119(97)00411-3. [DOI] [PubMed] [Google Scholar]
- 27.Nomura N, Miyajima N, Sazuka T, Tanaka A, Kawarabayashi Y, Sato S, Nagase T, Seki N, Ishikawa K, Tabata S. DNA Res. 1994;1:27–35. doi: 10.1093/dnares/1.1.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.